

Abstract
Gaucher disease (GD), the most common lysosomal storage disease, results from a deficiency of the lysosomal enzyme glucocerebrosidase. GD has been classified into 3 types, of which type 2 (the acute neuronopathic form) is most severe, presenting pre- or perinatally, or in the first few months of life. Traditionally, type 2 GD was considered to have the most uniform clinical phenotype when compared to other GD subtypes. However, case studies over time have demonstrated that type 2 GD, like types 1 and 3, manifests with a spectrum of phenotypes. This review includes case reports that illustrate the broad range of clinical presentations encountered in type 2 GD, as well as a discussion of associated manifestations, pathological findings, diagnostic techniques, and a review of current therapies. While type 2 GD is generally associated with severe mutations in the glucocerebrosidase gene, there is also significant genotypic heterogeneity observed.
Keywords: Gaucher disease, glucocerebrosidase, neuronopathic Gaucher disease, hydrops fetalis, genotype-phenotype correlation
Introduction
Gaucher disease (GD), the most common lysosomal storage disease, results from a deficiency of the lysosomal enzyme glucocerebrosidase. It is a disorder primarily affecting the reticular endothelial system, in which macrophages become engorged with stored lipid, giving rise to the characteristic appearance of Gaucher cells. Patients with GD are typically divided into three types, based on the presence or absence and rate of progression of neurologic manifestations [1]. In type 1, the non-neuronopathic form, clinical manifestations are restricted to the hematopoietic system, skeletal system and visceral organs. Types 2 and 3 are both neuronopathic forms affecting the central nervous system (CNS), however they exhibit differing rates of neurological deterioration. Type 2 GD (MIM 231000) describes the acute neuronopathic form [2].
Type 2 GD, originally described in 1927, is the rarest and most severe form of Gaucher disease [3]. Patients with type 2 GD typically present with symptoms either prenatally or during infancy, and usually die before the age of 3 years [2]. Type 2 GD makes up the minority of GD cases overall. In general, GD has an estimated frequency of 1 in 100,000 to 500,000 live births [4; 5]. Like other types of GD, type 2 GD is pan-ethnic in occurrence.
Traditionally, type 2 GD was considered to have the most uniform clinical phenotype when compared to other GD subtypes. However, case studies over time have demonstrated that type 2 GD also manifests along a phenotypic spectrum, similar to types 1 and 3. This review includes case reports that illustrate the phenotypic spectrum, with a discussion about the clinical and molecular features of type 2 GD.
Case Reports
The following case reports, selected from the literature, illustrate the range of variation in the clinical presentation of patients with type 2 GD. The age of presentation varies, from perinatal lethal forms to later-onset presentations. All of the cases demonstrate a rapid rate of neurological progression.
Case 1: [6]
A male fetus, resulting from a consanguineous mating, died in utero at 22 weeks of gestation. Upon autopsy, the fetus was found to have hydrops fetalis, hepatosplenomegaly, and multiple external abnormalities of the extremities, ears and nose. Decreased β-glucocerebrosidase activity was established in fetal fibroblasts.
Case 2: [7]
A neonate, born at 32 weeks of gestation to a non-consanguineous couple, displayed moderate ichthyosis with ectropion, some restriction of limb movements, hepatosplenomegaly, and thrombocytopenia. The patient’s clinical condition deteriorated with the development of apnea, a suspected infection, and jaundice, resulting in death at 3 weeks of age. Leukocyte enzyme assay showed a significant decrease in the levels of β-glucocerebrosidase activity.
Case 3: [8]
A male neonate, born at term to a non-consanguineous couple, was noted to have collodion-like skin at birth. The skin findings cleared by 2 weeks of age; however, he subsequently developed feeding difficulties and regression of motor milestones. Physical exam at 6 months of age was significant for spasticity, abnormal eye movements, an opisthotonic posture and hepatosplenomegaly. He died at 7 months of age. Fibroblast β-glucocerebrosidase activity was significantly decreased.
Case 4: [9]
A male neonate, born to a non-consanguineous couple following a pregnancy notable for intrauterine growth retardation, presented with facial dysmorphism, intense jaundice, massive hepatosplenomegaly and cutaneous hemorrhagic syndrome. Vitamin K replacement and multiple platelet transfusions partially improved his coagulopathy and thrombocytopenia. Neurological findings were noted at 2 months of age. Death occurred at 4 months following uncontrollable digestive tract bleeding. β-glucocerebrosidase activity in leukocytes was significantly decreased.
Case 5: [10]
A female infant with an uneventful perinatal course presented at 8 months of age with acute pneumonia and was noted to have splenomegaly, anemia, opisthotonus, and an absent gag reflex. Following demonstration of deficient β-glucocerebrosidase activity, she was treated with enzyme replacement therapy, alglucerase. A tracheostomy was performed due to frequent aspiration. Subsequently, she developed seizures refractory to epileptic treatment and died due to aspiration pneumonia at two and a half years of age.
Case 6: [11]
A previously healthy female infant, born to a non-consanguineous couple, was noted to have an oculomotor apraxia at 10 months of age. β-glucocerebrosidase activity was decreased in lymphocytes and Gaucher cells were seen on a bone marrow aspirate. The clinical picture was initially consistent with type 3 GD, however the neurological findings progressed rapidly, with bilateral abducens paralysis at 14 months and myoclonus at 16 months. A supranuclear gaze palsy with bilateral ptosis, a fixed downward gaze, absent vertical eye movements, and severely impaired swallowing were evident at 22 months. Splenic enlargement and growth retardation with gradual wasting increased steadily. A partial splenectomy was performed at 28 months but it failed to arrest the clinical deterioration. She died at 32 months of age.
Clinical Phenotypes (see Table 1)
Table 1.
Summary of clinical manifestations encountered in different forms of type 2 Gaucher disease
| Perinatal Lethal | Non-Perinatal Lethal | Intermediate | |
|---|---|---|---|
| Lifespan | In utero to 3 months of age | Up to 2 years of age | 2–6 years of age |
| Cutaneous | Hydrops fetalis, ichthyosis, disrupted lamaller bilayers in startum corneum | Ultrastructural alterations of stratum corneum on EM observed | NA |
| Pulmonary | Pulmonary hypoplasia | Recurrent aspiration | Recurrent aspiration, restrictive lung disease |
| Visceral | Hepatosplenomegaly, jaundice, GI bleeding | Hepatosplenomegaly, GI bleeding | Hepatosplenomegaly |
| Hematological | Purpura, thrombocyptopenia | Anemia, thrombocytopenia | Anemia, thrombocytopenia |
| Neurological | Arthrogryphosis, microcephaly, poor sucking, laryngeal stridor, neck hyperextension, opisthotonus, apnea | Strabismus, trismus, laryngeal stridor, neck hyperextension, esophageal dysmotility, unstable heart rate, limb hypotonia, expressionless facies, athetosis, epilepsy, myoclonus, apnea | Myoclonic epilepsy, progressive brainstem involvement, seizures |
| Ophthalmological | NA | Strabismus, oculomotor apraxia, absence/slowing of saccades | Strabismus, oculomotor apraxia, absence/slowing of saccades |
| Dysmorphology | Low-set ears, small nose with flat nasal bridge, anteverted nares | NA | NA |
| Global | Frequent prolonged stays in Neonatal Intensive Care Units | Fever, failure to thrive | Failure to thrive |
Perinatal lethal forms
In perinatal lethal forms of GD, the pregnancy is typically complicated by non-immune hydrops fetalis. Hydrops may cause the fetus to die in utero or to be delivered prematurely, resulting in death soon after. Although the pathophysiology of hydrops fetalis in patients with GD remains obscure, a few hypotheses exist. Some authors propose that hydrops fetalis is caused by anemia related to hypersplenism and Gaucher cells infiltrating the bone marrow, resulting in a high output congestive heart failure and eventual fetal and neonatal death [12]. An alternative hypothesis is that it is due to hypoproteinemia, resulting from massive liver involvement or vascular occlusion by Gaucher cells [13]. Dimmick and Applegrath [14] proposed that vascular occlusion by Gaucher cells is a plausible cause, with excessive erythroblastosis and hypoproteinemia as secondary causes. The supporting evidence includes the extensive presence of Gaucher cells in the hepatic sinusoids, hepatic veins and in the capillaries of the placental villi.
When hydrops is absent, the fetus usually reaches full term, and may present with distress that requires resuscitation, or with a rapidly worsening neurological condition beginning in the first week and leading to death within three months.
Neonatal ichthyosis
Neonatal ichthyosis is frequently encountered in type 2 GD. The abnormal skin is erythematous and shiny, and is present all over the body but predominately over the palms and soles or in flexural folds [10]. At times, the infant is considered to have the “collodian–baby” phenotype due to a cellophane-like membrane covering the skin (Figure 1). This dermopathy usually disappears within a few weeks. Histological and ultrastructural studies of skin from patients with type 2 GD, even without overt skin manifestations, reveal epidermal extracellular lamellar bilayer abnormalities [15]. These skin studies show findings similar to the mouse model of GD generated by targeted disruption of the mouse glucocerebrosidase gene [16]. The ichthyotic skin demonstrates an increased epidermal glycosylceramide to ceramide ratio, abnormal stratum corneum ultrastructure, and an increased transepidermal water loss [17]. It has been speculated that the altered cutaneous desquamation seen in patients with congenital ichthyosis could be a result of abnormal intercellular cohesion and/or induction of an abnormal skin permeability barrier leading to hyperproliferation [15].
Figure 1.
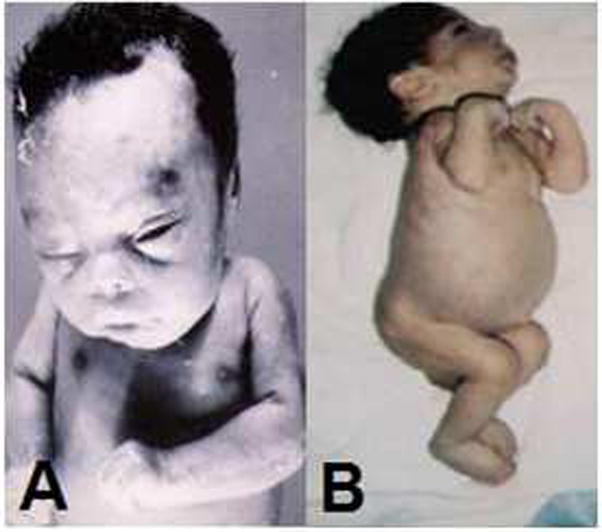
Two patients with symptoms of type 2 Gaucher disease, including (A) collodion skin, and (B) abdominal protrusion, due to hepatosplenomegaly, and opisthotonus
Dysmorphology
Overall, up to 35 % of fetuses diagnosed perinatally with type 2 GD are observed to be dysmorphic, with low-set ears, a small nose with a flat nasal bridge and anteverted nares. Given how similar the features are to those observed in fetal akinesia syndrome, it is presumed that either partial or full fetal akinesia sequence occurs in patients with perinatal lethal GD [18].
Neurological involvement
Typical neurological features in individuals with perinatal lethal forms are related to central nervous system degeneration, and include arthrogryphosis, microcephaly, and neonatal distress. The secondary progressive bulbar syndrome is represented by poor sucking, laryngeal stridor, neck hyperextension, opisthotonus and apnea [18].
Non-neurological involvement
Splenomegaly at birth is a major non-neurological sign in patients with perinatal lethal GD [18; 19] and is frequently associated with hepatomegaly (Figure 1). Jaundice, purpura with thrombocytopenia, and a deficiency of clotting factors have been reported. Causes of death related to non-neurological complications include pulmonary hypoplasia due to plural effusion, liver failure [18], and digestive tract bleeding [20; 21; 22].
Non-perinatal lethal forms
The clinical presentations of patients with non-perinatally lethal type 2 GD are all fairly similar, with the exception of the age of presentation and age of death. The prenatal history for most of the patients is unremarkable. The infants appear normal at birth, and the first signs of GD usually present before 9 months of age as visceral and/or neurological symptoms [23].
Neurologic involvement
Signs consistent with brain stem degeneration include hyperextension of the neck, strabismus, laryngeal stridor, trismus, rarely esophageal dysmotility [24], and unstable heart rate [25]. These signs may appear individually or in a cluster. Changes in motor function, such as limb hypertonia, brisk tendon reflexes and, rarely, expressionless facies or athetosis, may be related to brain stem dysfunction [26; 27]. Epilepsy, myoclonic epilepsy/myoclonus, and progressive microcephaly are less frequent.
It is difficult to evaluate cognition in patients with type 2 GD given the acuity of the disease, the young ages of the patients, and the severity of motor impairment. Reports range from decreased rate of gain [28], to regression of developmental milestones [23].
The association of oculomotor involvement with other signs of GD in the acute and chronic neuronopathic forms is quite characteristic. Strabismus is the most frequent sign of oculomotor involvement [23]. Other oculomotor signs less commonly observed in patients with acute neuronopathic involvement are oculomotor apraxia, and the absence or slowing of the horizontal saccades with preservation of vestibulogenic deviation and vertical gaze [26; 29; 30; 31; 32; 33; 34].
The major progressive neurological feature and most frequent cause of death is prolonged spontaneous apnea, occurring with increased frequency in the later stages of the disease [23].
Non-neurologic involvement
Visceral involvement primarily includes splenomegaly, usually associated with anemia, thrombocytopenia, and liver enlargement. Although hepatosplenomegaly is a common sign in type 2, complications such as portal hypertension, which was frequent in untreated type 3 GD, are rarely reported [20].
Skin involvement is less commonly observed as compared to patients with perinatal lethal forms.
Unlike other lysosomal storage diseases, fever is frequently observed in type 2 GD [23]. Fever may be a sign of a non-specific infection, or it may be present in the absence of infection and thus, resistant to antibiotic therapies. In these cases, type 2 GD appears to cause fever independently, suggesting an inflammatory component to the disease, possibly mediated by the activated glucosylceramide-laden macrophages.
Given the acuity of type 2 GD, the skeletal manifestations have not been well described. The skeletal complications seen in chronic GD types are primarily long term, such as growth retardation, osteoporosis, and fractures.
Pulmonary involvement is the most significant non-neuronopathic feature, and may be attributable to recurrent aspiration. Aspiration may be due to recurrent choking episodes related to neurological degeneration. Bilateral interstitial lung involvement, pulmonary hypertension, restrictive pulmonary insufficiency and intrapulmonary arterial-venous shunts are not frequently described in type 2 GD [23].
Failure to thrive may result from anorexia, nausea, vomiting and swallowing difficulties. Alternatively, failure to thrive may relate to increased resting energy expenditure [28]. Fatal gastrointestinal bleeding has been reported [35].
Intermediate phenotypes
Patients who survive beyond their second birthdays and whose clinical pictures fall between types 2 and 3, have been described as having intermediate phenotypic forms. Goker-Alpan et al. (2003) [36] reported nine such patients with severe neurologic disease and later ages of onset, with an average age at diagnosis of 12.6 months as compared to the average of 3.5 months in the typical cases studied by Mignot et al. [18]. The mean age of onset for type 3 was reported to be 4.2 years [37]. The skeletal and visceral involvement associated with this intermediate phenotype was mild to moderate, consistent with most cases of type 2 GD, and in contrast to the severe skeletal and visceral involvement of type 3. Similar to the cases with type 3, myoclonic epilepsy refractory to antiepileptic treatment was one of the most frequent findings, in contrast to what is typically seen in type 2. The study also found that most patients with the intermediate forms died of progressive brain stem involvement and aspiration pneumonia. The rate of disease progression in these patients was faster compared to patients with type 3, where death usually ensued in either adolescence or adulthood.
Overlap between neuronopathic forms of Gaucher disease
The ever-expanding spectrum of neurological involvement in patients with GD has compelled researchers and clinicians to describe patients with both acute and chronic neurologic GD as having neuronopathic GD, rather than restricting classification to types 2 or 3 [38]. Furthermore, patients with type 1 GD may present with neurological dysfunction, however the profile of neurological manifestation differs from that observed in patients with types 2 and 3. Patients with type 1 may show Parkinson disease symptoms and peripheral neuropathies [39]. In contrast, swallowing abnormalities are more common in patients with classical type 2. Dysfunction of major motor circuits, myoclonic epilepsy, oculomotor abnormalities, learning disabilities, and at times, intellectual deterioration are seen in classical type 3.
Davies et al. [40] developed a list of clinical signs and symptoms, along with a severity-scoring system, for patients with neuronopathic GD. It is important to note, however, that during the development of this severity scoring tool, authors excluded reports of patients with type 2, and focused primarily on the severity and progression of symptoms in patients with chronic GD only. Currently, two scales are used to assess the severity of involvement for patients with GD [41; 42]. Of note, these scales calculate all neurological involvement in one domain only, and are not sensitive enough to track neurological changes seen over time.
Pathological findings in patients with type 2 GD
Gaucher cells
Even though there is deficient activity of β-glucocerebrosidase in all cells of the body in GD, the clinical manifestations are primarily related to the macrophages that phagocytose senescent white and red blood cells. The phagocytic macrophages, intended to degrade large quantities of sphingolipids, are termed Gaucher cells and exhibit a characteristic morphology [43]. Microscopically, the cells are 20–100μm in size, have an eccentric nucleus, and a wrinkled tissue paper appearance of the cytoplasm [44; 45]. The contents of the cells react with periodic-acid Schiff (PAS) reagent [45], PAS leucofuchsin stain [46], acid phosphatase [47], and often ferritin. Electron micrographs of these cells show characteristic engorged lysosomes. They have a single limiting membrane surrounding a pale matrix with regular-appearing elongated structures of up to 5 μm in length, containing as many as 10–12 twisted fibrils [46].
Visceral pathology
The spleen is significantly enlarged, with grossly observed fibrosis, infarcts and nodules related to vascular malformations of the red pulp or extramedullary hematopoiesis [46]. While the size of the spleen increases up to 25-fold in patients with GD, including patients with type 1, glucosylceramide accounts for less than 2% of the additional tissue mass [48]. This disproportionality implies that although glucosylceramide accumulates significantly in GD, other biochemical pathways must be activated in GD, thus contributing to changes in the tissue.
The liver is usually grossly enlarged, and microscopy reveals Gaucher cells in the sinusoids. Glycolipids accumulate in the macrophages that line the walls of the sinusoids, but the hepatocytes are spared. Destructive changes, such as fibrosis, are occasionally observed [20; 46; 49].
The lungs frequently show extensive intra-alveolar accumulation of Gaucher cells [50]. Gaucher cells are also found in the cortical and medullary portions of the thymus, deep layers of the adrenal cortex, lymph nodes, and the lamina propria of the gastrointestinal tract.
In perinatal lethal forms, changes in the muscles of the pelvic girdle and lower limbs are also observed, with perimesial fibrosis and fiber size variation without fiber type grouping, pan fascicular atrophy, or clusters of hypertrophied type 1 fibers [49]. Fibrotic papillary muscles have been reported in one case of type 2 with hemosiderosis [51]. Skin shows compact orthokeratotic hyperkeratosis in many case reports.
Neuropathology
Despite the acute neurological involvement differentiating type 2 from other GD phenotypes, the gross histological appearance does not appear to be abnormal [52]. Microscopically, there is variable neuronal loss [2; 53; 54; 55], most prominent in the thalamus, brain stem, occipital cortex [56], and Purkinje fibers. The most consistent finding in the nervous system is the presence of Gaucher cells perivascularly in Virchow-Robin spaces and in cerebral grey matter, either individually or as clumps [32].
Neuronophagia, or destruction of nerve cells, is variably present. Neurons show intracytoplasmic striations, membrane-bound vacuoles, and cytosomes filled with flat parallel membranes [2; 53]. A variable degree of demyelination has been reported [2; 52; 56; 57].
While extreme widespread storage of glucosylceramide is a typically expected biochemical finding in lysosomal storage diseases, it is remarkably not found in most cases of GD. Even the cases where “extreme and widespread storage” has been reported, the cellular response to stored glucosylceramide is minimal [56].
It has been postulated that the small and focal lesions observed in postmortum brain samples from patients with GD type 2 may result more from dysfunction of surrounding cells rather than from the discrete pathological changes caused by minimal lipid accumulation [50]. Another possibility is that the glucosylceramide stored in the tissue macrophages either releases toxic substances, or causes failure of the protective mechanisms of these cells [58]. Since glucosphingolipids are potent inhibitors of protein kinase C activity [59] and could disrupt neuronal activity by interfering with signal transduction, they have also been thought to have neurotoxic effects in patients with type 2 GD [50]. Glucosylsphingosine is found at higher levels in the brains of the patients with type 2 and type 3 GD [60], suggesting a potential pathological role of this lipid in types 2 and 3 [61]. As compared to patients with type 2 GD, patients with type 3, who have more prolonged clinical courses, have pathological findings that show more extensive neuronal storage, as well as neuronal loss, neuronophagia and gliosis [53; 2].
Studies have been conducted at the cellular level in order to uncover how lysosomal accumulation of the substrate, glucosylceramide, mediates cellular pathology. The studies have looked at neuronal models of GD [62; 63; 64], chemically induced macrophage models [65], and postmortem brain samples from patients with type 2 GD [66]. The results have shown that changes in phospholipid metabolism and calcium homeostasis occur. They have also demonstrated that glucosylceramide can escape the lysosomes, and then interact with biochemical and cellular pathways located in other organelles.
Diagnosis
Neurodiagnostics
Analysis of the findings from structural and functional imaging in patients with type 2 GD has been conducted in order to establish a uniform method to assess clinical neurologic severity, to predict progression, and to optimize therapeutic intervention. Vellodi et al. [38] presented a clinical protocol for the minimal initial assessment of primary neurological involvement in GD, which includes brain imaging, neurophysiology, and neuropsychometry.
The tests designed for imaging structural CNS involvement have shown variable results. Cerebral CT and MRI findings in type 2 disease range from normal to mild cerebral atrophy [28; 34; 52; 67]. Dural thickening, which is considered to be specific for lysosomal storage diseases, has also been reported with contrast enhanced MRI imaging [68].
Electroencephalograms (EEG’s) may show disorganization of the background and variable foci of sharp waves and spikes [23]. However, the EEG findings may have little clinical significance [52; 67]. As would be expected, electrophysiological findings specific for brain stem neurodegenerative disease have abnormal responses with disappearance of III–V waves or with prolonged latencies [23; 67].
There is only a single report [32] regarding the noninvasive assessment of functional status. The single photon emission computed tomography (SPECT) in two patients revealed decreased perfusion of the brain initially, with progression that involved the entire cerebrum as the neurological manifestations intensified over a period of four months. Since there is an insufficient understanding of the functional and structural cerebral changes in this disease, more functional studies may result in a better understanding of the disease process.
Molecular diagnosis
The gene for human β-glucocerebrosidase (GBA) is located on chromosome 1q21. GBA contains 11 exons and 10 introns, covering 7.6 kb of sequence. A pseudogene for GBA, sharing 96% exonic sequence homology, is located 16kb downstream from the functional gene (Figure 2) [69; 70]. The 1q21 region is gene rich, with seven genes and two pseudogenes located in close proximity (GenBank accession number AF023263) [70]. The gene for metaxin (MTX) is located immediately downstream of the GBA pseudogene and is convergently transcribed, encoding a protein of 317 amino acids that appears to be part of a preprotein import complex in the outer membrane of mitochondria [71]. MTX also has a pseudogene located between the GBA gene and pseudogene, immediately adjacent to the 3′ end of the functional GBA gene [72]. These genes increase the likelihood of unequal pairing and recombination events in this region. Thus, polymerase chain reaction (PCR) amplification of genomic DNA for sequencing must selectively amplify the functional gene [73]. The first mutation described for patients with type 2 GD, Leu444Pro [74], remains among the most frequently encountered mutations, although over 50 different mutations have been described in patients with type 2 GD.
Figure 2.

The structure of the gene for human β-glucocerebrosidase (GBA) is shown, depicting is the 62-kb region surrounding GBA along chromosome 1q, with known genes and pseudogenes and their transcription directions. C1orf2, chromosome 1 open reading frame 2 (cote1); GBA, glucocerebrosidase; MTXP, metaxin 1 pseudogene; GBAP, glucocerebrosidase pseudogene; MTX1, metaxin 1; THBS3, thrombospondin 3.
In 2000, Stone et al. [73] summarized 33 different mutant alleles by characterizing all 62 alleles in 31 patients with type 2 GD. A few generalizations were observed that suggested genotype-phenotype correlations: (1) the most severe outcome of perinatal lethality due to hydrops fetalis often resulted from homozygosity for a recombinant allele; (2) the combination of one allele with the point mutation, Leu444Pro, and another mutation with a null or very severe allele seemed to be associated with classical type 2; (3) Leu444Pro was the most common mutation identified in patients with the intermediate phenotype [36]. None of the patients with type 2 have been described with the Asn370Ser mutation, commonly seen in type 1 GD. Various studies from other laboratories [75; 76; 77] have also associated homozygosity of Leu444Pro with neuronopathic GD, although not usually with type 2 GD. A unique mutant allele containing two distinct point mutations, Asp409His and His255Gln, also seems to be associated with the type 2 phenotype [78].
Different groups have attempted expression studies of human β-glucocerebrosidase isoforms from protein extracts taken from fibroblast cell lines with specific mutant alleles. The results range from absent to very weak expression, to expression of different isoforms [73; 79; 80; 81; 82; 83; 84]. Also, only a few point mutations have been assessed using functional studies.
In 2005, Goker-Alpan et al. [85] analyzed 32 non-type 2 patients who were all homozygous for the mutation Leu444Pro, and found the genotype to be associated with wide phenotypic variation. It was concluded that other factors, such as promoter mutations, modifier loci, environmental factors, and other non-genetic causes, must play roles in the observed variability. A genome-wide single nucleotide polymorphism (SNP) scan may help to highlight genetic modifiers.
Biochemical diagnosis
Documentation of a deficiency of β-glucocerebrosidase is the gold standard diagnostic measure for GD. Enzyme level is typically measured in leukocytes, amniocytes or fibroblasts cultured from the skin, and enzyme activity is usually assayed using fluorescent substrate 4-MU (4-methylumbelliferyl-β-glucoside). However, there is not a good correlation between the clinical phenotype and the amount of residual β-glucocerebrosidase activity [4].
Murine macrophage cell lines have demonstrated that β-glucocerebrosidase activity can be reduced to about 11–15% of the normal control level before increased storage of glucosylceramide occurs [86]. However, glucosylceramide does not accumulate in fibroblasts of patients with GD limiting their utility in some cell based studies.
The biochemical properties of β-glucocerebrosidase in patients with GD differ from those of control subjects in terms of pH optimum, KM, and heat stability [87]. Furthermore, the mutant enzyme studied in lymphoid cells from a patient with type 2 was found to be profoundly labile at pH 4 and 37 °C, in contrast to the residual enzyme activities from types 1 and 3, which were stable and similar to the enzyme in control subjects [88]. Monoclonal antibody studies have also demonstrated that, although the mutant enzyme from patients with type 2 has the same apparent molecular weight as enzyme from other patients with GD, an antigenic epitope is missing [89].
Surrogate biochemical markers
Even when enzyme activity is deficient, biochemical markers may be important for monitoring the disease course and the effect of medical therapy. For example, the dosage of costly enzyme replacement therapy, which can reverse visceral manifestations, can be modified according to disease severity and therapeutic response. Chitotriosidase is a marker that best correlates with disease severity. However, 6% of patients with GD lack serum chitotriosidase activity as a result of a mutation in the chitotriosidase gene [90], and the test is labor intensive and not standardized between laboratories. Increases in β-hexosamindase, tartarate resistant acid phosphatase, and/or angiotensinogen converting enzyme have been tracked, but the results are neither universal nor consistent [91]. Thus, research is currently being performed to identify more reliable surrogate markers, such as pulmonary and activation-regulated chemokine (PARC) [92] and CD163 [93], which reflect macrophage activity and which may help to monitor the severity and course of the disease.
Prenatal diagnosis
Except for one case report [51], GD has not been detected via routine non-invasive prenatal blood testing, including the triple test. However, prenatal ultrasound in the second trimester has been able to detect symptoms associated with perinatal lethal type 2 GD, such as fetal ascites, hepatosplenomegaly, and reduced fetal movements in association with polyhydramnios, thickened skin, a characteristically flattened nose, proptosis and a continuously opened mouth [94]. Cultured amniotic fluid cells collected in suspected cases clearly demonstrate deficiency of β-glucocerebrosidase activity [51]. Mutation analysis can also be performed on aminocytes or chorionic villi samples if the mutations in the family are known.
Bone marrow or tissue biopsy
Even though bone marrow, like other reticuloendothelial tissues, shows the presence of Gaucher cells, bone marrow biopsy is not indicated for the diagnosis of GD. The less invasive biochemical analysis of reduced enzyme activity in leukocytes or cell lines, in conjunction with the identification of mutations in GBA, is sufficient for diagnostic confirmation [95].
Ultrastructural electron microscopy of epidermal samples may have promise in the discrimination between type 2 GD and type 1 or type 3 [5; 96]. Skin samples from patients with type 2 GD show abnormal arrays of loosely packed lamellar body-derived sheets in the stratum corneum, as opposed to the normal lamellar bilayer structures found in the stratum corneum of control subjects and patients with types 1 and 3 GD [5]. These ultrastructural changes are found in patients with type 2 GD regardless of cutaneous clinical manifestations such as ichthyosis. These ultrastructural changes mirror lipid biochemical analyses of epidermal samples from patients with type 2 GD, which show a higher content of glucosylceramides than ceramides [15]. Control subjects and patients with type 1 or type 3 GD show a reversal of this ratio: they have a higher content of ceramides than glucosylceramides. However additional samples need to be evaluated to determine whether these complicated tests are truly predictive.
Therapy
The goals of treatment in any patient with GD are to reduce the glucosylceramide storage and to minimize the deleterious consequences caused by its accumulation. Enzyme replacement therapy achieves this by supplementing the defective enzyme with the active enzyme [75] using a recombinant form of glucocerebrosidase, Cerezyme (Genzyme Corporation, Cambridge, MA, USA). The modified enzyme is endocytosed and is subsequently delivered to lysosomes where it supplements the defective enzyme [75].
Enzyme replacement therapy (ERT) has been proven to be safe and effective in most patients with type 1 GD. Trials of ERT in patients with neuronopathic GD have demonstrated improvements in most visceral symptoms, similar to patients with type 1 GD. However, there have not been improvements in pulmonary manifestations [97; 98].
Most reports [36; 97; 99; 100] that have examined the effects of ERT on acute, intermediate and chronic neurological manifestations have concluded the therapy to be useful to treat systemic involvement, but ineffective in treating neurologic manifestations of the disease. Vellodi et al. [101] summarized the preliminary evidence, which showed that higher dosages of ERT (120U/kg intravenously every two weeks) have reversed, stabilized or slowed the progression of visceral involvement in cases with chronic neuronopathic GD. The same dosages have shown an increase in glucocerebrosidase activity in cerebrospinal fluid of patients with type 3 GD [102]. In one patient with type 2 GD, the levels of enzyme activity in cerebrospinal fluid (CSF) increased by 2.3-folds three hours after administration of higher dosages of ERT, but returned to pre-dose level within 24 hours. This CSF increase was only 0.08 % of that observed in plasma in a case with acute type 2 GD [103].
The most recent recommendations have stated that ERT has little effect on the progressive downhill course of acute neuronopathic GD, and may merely result in the prolongation of pain and suffering [38]. The almost negligible effect on neurological involvement is due to the inability of the recombinant enzyme to cross the blood-brain barrier [104] by systemic route of administration. In 2005, Lonser et al. [105] investigated the potential use of convection-enhanced delivery of Cerezyme to perfuse the brain and brain stem of rats with therapeutic levels of recombinant glucocerebrosidase. Enzyme activity substantially increased in the cortex and white matter compared to controls, without evidence of toxicity.
A follow-up study [106] performed two similar infusions in a human patient with type 2 GD at ages 10.5 months and 13 months. The first infusion was to the right paramedian frontal lobe, and the second was directed to the brainstem in the region of the right facial and abducens nuclei. Co-infusion of a gadolinium tracer allowed for real-time observation of glucocerebrosidase distribution. No toxicity was noted out to a follow-up point of 9 months, however the patient remained in his pre-infusion neurologic state. Other aggressive therapeutic interventions that have been considered include bone marrow transplantation, gene therapy and substrate reduction therapy. Bone marrow transplantation has been undertaken in patients with chronic neuronopathic GD, again with improvement primarily in visceral involvement [101; 107]. However, due to the significant morbidity and mortality associated with this procedure, it is not recommended as current management.
Counseling
Professional counseling, including genetic counseling, can enhance the quality of life for families at high risk for or with established neuronopathic GD. Local and national organizations are important for facilitating support of families with individuals affected by GD. Counseling regarding end of life planning, as well as bereavement counseling, should be offered to parents and siblings of individuals with acute neuronopathic GD. Newborn testing, and/or testing through amniocentesis, may be considered to assist with early diagnosis and planning of future pregnancies.
Discussion
Type 2 GD involves a rapidly progressive downhill course, though it presents with a wide phenotypic spectrum, ranging from perinatal lethal to later-onset forms. Correlation of genotype-phenotype is difficult, especially given how the clinical phenotypic spectrum has expanded. Thus, researchers have proposed using the broader term “neuronopathic GD” to describe types 2 and 3, which classically refer to acute and chronic presentations, respectively. Furthermore, the broad phenotypic spectrum calls for the need to look for genetic and environmental modifiers.
Despite the delineation of the type 2 subtype of GD in 1927, we still face major challenges in understanding the pathogenesis of this disease. For example, it is unclear as to whether the disease is due to the accumulation of glucosylceramide or due to another toxic metabolite, or to both. Our understanding of the disease could benefit from reevaluation of the metabolism of accumulated substrates in reticuloendothelial and neuronal tissues.
The absence of a therapy to halt, slow down or stabilize the rate of neurological deterioration in the patients with this rarest phenotype of GD remains a major challenge. The recognition of prenatal lethal forms of the disorder indicates that neurologic damage may already be present prenatally in other forms. Early experiences with ERT have confirmed our inability to alter or prevent neurologic damage in type 2 GD. A better understanding of the factors contributing to the phenotypic diversity in Gaucher disease may help to uncover other targets for therapeutic interventions.
Table 2.
Genotype–phenotype correlations in Type 2 Gaucher disease
| Patients with manifestations in utero | |||
|---|---|---|---|
| Ethnic background | Genotype | Clinical presentation | Reference |
| Afghan | recD/recD | Hydrops | [108] |
| Ashkenazi Jewish | recA/IVS 10+2 T>G | Hydrops | [109] |
| Turkish | c.533delC/c.533delC | Hydrops | [6] |
| Cape Verdean | His311Arg/His311Arg | Hydrops | [110] |
| North American | recB/recB | Hydrops | [10] |
| North American | recD/recD | Hydrops | [73] |
| Lebanese | recD/recD | Ichthyosis/Hydrops | [94] |
| Greek | c.[1505+1-1505+12ins 1505, G>A]//recNci | Ichthyosis/Hydrops | [111] |
| Pakistani | Cys16Ser/Cys16Ser | Hydrops | [112] |
| Patients diagnosed at birth | |||
|---|---|---|---|
| Ethnic background | Genotype | Clinical presentation | Reference |
| Dutch/Surinamese | Arg359X/Val398Phe | Ichthyosis | [110] |
| Australian | c.1263-1317del/Arg257Gln | Ichthyosis | [7] |
| Greek | recC/His255Gln | Hepatosplenomegaly | [113] |
| Lebanese | Arg120Trp/Ser196Pro | Ichthyosis | [114] |
| Lebanese | Ser196 Pro/Ser196Pro | Respiratory difficulty | [73] |
| North American | Phe259Leu/Asn188Lys | Opisthotonus | [73] |
| North American | c.1263-1317del/Arg285His | Unknown | [73] |
| Ashkenazi Jewish | Leu444Pro/Leu444Pro+Ala456Pro | Strabismus | [67] |
| Spanish | Leu444Pro+Glu326Lys/Pro182Leu | Ichthyosis | [115] |
| Patients identified after several months of age | |||
|---|---|---|---|
| Ethnic Background | Genotype | Age at presentation | Reference |
| African American | recE/Glu41Lys+Arg131Cys | 8 months | [116] |
| North American | Leu444Pro/Gly202Arg | 8 months | [10] |
| North American | Leu444Pro/Unknown | 7 months | [10] |
| North American | Leu444Pro/recF | 5 months | [10] |
| Mexican | Arg131Leu/Arg131Leu | 5 months | [8] |
| Swedish | Leu444Pro/c.330delAla | 3 months | [73] |
| Belgian | Gly202Arg/Gly202Arg | 6 months | [73] |
| North American | recG/Leu444Pro/Arg257Gln | 6 months | [73] |
| North American | Leu444Pro/IVS +1G>A | 4 months | [73] |
| North American | Gly202Arg/Gly202Arg | 5 months | [73] |
| Kenyan/Filipino | Leu444Pro/IVS +1G>A | 4 months | [73] |
| Chinese | Phe213Ile/Asn382Lys | 10 months | [117] |
| Chinese | Phe213Ile/Leu383Arg | 7 months | [117] |
| Chinese | Tyr363Cys/Unknown | 5 months | [117] |
| Chinese | Met416Val/c.1263-1317del55 | 8 months | [117] |
| Spanish | Leu444Pro/Ile260Thr | 6 months | [115] |
| Patient diagnosed near or after first birthday (intermediate phenotype) | ||||
|---|---|---|---|---|
| Ethnic Background | Genotype | Age at diagnosis | Age at death | Reference |
| Black | Leu444Pro/Leu444Pro | 20 months | 3 years | [36] |
| French | Rec Ncil/Val394Leu | 12 months | 6 years | [36] |
| North American | Leu444Pro/Tyr304Cys | 12 months | 2 years | [36] |
| North American | Phe213Ile/Ser107Leu | 11 months | 3.5 years | [36] |
| North American | Leu444Pro/Gly202Arg | 11 months | 4 years | [36] |
| Chinese | Pro122Leu/Val375Leu | 12 months | unknown | [117] |
| Chinese | Leu385Pro/Leu444Pro | 11 months | 1.5 years | [117] |
Genotypes are presented using traditional nomenclature. The various forms of rec refer to different recombinant alleles. RecA carries the pseudogene sequence beginning from intron 3, and the crossover site is in intron 3; recB carries the 55-bp deletion in exon 9, as well as point mutations Asp409His, Leu444Pro, Ala456Pro, and Val460Val, and the crossover site is in the intron 8 or the beginning of exon 9 region; recC carries mutations Asp409His, Leu444Pro, Ala456Pro, and Val460Val, and the crossover site is in exon 9; recD, E, F carry mutations Leu444Pro, Ala456Pro, and Val460Val, and the crossover sites include exon 9-intron 9, intron 9 or intron 9-exon 10, recG carries the pseudogene sequence beginning from the 3′ UTR or has a duplicated pseudogene with a fusion between MTX and pseudometaxin, and the crossover site is in the 3′UTR or in MTX gene; recNci carries mutations Leu444Pro, Ala456Pro and Val460Val, and the crossover site is at the end of exon 9 or intron 9.
Acknowledgments
Special thanks to Dr. Ehud Goldin for his constructive suggestions. This research was supported by the Division of Intramural Research of the National Human Genome Research Institute, and the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Knudson AG, Kaplan WD. Jewish Chronic Disease Hospital (Brooklyn New York N.Y.). Isaac Albert Research Institute. Genetics of the spingolipidoses. In: Aronson SM, Volk BW, editors. Cerebral sphingolipidoses; a symposium on Tay-Sachs’ disease and allied disorders. Academic Press; New York: 1962. p. xvii.p. 456. [Google Scholar]
- 2.Fredrickson DS, Sloan HR. Glycosylceremide lipidoses: Gaucher’s disease. In: Stanbury JB, Wyngaarden JB, Fredrickson DS, editors. The metabolic basis of inherited disease. McGraw-Hill; New York: 1972. p. xiv.p. 1778. [Google Scholar]
- 3.Oberling C, Woringer P. La maladie de Gaucher chez le nourrisson. Rev franc de pédiat. 1927;3:475–532. [Google Scholar]
- 4.Beutler E, Grabowski GA. Gaucher disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. pp. 3635–3668. [Google Scholar]
- 5.Tayebi N, Stone DL, Sidransky E. Type 2 gaucher disease: an expanding phenotype. Mol Genet Metab. 1999;68:209–19. doi: 10.1006/mgme.1999.2918. [DOI] [PubMed] [Google Scholar]
- 6.Tayebi N, Cushner SR, Kleijer W, Lau EK, Damschroder-Williams PJ, Stubblefield BK, Den Hollander J, Sidransky E. Prenatal lethality of a homozygous null mutation in the human glucocerebrosidase gene. Am J Med Genet. 1997;73:41–7. doi: 10.1002/(sici)1096-8628(19971128)73:1<41::aid-ajmg9>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 7.Lipson AH, Rogers M, Berry A. Collodion babies with Gaucher’s disease--a further case. Arch Dis Child. 1991;66:667. doi: 10.1136/adc.66.5.667-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fujimoto A, Tayebi N, Sidransky E. Congenital ichthyosis preceding neurologic symptoms in two sibs with type 2 Gaucher disease. Am J Med Genet. 1995;59:356–8. doi: 10.1002/ajmg.1320590315. [DOI] [PubMed] [Google Scholar]
- 9.Ben Turkia H, Tebib N, Azzouz H, Abdelmoula MS, Ben Chehida A, Caillaud C, Ben Dridi MF. Phenotypic continuum of type 2 Gaucher’s disease: an intermediate phenotype between perinatal-lethal and classic type 2 Gaucher’s disease. J Perinatol. 2009;29:170–2. doi: 10.1038/jp.2008.179. [DOI] [PubMed] [Google Scholar]
- 10.Tayebi N, Reissner KJ, Lau EK, Stubblefield BK, Klineburgess AC, Martin BM, Sidransky E. Genotypic heterogeneity and phenotypic variation among patients with type 2 Gaucher’s disease. Pediatr Res. 1998;43:571–8. doi: 10.1203/00006450-199805000-00003. [DOI] [PubMed] [Google Scholar]
- 11.Conradi N, Kyllerman M, Mansson JE, Percy AK, Svennerholm L. Late-infantile Gaucher disease in a child with myoclonus and bulbar signs: neuropathological and neurochemical findings. Acta Neuropathol. 1991;82:152–7. doi: 10.1007/BF00293959. [DOI] [PubMed] [Google Scholar]
- 12.Ginsburg SJ, Groll M. Hydrops fetalis due to infantile Gaucher’s disease. J Pediatr. 1973;82:1046–8. doi: 10.1016/s0022-3476(73)80441-x. [DOI] [PubMed] [Google Scholar]
- 13.Gillan JE, Lowden JA, Gaskin K, Cutz E. Congenital ascites as a presenting sign of lysosomal storage disease. J Pediatr. 1984;104:225–31. doi: 10.1016/s0022-3476(84)80997-x. [DOI] [PubMed] [Google Scholar]
- 14.Dimmick JE, Applegrath DA. Inborn metabolic diseases of the perinatal infant. In: Wigglesworth JS, Singer DB, editors. Textbook of fetal and perinatal pathology. Blackwell Scientific Publications; Boston: 1991. pp. 593–643. [Google Scholar]
- 15.Sidransky E, Fartasch M, Lee RE, Metlay LA, Abella S, Zimran A, Gao W, Elias PM, Ginns EI, Holleran WM. Epidermal abnormalities may distinguish type 2 from type 1 and type 3 of Gaucher disease. Pediatr Res. 1996;39:134–41. doi: 10.1203/00006450-199601000-00020. [DOI] [PubMed] [Google Scholar]
- 16.Tybulewicz VL, Tremblay ML, LaMarca ME, Willemsen R, Stubblefield BK, Winfield S, Zablocka B, Sidransky E, Martin BM, Huang SP, et al. Animal model of Gaucher’s disease from targeted disruption of the mouse glucocerebrosidase gene. Nature. 1992;357:407–10. doi: 10.1038/357407a0. [DOI] [PubMed] [Google Scholar]
- 17.Holleran WM, Ginns EI, Menon GK, Grundmann JU, Fartasch M, McKinney CE, Elias PM, Sidransky E. Consequences of beta-glucocerebrosidase deficiency in epidermis. Ultrastructure and permeability barrier alterations in Gaucher disease. J Clin Invest. 1994;93:1756–64. doi: 10.1172/JCI117160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mignot C, Gelot A, Bessieres B, Daffos F, Voyer M, Menez F, Fallet Bianco C, Odent S, Le Duff D, Loget P, Fargier P, Costil J, Josset P, Roume J, Vanier MT, Maire I, Billette de Villemeur T. Perinatal-lethal Gaucher disease. Am J Med Genet A. 2003;120A:338–44. doi: 10.1002/ajmg.a.20117. [DOI] [PubMed] [Google Scholar]
- 19.Zay A, Choy FY, Macleod P, Tan-Dy CR. Perinatal lethal Gaucher’s disease without prenatal complications. Clin Genet. 2008;73:191–5. doi: 10.1111/j.1399-0004.2007.00941.x. [DOI] [PubMed] [Google Scholar]
- 20.Barbier C, Devisme L, Dobbelaere D, Noizet O, Nelken B, Gottrand F. Neonatal cholestasis and infantile Gaucher disease: a case report. Acta Paediatr. 2002;91:1399–401. doi: 10.1111/j.1651-2227.2002.tb02841.x. [DOI] [PubMed] [Google Scholar]
- 21.Choulot JJ, Bargiarelli M, Saint-Martin J. Portal hypertension complicating Gaucher’s disease (author’s transl) Arch Fr Pediatr. 1981;38:267–8. [PubMed] [Google Scholar]
- 22.Lachmann RH, Wight DG, Lomas DJ, Fisher NC, Schofield JP, Elias E, Cox TM. Massive hepatic fibrosis in Gaucher’s disease: clinico-pathological and radiological features. QJM. 2000;93:237–44. doi: 10.1093/qjmed/93.4.237. [DOI] [PubMed] [Google Scholar]
- 23.Mignot C, Doummar D, Maire I, De Villemeur TB. Type 2 Gaucher disease: 15 new cases and review of the literature. Brain Dev. 2006;28:39–48. doi: 10.1016/j.braindev.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 24.Forster J, Chambers JP, Peters SP, Lee RE, Glew RH. Acute neuropathic Gaucher disease in a black infant. J Pediatr. 1978;93:823–4. doi: 10.1016/s0022-3476(78)81093-2. [DOI] [PubMed] [Google Scholar]
- 25.Cervos-Navarro J, Zimmer C. Light microscopic and ultrastructural study on CNS lesions in infantile Gaucher’s disease. Clin Neuropathol. 1990;9:310–3. [PubMed] [Google Scholar]
- 26.Bernard R, Payan H, Maestraggi P, Albouy E. Maladie De Gaucher Du Nourrisson a Manifestations Cerebrales Predominantes - Etude Anatomo-Clinique. Archives Francaises De Pediatrie. 1961;18:393. [Google Scholar]
- 27.Franchetti L, Rasore-Quartino A, Marini C. Gaucher’s disease in infants. Report of a case with typical neurological symptomatology. Pediatria (Napoli) 1968;76:969–79. [PubMed] [Google Scholar]
- 28.Prows CA, Sanchez N, Daugherty C, Grabowski GA. Gaucher disease: enzyme therapy in the acute neuronopathic variant. Am J Med Genet. 1997;71:16–21. [PubMed] [Google Scholar]
- 29.Campbell PE, Harris CM, Sirimanna T, Vellodi A. A model of neuronopathic Gaucher disease. J Inherit Metab Dis. 2003;26:629–39. doi: 10.1023/b:boli.0000005619.14180.5c. [DOI] [PubMed] [Google Scholar]
- 30.Grafe M, Thomas C, Schneider J, Katz B, Wiley C. Infantile Gaucher’s disease: a case with neuronal storage. Ann Neurol. 1988;23:300–3. doi: 10.1002/ana.410230315. [DOI] [PubMed] [Google Scholar]
- 31.Jedidi H, Hamza B, Chadly A. Gaucher’s disease in infants with predominant neurological manifestations. Tunis Med. 1966;44:85–95. [PubMed] [Google Scholar]
- 32.Lin DS, Lin SP, Liang DC, Ho CS, Wu MC. Technetium-99m-HmPAO brain SPECT in infantile Gaucher’s disease. Pediatr Neurol. 1999;20:66–9. doi: 10.1016/s0887-8994(98)00101-5. [DOI] [PubMed] [Google Scholar]
- 33.Meyer R. New case of pseudo-bulbar syndrome of the infant (Gaucher’s disease of the infant) Revue Neurologique. 1934;61:612–617. [Google Scholar]
- 34.Tsai LP, Sue WC, Hwu WL, Lin KH, Wang TR. Oculomotor apraxia in a case of Gaucher’s disease with homozygous T1448C mutation. Zhonghua Min Guo Xiao Er Ke Yi Xue Hui Za Zhi. 1996;37:52–5. [PubMed] [Google Scholar]
- 35.Hoffmann B, Schwahn B, Knobbe CB, Vogel M, Blohm M, Mayatepek E, Wendel U. Acute neuronopathic Gaucher disease complicated by fatal gastrointestinal bleeding. Neuropediatrics. 2006;37:163–5. doi: 10.1055/s-2006-924556. [DOI] [PubMed] [Google Scholar]
- 36.Goker-Alpan O, Schiffmann R, Park JK, Stubblefield BK, Tayebi N, Sidransky E. Phenotypic continuum in neuronopathic Gaucher disease: an intermediate phenotype between type 2 and type 3. J Pediatr. 2003;143:273–6. doi: 10.1067/S0022-3476(03)00302-0. [DOI] [PubMed] [Google Scholar]
- 37.Kraoua I, Sedel F, Caillaud C, Froissart R, Stirnemann J, Chaurand G, Flodrops H, Tari S, Gourfinkel-An I, Mathieu S, Belmatoug N, de Villemeur TB, Mignot C. A French experience of type 3 Gaucher disease: Phenotypic diversity and neurological outcome of 10 patients. Brain Dev. 2010 doi: 10.1016/j.braindev.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 38.Vellodi A, Tylki-Szymanska A, Davies EH, Kolodny E, Bembi B, Collin-Histed T, Mengel E, Erikson A, Schiffmann R. Management of neuronopathic Gaucher disease: revised recommendations. J Inherit Metab Dis. 2009;32:660–4. doi: 10.1007/s10545-009-1164-2. [DOI] [PubMed] [Google Scholar]
- 39.Cherin P, Rose C, de Roux-Serratrice C, Tardy D, Dobbelaere D, Grosbois B, Hachulla E, Jaussaud R, Javier RM, Noel E, Clerson P, Hartmann A. The neurological manifestations of Gaucher disease type 1: the French Observatoire on Gaucher disease (FROG) J Inherit Metab Dis. 2010;33:331–8. doi: 10.1007/s10545-010-9095-5. [DOI] [PubMed] [Google Scholar]
- 40.Davies EH, Surtees R, DeVile C, Schoon I, Vellodi A. A severity scoring tool to assess the neurological features of neuronopathic Gaucher disease. J Inherit Metab Dis. 2007;30:768–82. doi: 10.1007/s10545-007-0595-x. [DOI] [PubMed] [Google Scholar]
- 41.Di Rocco M, Giona F, Carubbi F, Linari S, Minichilli F, Brady RO, Mariani G, Cappellini MD. A new severity score index for phenotypic classification and evaluation of responses to treatment in type I Gaucher disease. Haematologica. 2008;93:1211–8. doi: 10.3324/haematol.12379. [DOI] [PubMed] [Google Scholar]
- 42.Zimran A, Kay A, Gelbart T, Garver P, Thurston D, Saven A, Beutler E. Gaucher disease. Clinical, laboratory, radiologic, and genetic features of 53 patients. Medicine (Baltimore) 1992;71:337–53. [PubMed] [Google Scholar]
- 43.Walkley SU. Cellular pathology of lysosomal storage disorders. Brain Pathol. 1998;8:175–93. doi: 10.1111/j.1750-3639.1998.tb00144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burns GF, Cawley JC, Flemans RJ, Higgy KE, Worman CP, Barker CR, Roberts BE, Hayhoe FG. Surface marker and other characteristics of Gaucher’s cells. J Clin Pathol. 1977;30:981–8. doi: 10.1136/jcp.30.10.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parkin JL, Brunning RD. Pathology of the Gaucher cell. Prog Clin Biol Res. 1982;95:151–75. [PubMed] [Google Scholar]
- 46.Lee RE. Pathology of Gaucher disease. In: Desnick RJ, Gatt S, Grabowski GA, editors. Gaucher disease: a century of delineation and research (Progress in clinical and biological research, volume 95) Alan R. Liss; New York: 1981. pp. 177–218. [Google Scholar]
- 47.Lam KW, Desnick RJ. Biochemical properties of the tartrate-resistant acid phosphatase activity in Gaucher disease. Prog Clin Biol Res. 1982;95:267–78. [PubMed] [Google Scholar]
- 48.Cox TM. Gaucher disease: understanding the molecular pathogenesis of sphingolipidoses. J Inherit Metab Dis. 2001;24(Suppl 2):106–21. doi: 10.1023/a:1012496514170. discussion 87–8. [DOI] [PubMed] [Google Scholar]
- 49.Finn LS, Zhang M, Chen SH, Scott CR. Severe type II Gaucher disease with ichthyosis, arthrogryposis and neuronal apoptosis: molecular and pathological analyses. Am J Med Genet. 2000;91:222–6. [PubMed] [Google Scholar]
- 50.Sidransky E. New perspectives in type 2 Gaucher disease. Adv Pediatr. 1997;44:73–107. [PubMed] [Google Scholar]
- 51.Sharma R, Hudak ML, Perszyk AA, Premachandra BR, Li H, Monteiro C. Perinatal lethal form of Gaucher’s disease presenting with hemosiderosis. Am J Perinatol. 2000;17:201–6. doi: 10.1055/s-2000-9419. [DOI] [PubMed] [Google Scholar]
- 52.Kaga K, Ono M, Yakumaru K, Owada M, Mizutani T. Brainstem pathology of infantile Gaucher’s disease with only wave I and II of auditory brainstem response. J Laryngol Otol. 1998;112:1069–73. doi: 10.1017/s0022215100142483. [DOI] [PubMed] [Google Scholar]
- 53.Adachi M, Wallace BJ, Schneck L, Volk BW. Fine structure of central nervous system in early infantile Gaucher’s disease. Arch Pathol. 1967;83:513–26. [PubMed] [Google Scholar]
- 54.Lloyd OC, Norman RM, Urich H. The neuropathology of infantile Gaucher’s disease. J Pathol Bacteriol. 1956;72:121–31. doi: 10.1002/path.1700720116. [DOI] [PubMed] [Google Scholar]
- 55.Peters SP, Lee RE, Glew RH. Gaucher’s disease, a review. Medicine (Baltimore) 1977;56:425–42. doi: 10.1097/00005792-197709000-00004. [DOI] [PubMed] [Google Scholar]
- 56.Leech RW, Shuman RM, Putnam WD, Rance F, Jewett TT. Gaucher’s disease: a case history with extensive lipid storage in the brain. Am J Clin Pathol. 1985;83:516–9. doi: 10.1093/ajcp/83.4.516. [DOI] [PubMed] [Google Scholar]
- 57.Kaga M, Azuma C, Imamura T, Murakami T, Kaga K. Auditory brainstem response (ABR) in infantile Gaucher’s disease. Neuropediatrics. 1982;13:207–10. doi: 10.1055/s-2008-1059624. [DOI] [PubMed] [Google Scholar]
- 58.Brady RO, Barranger JA. Glucosylceramide lipidosis: Gaucher’s disease. In: Stanbury JB, Wyngaarden JB, Fredrickson DS, Goldstein JL, Brown MS, editors. The metabolic basis of inherited disease. McGraw-Hill; New York: 1983. pp. 842–856. [Google Scholar]
- 59.Hannun YA, Bell RM. Lysosphingolipids inhibit protein kinase C: implications for the sphingolipidoses. Science. 1987;235:670–4. doi: 10.1126/science.3101176. [DOI] [PubMed] [Google Scholar]
- 60.Orvisky E, Park JK, LaMarca ME, Ginns EI, Martin BM, Tayebi N, Sidransky E. Glucosylsphingosine accumulation in tissues from patients with Gaucher disease: correlation with phenotype and genotype. Mol Genet Metab. 2002;76:262–70. doi: 10.1016/s1096-7192(02)00117-8. [DOI] [PubMed] [Google Scholar]
- 61.Suzuki K. Twenty five years of the “psychosine hypothesis”: a personal perspective of its history and present status. Neurochem Res. 1998;23:251–9. doi: 10.1023/a:1022436928925. [DOI] [PubMed] [Google Scholar]
- 62.Bodennec J, Pelled D, Riebeling C, Trajkovic S, Futerman AH. Phosphatidylcholine synthesis is elevated in neuronal models of Gaucher disease due to direct activation of CTP: phosphocholine cytidylyltransferase by glucosylceramide. FASEB J. 2002;16:1814–6. doi: 10.1096/fj.02-0149fje. [DOI] [PubMed] [Google Scholar]
- 63.Korkotian E, Schwarz A, Pelled D, Schwarzmann G, Segal M, Futerman AH. Elevation of intracellular glucosylceramide levels results in an increase in endoplasmic reticulum density and in functional calcium stores in cultured neurons. J Biol Chem. 1999;274:21673–8. doi: 10.1074/jbc.274.31.21673. [DOI] [PubMed] [Google Scholar]
- 64.Lloyd-Evans E, Pelled D, Riebeling C, Bodennec J, de-Morgan A, Waller H, Schiffmann R, Futerman AH. Glucosylceramide and glucosylsphingosine modulate calcium mobilization from brain microsomes via different mechanisms. J Biol Chem. 2003;278:23594–9. doi: 10.1074/jbc.M300212200. [DOI] [PubMed] [Google Scholar]
- 65.Trajkovic-Bodennec S, Bodennec J, Futerman AH. Phosphatidylcholine metabolism is altered in a monocyte-derived macrophage model of Gaucher disease but not in lymphocytes. Blood Cells Mol Dis. 2004;33:77–82. doi: 10.1016/j.bcmd.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 66.Pelled D, Trajkovic-Bodennec S, Lloyd-Evans E, Sidransky E, Schiffmann R, Futerman AH. Enhanced calcium release in the acute neuronopathic form of Gaucher disease. Neurobiol Dis. 2005;18:83–8. doi: 10.1016/j.nbd.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 67.Aviner S, Garty BZ, Rachmel A, Baris HN, Sidransky E, Shuffer A, Attias J, Yaniv Y, Cohen IJ. Type 2 Gaucher disease occurs in Ashkenazi Jews but is surprisingly rare. Blood Cells Mol Dis. 2009;43:294–7. doi: 10.1016/j.bcmd.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chang YC, Huang CC, Chen CY, Zimmerman RA. MRI in acute neuropathic Gaucher’s disease. Neuroradiology. 2000;42:48–50. doi: 10.1007/s002340050012. [DOI] [PubMed] [Google Scholar]
- 69.Horowitz M, Wilder S, Horowitz Z, Reiner O, Gelbart T, Beutler E. The human glucocerebrosidase gene and pseudogene: structure and evolution. Genomics. 1989;4:87–96. doi: 10.1016/0888-7543(89)90319-4. [DOI] [PubMed] [Google Scholar]
- 70.Winfield SL, Tayebi N, Martin BM, Ginns EI, Sidransky E. Identification of three additional genes contiguous to the glucocerebrosidase locus on chromosome 1q21: implications for Gaucher disease. Genome Res. 1997;7:1020–6. doi: 10.1101/gr.7.10.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Armstrong LC, Komiya T, Bergman BE, Mihara K, Bornstein P. Metaxin is a component of a preprotein import complex in the outer membrane of the mammalian mitochondrion. J Biol Chem. 1997;272:6510–8. doi: 10.1074/jbc.272.10.6510. [DOI] [PubMed] [Google Scholar]
- 72.Long GL, Winfield S, Adolph KW, Ginns EI, Bornstein P. Structure and organization of the human metaxin gene (MTX) and pseudogene. Genomics. 1996;33:177–84. doi: 10.1006/geno.1996.0181. [DOI] [PubMed] [Google Scholar]
- 73.Stone DL, Tayebi N, Orvisky E, Stubblefield B, Madike V, Sidransky E. Glucocerebrosidase gene mutations in patients with type 2 Gaucher disease. Hum Mutat. 2000;15:181–8. doi: 10.1002/(SICI)1098-1004(200002)15:2<181::AID-HUMU7>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 74.Tsuji S, Choudary PV, Martin BM, Stubblefield BK, Mayor JA, Barranger JA, Ginns EI. A mutation in the human glucocerebrosidase gene in neuronopathic Gaucher’s disease. N Engl J Med. 1987;316:570–5. doi: 10.1056/NEJM198703053161002. [DOI] [PubMed] [Google Scholar]
- 75.Grabowski GA, Hopkin RJ. Enzyme therapy for lysosomal storage disease: principles, practice, and prospects. Annu Rev Genomics Hum Genet. 2003;4:403–36. doi: 10.1146/annurev.genom.4.070802.110415. [DOI] [PubMed] [Google Scholar]
- 76.Koprivica V, Stone DL, Park JK, Callahan M, Frisch A, Cohen IJ, Tayebi N, Sidransky E. Analysis and classification of 304 mutant alleles in patients with type 1 and type 3 Gaucher disease. American Journal of Human Genetics. 2000;66:1777–86. doi: 10.1086/302925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhao H, Keddache M, Bailey L, Arnold G, Grabowski G. Gaucher’s disease: identification of novel mutant alleles and genotype-phenotype relationships. Clin Genet. 2003;64:57–64. doi: 10.1034/j.1399-0004.2003.00100.x. [DOI] [PubMed] [Google Scholar]
- 78.Michelakakis H, Moraitou M, Dimitriou E, Santamaria R, Sanchez G, Gort L, Chabas A, Grinberg D, Dassopoulou M, Fotopoulos S, Vilageliu L. Homozygosity for the double D409H+H255Q allele in type II Gaucher disease. J Inherit Metab Dis. 2006;29:591. doi: 10.1007/s10545-006-0316-x. [DOI] [PubMed] [Google Scholar]
- 79.Torralba MA, Perez-Calvo JI, Pastores GM, Cenarro A, Giraldo P, Pocovi M. Identification and characterization of a novel mutation c.1090G>T (G325W) and nine common mutant alleles leading to Gaucher disease in Spanish patients. Blood Cells Mol Dis. 2001;27:489–95. doi: 10.1006/bcmd.2001.0410. [DOI] [PubMed] [Google Scholar]
- 80.Montfort M, Chabas A, Vilageliu L, Grinberg D. Functional analysis of 13 GBA mutant alleles identified in Gaucher disease patients: Pathogenic changes and “modifier” polymorphisms. Hum Mutat. 2004;23:567–75. doi: 10.1002/humu.20043. [DOI] [PubMed] [Google Scholar]
- 81.Pasmanik-Chor M, Madar-Shapiro L, Stein EO, Aerts H, Gatt S, Horowitz M. Expression of mutated glucocerebrosidase alleles in human cells. Hum Mol Genet. 1997;6:887–95. doi: 10.1093/hmg/6.6.887. [DOI] [PubMed] [Google Scholar]
- 82.Miocic S, Filocamo M, Dominissini S, Montalvo AL, Vlahovicek K, Deganuto M, Mazzotti R, Cariati R, Bembi B, Pittis MG. Identification and functional characterization of five novel mutant alleles in 58 Italian patients with Gaucher disease type 1. Hum Mutat. 2005;25:100. doi: 10.1002/humu.9301. [DOI] [PubMed] [Google Scholar]
- 83.Grace ME, Newman KM, Scheinker V, Berg-Fussman A, Grabowski GA. Analysis of human acid beta-glucosidase by site-directed mutagenesis and heterologous expression. J Biol Chem. 1994;269:2283–91. [PubMed] [Google Scholar]
- 84.Hodanova K, Melkova Z, Horowitz M, Hrebicek M. Transient expression of wild-type and mutant glucocerebrosidases in hybrid vaccinia expression system. Eur J Hum Genet. 2003;11:369–74. doi: 10.1038/sj.ejhg.5200974. [DOI] [PubMed] [Google Scholar]
- 85.Goker-Alpan O, Hruska KS, Orvisky E, Kishnani PS, Stubblefield BK, Schiffmann R, Sidransky E. Divergent phenotypes in Gaucher disease implicate the role of modifiers. J Med Genet. 2005;42:e37. doi: 10.1136/jmg.2004.028019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schueler UH, Kolter T, Kaneski CR, Zirzow GC, Sandhoff K, Brady RO. Correlation between enzyme activity and substrate storage in a cell culture model system for Gaucher disease. J Inherit Metab Dis. 2004;27:649–58. doi: 10.1023/b:boli.0000042959.44318.7c. [DOI] [PubMed] [Google Scholar]
- 87.Michelin K, Wajner A, Goulart Lda S, Fachel AA, Pereira ML, de Mello AS, Souza FT, Pires RF, Giugliani R, Coelho JC. Biochemical study on beta-glucosidase in individuals with Gaucher’s disease and normal subjects. Clin Chim Acta. 2004;343:145–53. doi: 10.1016/j.cccn.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 88.Maret A, Salvayre R, Troly M, Douste-Blazy L. Acid lability of the mutated glucosylceramide-beta-glucosidase in a lymphoid cell line from type 2 Gaucher disease. Enzyme. 1990;43:99–106. doi: 10.1159/000468712. [DOI] [PubMed] [Google Scholar]
- 89.Ginns EI, Tegelaers FP, Barneveld R, Galjaard H, Reuser AJ, Brady RO, Tager JM, Barranger JA. Determination of Gaucher’s disease phenotypes with monoclonal antibody. Clin Chim Acta. 1983;131:283–7. doi: 10.1016/0009-8981(83)90097-9. [DOI] [PubMed] [Google Scholar]
- 90.Boot RG, Renkema GH, Verhoek M, Strijland A, Bliek J, de Meulemeester TM, Mannens MM, Aerts JM. The human chitotriosidase gene. Nature of inherited enzyme deficiency. J Biol Chem. 1998;273:25680–5. doi: 10.1074/jbc.273.40.25680. [DOI] [PubMed] [Google Scholar]
- 91.Aerts JM, Hollak CE. Plasma and metabolic abnormalities in Gaucher’s disease. Baillieres Clin Haematol. 1997;10:691–709. doi: 10.1016/s0950-3536(97)80034-0. [DOI] [PubMed] [Google Scholar]
- 92.Deegan PB, Moran MT, McFarlane I, Schofield JP, Boot RG, Aerts JM, Cox TM. Clinical evaluation of chemokine and enzymatic biomarkers of Gaucher disease. Blood Cells Mol Dis. 2005;35:259–67. doi: 10.1016/j.bcmd.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 93.Moller HJ, de Fost M, Aerts H, Hollak C, Moestrup SK. Plasma level of the macrophage-derived soluble CD163 is increased and positively correlates with severity in Gaucher’s disease. Eur J Haematol. 2004;72:135–9. doi: 10.1046/j.0902-4441.2003.00193.x. [DOI] [PubMed] [Google Scholar]
- 94.Rowlands S, Murray H. Prenatal ultrasound findings in a fetus diagnosed with Gaucher’s disease (type 2) at birth. Prenat Diagn. 1997;17:765–9. doi: 10.1002/(sici)1097-0223(199708)17:8<765::aid-pd122>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 95.Staretz-Chacham O, Choi JH, Wakabayashi K, Lopez G, Sidransky E. Psychiatric and behavioral manifestations of lysosomal storage disorders. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2010 doi: 10.1002/ajmg.b.31097. In Press. [DOI] [PubMed] [Google Scholar]
- 96.Prasad A, Kaye EM, Alroy J. Electron microscopic examination of skin biopsy as a cost-effective tool in the diagnosis of lysosomal storage diseases. J Child Neurol. 1996;11:301–8. doi: 10.1177/088307389601100408. [DOI] [PubMed] [Google Scholar]
- 97.Bove KE, Daugherty C, Grabowski GA. Pathological findings in Gaucher disease type 2 patients following enzyme therapy. Hum Pathol. 1995;26:1040–5. doi: 10.1016/0046-8177(95)90097-7. [DOI] [PubMed] [Google Scholar]
- 98.Altarescu G, Hill S, Wiggs E, Jeffries N, Kreps C, Parker CC, Brady RO, Barton NW, Schiffmann R. The efficacy of enzyme replacement therapy in patients with chronic neuronopathic Gaucher’s disease. J Pediatr. 2001;138:539–47. doi: 10.1067/mpd.2001.112171. [DOI] [PubMed] [Google Scholar]
- 99.Bembi B, Zanatta M, Carrozzi M, Baralle F, Gornati R, Berra B, Agosti E. Enzyme replacement treatment in type 1 and type 3 Gaucher’s disease. Lancet. 1994;344:1679–82. doi: 10.1016/s0140-6736(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 100.Erikson A. Gaucher disease--Norrbottnian type (III). Neuropaediatric and neurobiological aspects of clinical patterns and treatment. Acta Paediatr Scand Suppl. 1986;326:1–42. [PubMed] [Google Scholar]
- 101.Vellodi A, Bembi B, de Villemeur TB, Collin-Histed T, Erikson A, Mengel E, Rolfs A, Tylki-Szymanska A. Management of neuronopathic Gaucher disease: a European consensus. J Inherit Metab Dis. 2001;24:319–27. doi: 10.1023/a:1010514614570. [DOI] [PubMed] [Google Scholar]
- 102.Schiffmann R, Heyes MP, Aerts JM, Dambrosia JM, Patterson MC, DeGraba T, Parker CC, Zirzow GC, Oliver K, Tedeschi G, Brady RO, Barton NW. Prospective study of neurological responses to treatment with macrophage-targeted glucocerebrosidase in patients with type 3 Gaucher’s disease. Ann Neurol. 1997;42:613–21. doi: 10.1002/ana.410420412. [DOI] [PubMed] [Google Scholar]
- 103.Migita M, Hamada H, Fujimura J, Watanabe A, Shimada T, Fukunaga Y. Glucocerebrosidase level in the cerebrospinal fluid during enzyme replacement therapy--unsuccessful treatment of the neurological abnormality in type 2 Gaucher disease. Eur J Pediatr. 2003;162:524–5. doi: 10.1007/s00431-001-0859-7. [DOI] [PubMed] [Google Scholar]
- 104.Desnick RJ, Schuchman EH. Enzyme replacement and enhancement therapies: lessons from lysosomal disorders. Nat Rev Genet. 2002;3:954–66. doi: 10.1038/nrg963. [DOI] [PubMed] [Google Scholar]
- 105.Lonser RR, Walbridge S, Murray GJ, Aizenberg MR, Vortmeyer AO, Aerts JM, Brady RO, Oldfield EH. Convection perfusion of glucocerebrosidase for neuronopathic Gaucher’s disease. Ann Neurol. 2005;57:542–8. doi: 10.1002/ana.20444. [DOI] [PubMed] [Google Scholar]
- 106.Lonser RR, Schiffman R, Robison RA, Butman JA, Quezado Z, Walker ML, Morrison PF, Walbridge S, Murray GJ, Park DM, Brady RO, Oldfield EH. Image-guided, direct convective delivery of glucocerebrosidase for neuronopathic Gaucher disease. Neurology. 2007;68:254–61. doi: 10.1212/01.wnl.0000247744.10990.e6. [DOI] [PubMed] [Google Scholar]
- 107.Ringden O, Groth CG, Erikson A, Granqvist S, Mansson JE, Sparrelid E. Ten years’ experience of bone marrow transplantation for Gaucher disease. Transplantation. 1995;59:864–70. [PubMed] [Google Scholar]
- 108.Sidransky E, Tayebi N, Stubblefield BK, Eliason W, Klineburgess A, Pizzolato GP, Cox JN, Porta J, Bottani A, DeLozier-Blanchet CD. The clinical, molecular, and pathological characterisation of a family with two cases of lethal perinatal type 2 Gaucher disease. J Med Genet. 1996;33:132–6. doi: 10.1136/jmg.33.2.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Reissner K, Tayebi N, Stubblefield BK, Koprivica V, Blitzer M, Holleran W, Cowan T, Almashanu S, Maddalena A, Karson EM, Sidransky E. Type 2 Gaucher disease with hydrops fetalis in an Ashkenazi Jewish family resulting from a novel recombinant allele and a rare splice junction mutation in the glucocerebrosidase locus. Mol Genet Metab. 1998;63:281–8. doi: 10.1006/mgme.1998.2675. [DOI] [PubMed] [Google Scholar]
- 110.Stone DL, van Diggelen OP, de Klerk JB, Gaillard JL, Niermeijer MF, Willemsen R, Tayebi N, Sidransky E. Is the perinatal lethal form of Gaucher disease more common than classic type 2 Gaucher disease? Eur J Hum Genet. 1999;7:505–9. doi: 10.1038/sj.ejhg.5200315. [DOI] [PubMed] [Google Scholar]
- 111.Michelakakis H, Dimitriou E, Moraitou M, Valari M, Yatrakou E, Mitsiadi V, Cozar M, Vilageliu L, Grinberg D, Karachristou K. Perinatal lethal form of Gaucher disease. Clinical and molecular characterization of a Greek case. Blood Cells Mol Dis. 2010;44:82–3. doi: 10.1016/j.bcmd.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 112.Church HJ, Cooper A, Stewart F, Thornton CM, Wraith JE. Homozygous loss of a cysteine residue in the glucocerebrosidase gene results in Gaucher’s disease with a hydropic phenotype. Eur J Hum Genet. 2004;12:975–8. doi: 10.1038/sj.ejhg.5201251. [DOI] [PubMed] [Google Scholar]
- 113.Sidransky E, Sherer DM, Ginns EI. Gaucher disease in the neonate: a distinct Gaucher phenotype is analogous to a mouse model created by targeted disruption of the glucocerebrosidase gene. Pediatr Res. 1992;32:494–8. doi: 10.1203/00006450-199210000-00023. [DOI] [PubMed] [Google Scholar]
- 114.Lui K, Commens C, Choong R, Jaworski R. Collodion babies with Gaucher’s disease. Arch Dis Child. 1988;63:854–6. doi: 10.1136/adc.63.7.854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chabas A, Gort L, Diaz-Font A, Montfort M, Santamaria R, Cidras M, Grinberg D, Vilageliu L. Perinatal lethal phenotype with generalized ichthyosis in a type 2 Gaucher disease patient with the [L444P;E326K]/P182L genotype: effect of the E326K change in neonatal and classic forms of the disease. Blood Cells Mol Dis. 2005;35:253–8. doi: 10.1016/j.bcmd.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 116.Sinclair G, Choy FY, Humphries L. A novel complex allele and two new point mutations in type 2 (acute neuronopathic) Gaucher disease. Blood Cells Mol Dis. 1998;24:420–7. doi: 10.1006/bcmd.1998.0210. [DOI] [PubMed] [Google Scholar]
- 117.Tang NL, Zhang W, Grabowski GA, To KF, Choy FY, Ma SL, Shi HP. Novel mutations in type 2 Gaucher disease in Chinese and their functional characterization by heterologous expression. Hum Mutat. 2005;26:59–60. doi: 10.1002/humu.9348. [DOI] [PubMed] [Google Scholar]