

Abstract
Background. Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) is an inherited cardiac disease with reduced penetrance and a highly variable expression. Mutations in the gene encoding the plakophilin-2 gene (PKP2) are detected in about 50% of ARVC/D patients. The p.Arg79X mutation in PKP2 has been identified in Europe and North America and has been functionally characterised. We evaluated the prevalence of the p.Arg79X mutation in PKP2 in the Dutch population.
Methods. Twelve index patients and 41 family members were evaluated in three university hospitals in the Netherlands. The diagnosis of ARVC/D was established according to the recently revised Task Force Criteria. Segregation of the p.Arg79X mutation was studied and haplotypes were reconstructed to determine whether the p.Arg79X mutation was a recurrent or a founder mutation.
Results. The p.Arg79X mutation in PKP2 was identified in 12 index patients. Haplotype analysis revealed a shared haplotype among Dutch p.Arg79X mutation carriers, indicating a common founder. Six index patients (50%) had a first- or second-degree relative who had died of sudden cardiac death below 40 years of age. At age 60, only 60% of the mutation carriers had experienced any symptoms. There was no significant difference in symptom-free survival and event-free survival between men and women.
Conclusion. We have identified the largest series of patients with the same desmosome gene mutation in ARVC/D reported to date. This p.Arg79X mutation in PKP2 is a founder mutation in the Dutch population. The phenotypes of PKP2 p.Arg79X mutation carriers illustrate the clinical variability and reduced penetrance often seen in ARVC/D. (Neth Heart J 2010;18:583–91.)
Keywords: Cardiomyopathy, ARVC/D, Genetics, PKP2, Founder Mutation
Arrhythmogenic right ventricular cardiomyopathy/ dysplasia (ARVC/D) is an inherited cardiac disease, characterised by fibrofatty replacement of cardiomyocytes, primarily in the right ventricle (figure 1A).1,2 The clinical presentation of ARVC/D is highly variable, ranging from asymptomatic to sudden cardiac death (SCD) and/or heart failure, even at a relatively young age.3,4 The clinical diagnosis of ARVC/D is based upon criteria originally proposed by an international Task Force in 1994;5 these have recently been modified to improve diagnostic sensitivity and maintain diagnostic specificity.6 The modified Task Force Criteria (TFC) include quantitative parameters for the imaging studies and tissue characterisation, revised ECG criteria, and genetic status. Applying these modified TFC to a Dutch cohort of patients with proven ARVC/D, their family members, and to patients with probable ARVC/D, produced a major increase in the diagnostic yield of ARVC/D, especially due to the revised ECG criteria and the identification of pathogenic mutations.7 The estimated prevalence of ARVC/D ranges from 1:1000 to 1:5000, with men being more frequently affected than women. ARVC/D is familial in up to 50% of cases.1,8–12
Since the identification of mutations in the genes encoding the desmosomal proteins desmoplakin (DSP)13 and plakophilin-2 (PKP2),14–17 followed by mutations in desmocollin-2 (DSC2),18 desmoglein-2 (DSG2)19 and plakoglobin (JUP),20 it has been recognised that ARVC/D is mainly a disorder of the cardiac desmosome (figure 2), a cell adhesion complex residing in the intercalated disk of cardiomyocytes. Comprehensive screening of these genes encoding the proteins of this complex leads to the identification of a pathogenic mutation in approximately 40 to 60% of ARVC/D patients. 7,21-23 PKP2 has the highest yield from mutational screening, being present in up to 55% of patients in the Netherlands and the USA in groups fulfilling the TFC.7,14–17
Figure 1.
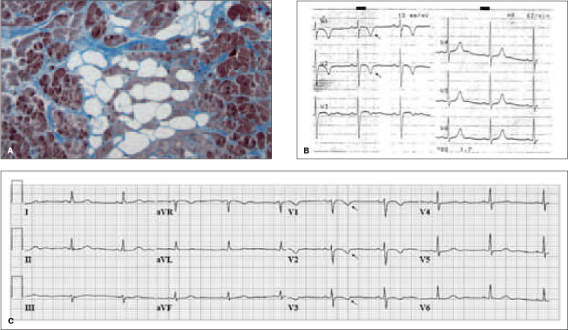
A) Histology: high-powered visualisation of fatty infiltration (white colour) and fibrous tissue (blue colour) surrounding atrophic cardiomyocytes and indicative of ARVC/D. B) ECG of patient G1-III:1, obtained 8 months before the occurrence of SCD at the age of 27 years. The ECG showed inverted T waves in leads V1 and V2 (single arrow). C) ECG of patient G1-II:1, obtained at age 56 years. The ECG showed inverted T waves in leads V1 to 3 (single arrow).
The detection of a pathogenic mutation in a proband diagnosed with ARVC/D has important implications for family members. Cascade screening will identify previously unknown mutation carriers, enabling timely diagnosis and facilitating prevention of subsequent complications, thereby reducing morbidity and mortality. Excluding a pathogenic mutation in a family member justifies dismissal from regular cardiological follow-up.24
To date, more than 140 different pathogenic mutations have been reported in ARVC/D patients. 25 In this series in the Netherlands Heart Journal on recurrent and founder mutations in the Netherlands, we describe the largest series of Dutch patients with the same desmosome gene mutation, the p.Arg79X mutation in PKP2. In addition, we discuss patients carrying this mutation by comparing them to patients with the same mutation described in the literature, and we illustrate the clinical variability linked to this mutation. We also discuss functional analyses that have been performed to clarify the molecular and cellular consequences of the p.Arg79X mutation in PKP2.
Patients and Methods
Clinical evaluation and diagnostic criteria
Twelve index patients carrying the p.Arg79X mutation were evaluated in one of the three university hospitals working on this study in the Netherlands. A total of 41 additional family members were also clinically and/or genetically evaluated. A history was taken from all the index patients, and they were evaluated by physical examination, 12-lead ECG, 24-hour Holter monitoring, exercise testing, and two-dimensional transthoracic echocardiography. In addition, MRI, nuclear scintigraphy, signal-averaged ECG (SA-ECG), left and right ventricular cineangiography, electrophysiology study, and/or a right ventricular endomyocardial biopsy were performed in a subset of index patients.
The diagnosis of ARVC/D in index patients was established according to the modified TFC.6 A diagnosis of ARVC/D was considered definite if a patient fulfilled two major, one major and two minor, or four minor criteria. For each category, only one major or one minor criterion can be included. A diagnosis of ARVC/D was considered probable if only one major and one minor, or three minor criteria from different categories were present. ARVC/D was considered proven familial if at least one additional family member was found to fulfil the TFC. Endpoints/events in follow-up were death due to a cardiac cause (including SCD) and an appropriate discharge of an implantable cardioverter defibrillator (ICD). These events, and ventricular tachycardia and syncope, were considered as symptoms for the Kaplan-Meier analysis.
The postal codes of all the p.Arg79X mutation carriers and of all the individuals who underwent PKP2 sequence analysis in the Netherlands were analysed to study their geographical distribution.
Figure 2.
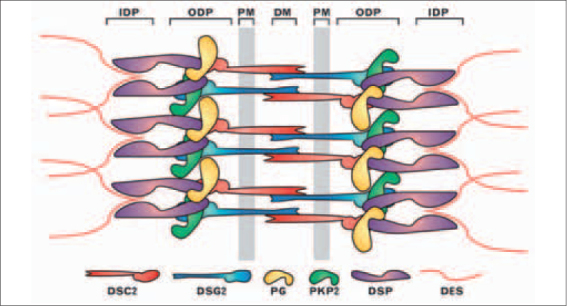
Schematic representation of the molecular organisation of cardiac desmosomes. A model of the relative organisation of major desmosome components is presented. The plasma membrane-bound (PM) desmocollin 2 (DSC2) and desmoglein 2 (DSG2) proteins interact via their extracellular domains at the dense midline (DM) in the extracellular space between adjacent cells. Their cytoplasmic domains interact with plakoglobin (PG) and plakophilin 2 (PKP2) in the outer dense plaque (ODP). Also in the ODP, PKP2 and plakoglobin interact with the N-terminal domain of desmoplakin (DSP). The C-terminus of desmoplakin anchors intermediate filaments, mainly desmin (DES), at the inner dense plaque (IDP).24
Genetic evaluation
DNA for PKP2 sequence analysis was isolated from peripheral blood samples according to standard protocols. PKP2 primer sequences were obtained from Gerull et al.,14 and mutational analysis was performed in all index patients, as described previously. 17 Sequence analysis of DSC2, DSG2, DSP and JUP was performed in six index patients, and in eight of twelve families we were able to study the segregation of the p.Arg79X mutation.
Haplotype analysis
To determine whether the p.Arg79X mutation is recurrent or has a common founder, we previously performed haplotype analysis in five p.Arg79X index patients, using five repeat markers within a region of 300,000 bp, including the entire genomic region of PKP2.17 For patients from the other seven families, the haplotypes were reconstructed using the same markers.
Statistical analysis
Kaplan-Meier analysis was used to determine the cumulative symptom-free and event-free survival in ARVC/D patients with the p.Arg79X mutation in PKP2. The log-rank test was used to compare the results for men versus women, with values of p<0.05 being considered significant. All data were analysed with the Statistical Package for Social Sciences (SPSS version 16.0; SPSS, Inc., Chicago, Ill).
Results
Clinical evaluation and diagnostic criteria
The age at initial presentation, diagnostic criteria, and follow-up of ARVC/D probands and their family members are presented in table 1 (see also figure 3). Eleven index patients were diagnosed with definite ARVC/D. In patient G1-III:1, retrospective analysis of the ECG after SCD at the age of 27 years, showed inverted T waves in V1 and V2 (figure 1B), which were also found in his mother’s ECG (G1-II:1, see figure 1C). One other index patient (A45-IV:8) was diagnosed with ARVC/D after SCD at the age of 25 years. Another index patient (A44-II:4) was diagnosed with probable ARVC/D.
Three of twelve index patients were women, with an average age at initial presentation of 37 years (22-66; median 29). Nine of the twelve index patients presented with ventricular tachycardia (VT), one with syncope and two were diagnosed at autopsy.
In six families, at least one relative of the index patient fulfilled the TFC for the diagnosis of ARVC/D. Six index patients (50%) had a first- or second-degree relative with SCD (range 15 to 55 years).
Figure 4 shows the symptom-free and event-free survival for p.Arg79X mutation carriers; there is no statistically significant difference between men and women. At age 40, only 32% of the mutation carriers (37% of men, 14% of women) had experienced any symptoms; at age 60 this number had increased to 60% (65% of men, 54% of women). Notably, cascade screening identified three mutation carriers aged 70 or over (two males aged 70 and 80, one female aged 87), who had never experienced any symptoms. Heart failure was not a prominent feature in the p.Arg79X mutation carriers.
Immunohistochemical analysis of plakoglobin, based on the use of immunoperoxidase as described by Asimaki et al.,26 was performed on myocardial samples of patient III:1 from family G39. When compared with that of a myocardial sample of a control heart, a strong decrease of plakoglobin expression at the desmosomes, in both the right and left ventricle, was detected.
Genetic evaluation
The c.235C>T mutation in exon 2 was identified in all 12 index patients, resulting at the protein level in a premature stop codon at position 79 (p.Arg79X). The p.Arg79X mutation occurs in 5% of all Dutch patients fulfilling the TFC (data not shown). The segregation of the p.Arg79X mutation was studied in eight families (figure 3); we found that all family members with definite or probable ARVC/D for whom DNA was available carried the mutation. So far, comprehensive analysis of all five desmosomal genes was performed in six index patients (G01, G06, A01, A04, A25, U2); no additional mutations were found in DSC2, DSG2, DSP and JUP in these patients.
Haplotype analysis
Haplotype analysis revealed a shared haplotype among all p.Arg79X mutation carriers (table 2). These data strongly suggest that the p.Arg79X mutation originated from a common founder rather than being recurrent. The geographical distribution of index patients carrying the p.Arg79X mutation in PKP2 and the number of PKP2 analyses performed in the Netherlands are shown in figure 5. The distribution of the index patients suggests that the mutation originated from the northern region of the Netherlands.
Table 1.
Clinical characteristics of ARVC/D probands and family members with the p.Arg79X mutatio
| Task force criteria for the diagnosis of ARVC/D | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family | Patient | p.Arg79X | Gender | Age at onset | Presentation | Structural alteration | Tissue characterisation | Abnormal repolarisation | Abnormal depolarisation | Arrhythmias | Family history | TFC (maj/min) | Follow-up, age |
| G01 | III:1|| | * | M | 27y | SCD | ++ | + | + | + | ++ | 2/3 || | SCD, 27y | |
| II:1 | * | F | 53y | Fam.scr. 48y | ++ | + | + | ++ | 2/2 | ||||
| II:3 | * | F | Fam.scr. 50y | ++ | ++ | 2/0 | |||||||
| II:4 | * | F | Fam.scr. 46y | ++ | 1/0 | ||||||||
| III:3 | * | M | Fam.scr. 29y | ++ | ++ | 2/0 | |||||||
| G06 | II:2 | * | M | 43y | VT | ++ | ++ | + | ++ | 3/1 | |||
| II:1 | † | F | 15y | SCD | SCD,15y | ||||||||
| II:3 | * | F | 40y | Fam.scr. 40y | ++ | + | ++ | 2/1 | |||||
| G39 | I:1 | † | M | 66y | VT | ++ | + | ++ | 2/1 | Death, 73y | |||
| II:3 | * | M | 65y | Fam.scr. 48y | + | ++ | ++ | 1/2 | ICD, 66y | ||||
| III:1 | * | M | 21y | SCD | ++ | ++ | ++ | 3/0 | SCD, 21y | ||||
| G40 | II:1 | * | M | 56y | VT | ++ | ++ | 2/0 | ICD, 57y | ||||
| G41 | II:3 | * | M | 28y | VT | ++ | + | ++ | 2/1 | ICD, 39y | |||
| II:2 | † | M | 29y | VT | + | ++ | 1/1 | ~ICD, 36y | |||||
| III:2 | * | M | Fam.scr. 06y | ++ | 1/0 | ||||||||
| A01 | III:2 | * | F | 40y | VT | ++ | ++ | + | ++ | ++ | 4/1 | ||
| II:2‡ | * | F | Fam.scr. 87y | ++ | 1/0 | ||||||||
| II:3‡ | * | M | Fam.scr. 80y | ++ | 1/0 | ||||||||
| A04 | III:2 | * | M | 29y | VT | ++ | ++ | ++ | 3/0 | ICD, 33y | |||
| II:3‡ | * | M | Fam.scr. 70y | ++ | 1/0 | ||||||||
| A25 | II:2 | * | M | 28y | VT | ++ | + | ++ | 1/2 | ICD, 28y | |||
| II:1 | * | F | 50y | Syncope | ++ | + | ++ | 2/1 | |||||
| II:3§ | † | M | Unknown | Death, 25y | |||||||||
| II:4 | * | M | Fam.scr. 54y | ++ | 1/0 | ||||||||
| III:1 | * | M | Fam.scr. 31y | ++ | 1/0 | ||||||||
| III:2‡ | * | M | Fam.scr. 28y | ++ | 1/0 | ||||||||
| III:6 | * | F | Fam.scr. 25y | ++ | 1/0 | ||||||||
| III:7 | * | M | Fam.scr. 19y | ++ | 1/0 | ||||||||
| A43 | II:1 | * | F | 55y | Syncope | ++ | ++ | + | ++ | 3/1 | |||
| III:2 | * | F | Fam.scr. 34y | ++ | 1/0 | ||||||||
| A44 | II:4 | * | M | 25y | VT | + | ++ | 1/1 | |||||
| I:2§ | * | F | Fam.scr. 84y | ++ | 1/0 | ||||||||
| II:1 | † | F | 29y | SCD | SCD, 29y | ||||||||
| A45 | IV:8 || | * | F | 25y | SCD | + | ++ | 1/1 || | SCD, 25y | ||||
| II:1 | † | F | 51y | SCD | SCD, 51y | ||||||||
| II:2 | † | M | 46y | SCD | SCD, 46y | ||||||||
| III:1 | † | M | 20y | SCD | SCD, 20y | ||||||||
| III:4 | * | F | Fam.scr. 80y | ++ | 1/0 | ||||||||
| III:8§ | * | F | Unknown | ++ | 1/0 | Death, 67y | |||||||
| III:9 | * | M | Fam.scr. 70y | + | ++ | 1/1 | |||||||
| III:10 | † | M | 55y | SCD | SCD, 55y | ||||||||
| III:11 | * | F | Fam.scr. 67y | ++ | ++ | 2/0 | |||||||
| IV:3 | * | F | Fam.scr. 54y | ++ | 1/0 | ||||||||
| IV:5 | * | M | Fam.scr. 45y | ++ | 1/0 | ||||||||
| IV:6 | * | M | Fam.scr. 38y | ++ | ++ | 2/0 | ICD, 38y | ||||||
| IV:9§ | * | F | Unknown | ++ | 1/0 | ||||||||
| IV:11 | * | F | Fam.scr. 43y | ++ | 1/0 | ||||||||
| U02 | II:4 | * | M | 22y | VT | ++ | ++ | ++ | ++ | ++ | 5/0 | SCD, 57y | |
| II:1 | * | F | 64y | Fam.scr. 60y | + | ++ | ++ | + | ++ | 3/2 | ICD, 64y | ||
| II:2 | * | M | 48y | SCD | ++ | 1/0 | SCD, 48y | ||||||
| III:1 | * | F | Fam.scr. 29y | + | ++ | 1/1 | |||||||
| III:2 | * | F | Fam.scr. 26y | ++ | ++ | 2/0 | |||||||
| III:4 | * | F | Fam.scr. 25y | ++ | + | ++ | 2/1 | ||||||
* indicates proven p.Arg79X mutation carrier, † likely p.Arg79X mutation carrier, ‡ asymptomatic, no cardiac examination performed, § no clinical data available, || diagnosed at autopsy, ++ = major criterion, + = minor criterion, TFC = number of major/minor criteria respectively, SCD=sudden cardiac death, Fam.scr.= family screening, VT=ventricular tachycardia, ICD = implantable cardioverter defibrillator, ~ICD = appropriate ICD discharge. Index patients are in bold. Family numbers correspond with Figure 3. Only the presence of a feature is indicated. End points are in italics.
Figure 3.
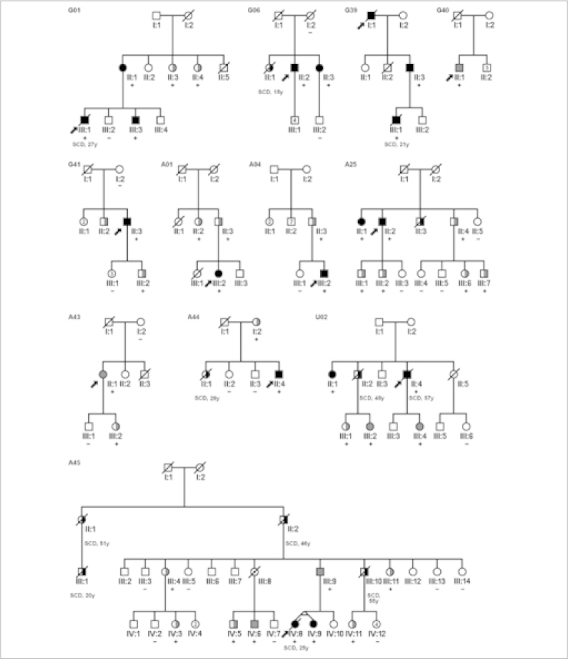
Pedigrees of the 12 families with the p.Arg79X mutation in PKP2. Squares indicate male family members, circles indicate female family members, slashes indicate deceased family members, and arrows indicate index patients. Solid black symbols indicate proven ARVC/D, solid grey symbols indicate probable ARVC/D, half-filled black symbols indicate SCD, half-filled grey symbols indicate mutation carriers identified through family screening, dotted symbols indicate obligate carriers, and open symbols indicate unaffected family members. Genotype results are indicated by (+) = p.Arg79X present and (−) = p.Arg79X absent.
Figure 4.
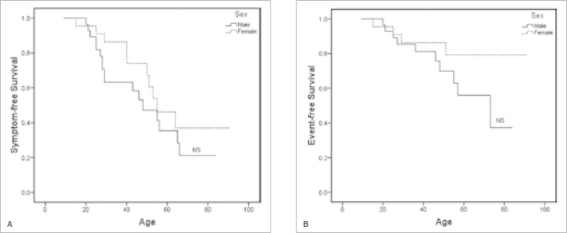
Kaplan-Meier survival analysis of p.Arg79X mutation carriers. A) Symptom-free survival. B) Event-free survival. Death due to a cardiac cause and an appropriate ICD discharge are considered as events. These events as well as ventricular tachycardia and syncope are considered to be symptoms.
Table 2.
Haplotype (in bold) associated with the p.Arg79X mutation in the Dutch population.
| Index patient | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Position | G01 | G06 | G39 | G40 | G41 | A01 | A04 | A25 | A43 | A44 | A45* | U02 | |||||||||||||
| 32.700k | 3 | 5 | 5 | 5 | 1 | 5 | 2 | 5 | 4 | 5 | 5 | 5 | 1 | 5 | 4 | 5 | 2 | 5 | 4 | 5 | 1 | 5 | 4 | 5 | |
| 32.830k | Exon 14 | 2 | 6 | 1 | 6 | 5 | 6 | 4 | 6 | 2 | 6 | 2 | 6 | 7 | 6 | 5 | 6 | 3 | 6 | 7 | 6 | 7 | 6 | 2 | 6 |
| c.235C>T | Exon 02 | C | T | C | T | C | T | C | T | C | T | C | T | C | T | C | T | C | T | C | T | C | T | C | T |
| 32.940k | Exon 01 | 1 | 2 | 3 | 2 | 1 | 2 | 3 | 2 | 1 | 2 | 1 | 2 | 1 | 2 | 1 | 2 | 1 | 2 | 1 | 2 | 1 | 2 | 1 | 2 |
| 32.970k | 1 | 2 | 5 | 2 | 4 | 2 | 3 | 2 | 3 | 2 | 4 | 2 | 4 | 2 | 4 | 2 | 4 | 2 | 4 | 2 | 4 | 2 | 1 | 2 | |
| 33.000k | 1 | 2 | 2 | 2 | 1 | 2 | 10 | 2 | 8 | 1 | 4 | 2 | 8 | 2 | 2 | 2 | 2 | 2 | 8 | 2 | 9 | 2 | 6 | 2 | |
The first column shows the genomic position of the markers and the c.235C>T, p.Arg79X mutation on chromosome 12. For each marker, the shortest haplotype is set at 1. The columns in normal and bold font represent the two inherited haplotypes, one from each parent. Note that the index patient of family G41 has a different allele for the 33.000k marker.*Haplotype analysed in IV:3 (not the index patient).
Discussion
We identified the p.Arg79X mutation in PKP2 in 12 index patients diagnosed with ARVC/D or probable ARVC/D; it is thus one of the most commonly detected PKP2 mutations in the Netherlands. Haplotype analysis strongly suggested a common founder. Gerull et al. identified the same mutation in six out of 120 probands of Western European descent, but could not identify shared haplotypes among these patients.14 In addition, Dalal et al. identified the same mutation in two female ARVC/D patients of North American Caucasian origin, but gave no further information regarding haplotype analysis.16,23 These observations suggest that the PKP2 p.Arg79X mutation is recurrent and that a founder effect is present in the Dutch population. A founder effect was also identified for three other PKP2 mutations in the same Dutch cohort (p.Gln133X, p.Cys796Arg, and c.2489+1G>A).17 However, no other studies have reported founder mutations, although a few recurrent PKP2 mutations have been identified (e.g. p.Ser50fsX110, p.Val837fsX930, and c.2146-1G>C).14–16 The fact that other groups might have studied a more heterogeneous population may account for the lack of founder mutations in their cohorts.
Gerull et al. found the p.Arg79X mutation in six male index patients. All had a history of ventricular tachycardias, two had experienced cardiac arrest, but only one had a positive family history.14 The first patient from North America described by Dalal et al. was a female athlete who presented with syncope at age 28. Cardiac examination revealed mild global RV dilatation, T-wave inversions in leads V1-V3, late potentials on SA-ECG, a left bundle branch block type VT and >1000 ventricular extrasystoles/24 hours. She had a negative family history.16 Their second patient was also a female but her age at onset and initial presentation were not reported.23 In agreement with these observations, we found that nine of our twelve index patients were male and that VTs were present in the majority of cases (9/12). In our cohort, the effect of the p.Arg79X mutation in PKP2 was characterised by a remarkably high clinical variability, typical of ARVC/D; 13% (7/53) of our likely or proven mutation carriers died of SCD before age 30, while 40% did not experience any symptoms at age 60. In the mutation carriers with symptoms, VTs and SCD were prominent, although heart failure was not.
Figure 5.
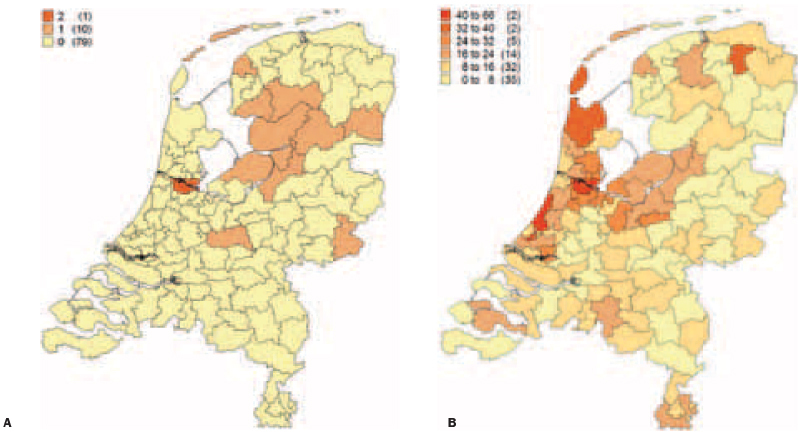
Postal code maps of the Netherlands. A) Distribution of index patients carrying the p.Arg79X in PKP2 in the Netherlands. B) Overview of PKP2 analyses in ARVC/D patients in the Netherlands. The number of p.Arg79X mutation carriers or PKP2 analyses is shown per region (in parenthesis: the number of regions, 90 in total). On average, each region contains 180,000 inhabitants.
A possible explanation for this high variability may be the co-occurrence of more than one mutation in the desmosomal genes, which has been recognised in up to 8% of ARVC/D patients.22,23,27 These mutations can be present in different genes (digenic heterozygosity) or on different alleles of the same gene (compound heterozygosity). Theoretically, digenic or compound heterozygosity could explain the severe end of the phenotypic spectrum of the p.Arg79X mutation carriers. However, we found no second mutation in the six index patients we analysed comprehensively for the five genes encoding desmosomal proteins (in addition to PKP2: DSC2, DSG2, DSP, and JUP).
Although the number of identified mutations in PKP2 and other ARVC/D-related genes is still rapidly increasing, data on the functional effects at the cellular and molecular level are scarce. For the p.Arg79X mutation in PKP2, however, the consequences of the expression of the mutation have been studied, using neonatal rat ventricular myocytes.28 This showed that the mutant protein failed to localise to the cell membrane, in contrast to endogenous PKP2 and DSP. The major cardiac gap junction protein connexin-43 (Cx43) was also reduced in this model. This was also true for HSP90, a protein that interacts with Cx43 and is known to be essential for the survival of cardiomyocytes. 29,30 Finally, the expression of p.Arg79X led to the inability of the mutated PKP2 protein to interact with both DSP and Cx43. Together, these results suggest that the p.Arg79X mutation leads to a loss of myocardial integrity characteristic of ARVC/D.28
Finally, Asimaki et al. showed that immunohistochemical analysis of conventional endomyocardial biopsy samples appears to be both a highly sensitive and specific diagnostic test for ARVC/D, as was demonstrated in a group of 11 ARVC/D patients.26 Reduced immunoreactive signal levels of plakoglobin and Cx43 were found to be a consistent feature in patients with ARVC/D. Although we found reduced signal levels for plakoglobin in one of the p.Arg79X mutation carriers, this was not a consistent finding in another series of patients (data not shown) and further studies are required to establish the possible role of immunohistochemical analysis in patients with ARVC/D.
Conclusions
In the majority of cases, ARVC/D is caused by mutations in genes encoding desmosomal proteins, especially PKP2. The PKP2 p.Arg79X mutation is a recurrent mutation in Europe and North America. We identified the p.Arg79X mutation in 12 Dutch index patients and showed that they share the same haplotype, strongly suggesting a founder effect in the Dutch population. The PKP2 p.Arg79X mutation carriers comprise the largest series of patients with the same desmosome gene mutation reported so far, and we illustrate that the clinical phenotype is highly variable, varying from SCD to non-penetrance.
Acknowledgements
We would like to thank Pieter Postema for his help in composing the postal code maps of the Netherlands, Ludolf Boven for technical assistance, and Jackie Senior for editing this manuscript. This study was financially supported by the Netherlands Heart Foundation (grants 2003B062, 2007B132 and 2007B139) and the Interuniversity Cardiology Institute of the Netherlands (project 06901).
References
- 1.Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, et al. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65:384–398. doi: 10.1161/01.cir.65.2.384. [DOI] [PubMed] [Google Scholar]
- 2.Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318:129–133. doi: 10.1056/NEJM198801213180301. [DOI] [PubMed] [Google Scholar]
- 3.Calkins H. Arrhythmogenic right-ventricular dysplasia/cardiomyopathy. Curr Opin Cardiol. 2006;21:55–63. doi: 10.1097/01.hco.0000198984.70884.4d. [DOI] [PubMed] [Google Scholar]
- 4.Marcus FI, Fontaine G. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: a review. Pacing Clin Electrophysiol. 1995;18:1298–1314. doi: 10.1111/j.1540-8159.1995.tb06971.x. [DOI] [PubMed] [Google Scholar]
- 5.McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine G, et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J. 1994;71:215–218. doi: 10.1136/hrt.71.3.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121:1533–1541. doi: 10.1161/CIRCULATIONAHA.108.840827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cox MG, van der Smagt JJ, Noorman M, Wiesfeld AC, Volders PG, van Langen IM, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy diagnostic task force criteria: impact of new task force criteria. Circ Arrhythm Electrophysiol. 2010;3:126–133. doi: 10.1161/CIRCEP.109.927202. [DOI] [PubMed] [Google Scholar]
- 8.Nava A, Bauce B, Basso C, Muriago M, Rampazzo A, Villanova C, et al. Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2000;36:2226–2233. doi: 10.1016/S0735-1097(00)00997-9. [DOI] [PubMed] [Google Scholar]
- 9.Hamid MS, Norman M, Quraishi A, Firoozi S, Thaman R, Gimeno JR, et al. Prospective evaluation of relatives for familial arrhythmogenic right ventricular cardiomyopathy/dysplasia reveals a need to broaden diagnostic criteria. J Am Coll Cardiol. 2002;40:1445–1450. doi: 10.1016/S0735-1097(02)02307-0. [DOI] [PubMed] [Google Scholar]
- 10.Peters S, Trummel M, Meyners W. Prevalence of right ventricular dysplasia-cardiomyopathy in a non-referral hospital. Int J Cardiol. 2004;97:499–501. doi: 10.1016/j.ijcard.2003.10.037. [DOI] [PubMed] [Google Scholar]
- 11.Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373:1289–1300. doi: 10.1016/S0140-6736(09)60256-7. [DOI] [PubMed] [Google Scholar]
- 12.Marcus FI, Zareba W, Calkins H, Towbin JA, Basso C, Bluemke DA, et al. Arrhythmogenic right ventricular cardiomyopathy/ dysplasia clinical presentation and diagnostic evaluation: results from the North American Multidisciplinary Study. Heart Rhythm. 2009;6:984–992. doi: 10.1016/j.hrthm.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rampazzo A, Nava A, Malacrida S, Beffagna G, Bauce B, Rossi V, et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2002;71:1200–1206. doi: 10.1086/344208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. 2004;36:1162–1164. doi: 10.1038/ng1461. [DOI] [PubMed] [Google Scholar]
- 15.Syrris P, Ward D, Asimaki A, Sen-Chowdhry S, Ebrahim HY, Evans A, et al. Clinical expression of plakophilin-2 mutations in familial arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113:356–364. doi: 10.1161/CIRCULATIONAHA.105.561654. [DOI] [PubMed] [Google Scholar]
- 16.Dalal D, Molin LH, Piccini J, Tichnell C, James C, Bomma C, et al. Clinical features of arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in plakophilin-2. Circulation. 2006;113:1641–1649. doi: 10.1161/CIRCULATIONAHA.105.568642. [DOI] [PubMed] [Google Scholar]
- 17.van Tintelen JP, Entius MM, Bhuiyan ZA, Jongbloed R, Wiesfeld AC, Wilde AA, et al. Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/ cardiomyopathy. Circulation. 2006;113:1650–1658. doi: 10.1161/CIRCULATIONAHA.105.609719. [DOI] [PubMed] [Google Scholar]
- 18.Heuser A, Plovie ER, Ellinor PT, Grossmann KS, Shin JT, Wichter T, et al. Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2006;79:1081–1088. doi: 10.1086/509044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pilichou K, Nava A, Basso C, Beffagna G, Bauce B, Lorenzon A, et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113:1171–1179. doi: 10.1161/CIRCULATIONAHA.105.583674. [DOI] [PubMed] [Google Scholar]
- 20.Asimaki A, Syrris P, Wichter T, Matthias P, Saffitz JE, McKenna WJ. A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2007;81:964–973. doi: 10.1086/521633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sen-Chowdhry S, Syrris P, McKenna WJ. Role of genetic analysis in the management of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2007;50:1813–1821. doi: 10.1016/j.jacc.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 22.Bhuiyan ZA, Jongbloed JD, van der Smagt J, Lombardi PM, Wiesfeld AC, Nelen M, et al. Desmoglein-2 and desmocollin-2 mutations in Dutch arrhythmogenic right ventricular dysplasia/ cardiomoypathy patients: Results from a multicenter study. Circ Cardiovasc Genet. 2009;2:418–427. doi: 10.1161/CIRCGENETICS.108.839829. [DOI] [PubMed] [Google Scholar]
- 23.den Haan AD, Tan B, Zikusoka M, Ibanez Llado L, Jain R, Daly A, et al. Comprehensive Desmosome Mutation Analysis in North Americans with Arrhythmogenic Right Ventricular Dysplasia/ Cardiomyopathy. Circ Cardiovasc Genet. 2009;2:428–435. doi: 10.1161/CIRCGENETICS.109.858217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Tintelen JP, Hofstra RM, Wiesfeld AC, van den Berg MP, Hauer RN, Jongbloed JD. Molecular genetics of arrhythmogenic right ventricular cardiomyopathy: emerging horizon? Curr Opin Cardiol. 2007;22:185–192. doi: 10.1097/HCO.0b013e3280d942c4. [DOI] [PubMed] [Google Scholar]
- 25.van der Zwaag PA, Jongbloed JD, van den Berg MP, van der Smagt JJ, Jongbloed R, Bikker H, et al. A genetic variants database for arrhythmogenic right ventricular dysplasia/cardiomyopathy. Hum Mutat. 2009;30:1278–83; available from http://www.arvcdatabase.info. [DOI] [PubMed]
- 26.Asimaki A, Tandri H, Huang H, Halushka MK, Gautam S, Basso C, et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009;360:1075–1084. doi: 10.1056/NEJMoa0808138. [DOI] [PubMed] [Google Scholar]
- 27.Xu T, Yang Z, Vatta M, Rampazzo A, Beffagna G, Pillichou K, et al. Compound and Digenic Heterozygosity Contributes to Arrhythmogenic Right Ventricular Cardiomyopathy. J Am Coll Cardiol. 2010;55:587–597. doi: 10.1016/j.jacc.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joshi-Mukherjee R, Coombs W, Musa H, Oxford E, Taffet S, Delmar M. Characterization of the molecular phenotype of two arrhythmogenic right ventricular cardiomyopathy (ARVC)-related plakophilin-2 (PKP2) mutations. Heart Rhythm. 2008;5:1715–1723. doi: 10.1016/j.hrthm.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yun BG, Matts RL. Differential effects of Hsp90 inhibition on protein kinases regulating signal transduction pathways required for myoblast differentiation. Exp Cell Res. 2005;307:212–223. doi: 10.1016/j.yexcr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 30.Rodriguez-Sinovas A, Boengler K, Cabestrero A, Gres P, Morente M, Ruiz-Meana M, et al. Translocation of connexin 43 to the inner mitochondrial membrane of cardiomyocytes through the heat shock protein 90-dependent TOM pathway and its importance for cardioprotection. Circ Res. 2006;99:93–101. doi: 10.1161/01.RES.0000230315.56904.de. [DOI] [PubMed] [Google Scholar]