

Lead is a ubiquitous environmental contaminant; nearly 5% of American children are affected by lead poisoning (a blood lead level (BLL) of 10 μgdL−1 or higher).[1] Even lower BLLs have been shown to cause many subtle health effects in children. Lead, which is found in paint and soil, causes toxicity by several possible mechanisms. Pb2+ interacts with several zinc enzymes or proteins (such as carbonic anhydrase, acetylcholine esterase, Cys2His2 “zinc-finger” proteins, and acid phophatases)[2,3] and calcium ion binding proteins (calmodulin, calbindin, and troponin C).[4] Inhibition of protein function is induced by alternative coordination number and structural preferences.[5,6] Pb2+ is a chemically interesting toxin in that it can replace calcium and sometimes zinc in “hard” active sites that are oxygen/nitrogen rich; it can also attack softer ligands, such as all-sulfur-containing zinc ion coordination sites. Among the sulfur-rich targets for Pb2+ are glutathione and metallothioneines, which cause perturbations of essential metal ion homeostasis.
Aminolevulinic acid dehydratase (ALAD), a zinc-dependent enzyme, is inhibited by a femtomolar concentrations of Pb2+.[2] ALAD is found in yeast and mammals and is involved in the second step of heme biosynthesis. Pb2+-poisoned ALAD blocks the synthesis of hemoglobin, causing anemia in mammals. Furthermore, toxic levels of aminolevulinic acid can result. The crystal structure of ALAD contains an unusual Zn(Cys)3H2O site, where Zn2+ is substituted by Pb2+ in a trigonal pyramidal geometry.[7] The high affinity of Pb2+ to cysteine thiolates is presumably due to the high enthalpy of Pb–S bond formation and the preferred PbS3 coordination geometry in thiolate-rich sites of proteins.[8] A number of peptides[9–11] and small-molecule synthetic models[12] have been used to understand the chemistry of the PbII-poisoned ALAD. UV/Vis and EXAFS studies on the metalloregulatory protein Pb-PbrR691 and Pb2+ model compounds reveal that Pb2+ binds in a PbS3 environment.[13]
Heteronuclear magnetic resonance spectroscopy with nuclei such as 43Ca, 113Cd, and 199Hg has been a powerful tool for studying the active site structures of metalloenzymes and their model compounds.[14–21] Similarly, lead provides an NMR active nucleus (207Pb, nuclear spin I = 1/2) with a natural abundance of 22.6% and a relatively good receptivity (11.7 times higher than that of 13C).[22] However, owing to the wide chemical shift range (over 16000 ppm), the use of 207Pb NMR spectroscopy is non-trivial.[8,22–24]
Recently, Vogel and co-workers utilized 207Pb NMR spectroscopy (using isotopically enriched 207Pb) to study Pb2+ binding to the Ca2+ site of calcium-binding proteins, including calmodulin (CaM).[25] To our knowledge, this is the sole example of 207Pb NMR as a probe in metalloproteins. Of great importance, there are no reported 207Pb spectra for sulfur-rich metalloproteins. A number of small synthetic molecules with or without mixed O, S, and N donor ligands (for example S2O2, S2N2, N2O4, N3O3, N4, N6) have been characterized using this technique.[22–24,26–28] The 207Pb NMR signal for the thiol-rich binding sites should be shifted further downfield than that of oxygen- and nitrogen-rich calcium-binding sites. Thus, we can distinguish PbS3 versus PbS3O coordination environments very easily by using 207Pb NMR.[22] The coordination number and geometry of the Pb2+ ion can also be examined.[29] Dean, Payne, Christou, and their co-workers have synthesized [Ph4As][Pb(SPh)3] and characterized complexes in non-aqueous media using 207Pb NMR spectroscopy.[30–32] Despite these studies, no significant advancement of 207Pb NMR has been accomplished to explore the thiolate-rich proteins scaffolds. Herein, we present the 207Pb NMR for a physiologically relevant coordination environment of thiolate-rich metallopeptides in the preferred homoleptic trigonal pyramidal geometry for PbII ions by utilizing three-strand coiled-coil peptides. To our knowledge, this is the first report of 207Pb NMR spectroscopy used in a Cys3 motif that can be a direct probe for the thiol-rich metalloenzymes, such as ALAD, which are directly implicated in human lead poisoning.
We have utilized new three-strand coiled-coil (3-SCC) peptides (CoilSer and TRI family) to obtain insight into how toxic metals, such as Hg2+, As3+, Cd2+, and Pb2+, bind in thiol-rich sites of metalloenzymes.[19–21,33–38] These α-helical peptide families have heptad repeats of seven amino acid residues that contain hydrophobic leucine residues in the a (first) and d (fourth) positions (Table 1).[39] The resultant 3-SCC has all of the hydrophobic leucine residues packed on the interior of the 3-SCC and hydrophilic residues (e and g) on the exterior, forming salt bridges that stabilize the coiled coil. A metal binding site can be created by the substitution of a leucine by cysteine in the a or d positions of the heptad repeat unit to give a metal binding site within the hydrophobic core of the peptide trimer.[35,39] The sulfur atoms in an a site are oriented towards the interior of the coiled coil and preorganized for metal binding, whereas the sulfur atoms in a d site point away from the interior towards the helical interface, creating a relatively larger cavity (Figure 1). Metal ions such as Cd2+, Hg2+, and As3+ are preferentially bound to the a site, whereas larger metal ions such as Pb2+ prefer the d site.[40]
Table 1.
Sequence and name of the peptides used in this study.
| Peptides | Sequence |
|---|---|
| abcdefg abcdefg abcdefg abcdefg | |
| CSL12C | Ac-E WEALEKK LAACESK LQALEKK LEALEHG-NH2 |
| CSL16C | Ac-E WEALEKK LAALESK CQALEKK LEALEHG-NH2 |
| CSL12AL16C | Ac-E WEALEKK LAAAESK CQALEKK LEALEHG-NH2 |
| BabyL12C | Ac-G LKALEEK LKACEEK LKALEEK G-NH2 |
| GrandL12AL16CL26C | Ac-G LKALEEK LKAAEEK CKALEEK LKACEEK LKALEEK G-NH2 |
Figure 1.
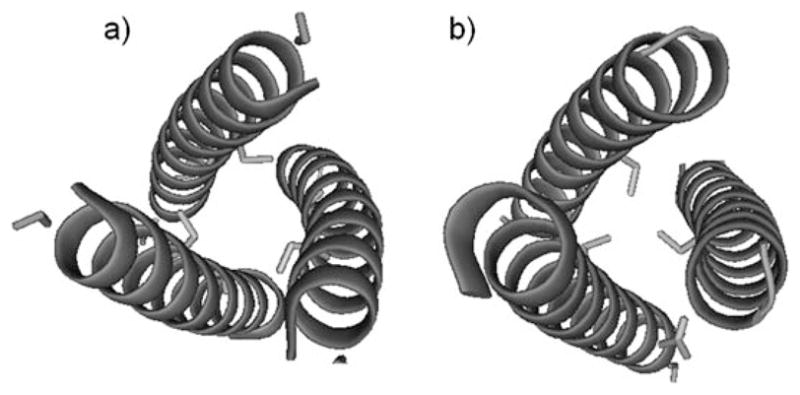
Pymol representation showing the orientation of cysteine residues. a) CoilSerL12C, d site; b) CoilSerL16C, a site. Cysteine side chains are shown as sticks and peptide α helices are shown as coils. PDB code: 3H5F.[45]
The use of these well-defined peptides provides several advantages for detecting the 207Pb signal in an all-thiolate (homoleptic) environment that mimics ALAD. In contrast to small organic lead thiolate complexes, the designed peptides are highly soluble and stable in water. Therefore, 207Pb NMR studies at relatively high concentrations (10–12 mM) and physiological pH is successful without peptide aggregation or Pb(OH)2 precipitation. Similar preparations were unsuccessful for cysteine, which was due to the precipitation of a PbCys3 complex that can only be dissolved at high pH (>12). Thus, a solely PbS3 coordination environment cannot be attained by cysteine at physiological pH.
Binding studies of Pb2+ to TRIL12C and TRIL16C were previously monitored by UV/Vis, EXAFS, and CD spectroscopy[35] and shown to have high affinity (>108M−1) with the peptides studied herein. The presence of a characteristic ligand-to-metal charge-transfer (LMCT) band at about 345 nm (ε ≈ 3500Lmol−1cm−1) is indicative of PbS3 in a trigonal pyramidal geometry. Recent EXAFS studies by Matzapetakis et al. identified a three-coordinate Pb2+ site in Pb(TRIL16C)3− with Pb–S scatters at 2.63 Å. Similar results have been reported by Giedroc and co-workers for the preference of Pb2+ for a PbS3 coordination environment in the metalloregulatory protein CadC.[41,42] This data compares well with Pb–S scatters found for the lead-inhibited active site of ALAD. Therefore, the metallopeptides described herein are in close approximation to the Pb2+-inhibited active site of ALAD and related lead-binding proteins.
The natural-abundance 207Pb NMR spectra of all of the metal-lopeptides with a single binding site had a single lead signal at 5500–5800 ppm with broad linewidths (15–25 pm) (Figure 2). Similar broad signals have been reported in protein NMR studies with 199Hg and 205Tl, which may be due to nuclear relaxation by chemical shift anisotropy (CSA).[43,44] The peptides with a d metal binding site have downfield chemical shifts relative to those of the a site peptides. Several interesting trends can be extracted from these data: similar chemical shifts for the peptides having a d site are seen independent of the length of the peptide or the intrinsic stability of the aggregate, Pb-(BabyL12C)3− (δ = 5786 ppm, w1/2 = 20 ppm) and Pb-(CSL12C)3− (δ = 5814 ppm, w1/2 = 18 ppm). These chemical shifts are similar to the previously reported trigonal pyramidal PbS3 structure of a small synthetic organic compound [Ph4As][Pb(SPh)3] (δ = 5828 ppm).[31] The possibility of formation of nitrogen- or oxygen-bound species can be ruled out as a distinct upfield chemical shift has been observed for mixed-donor ligand types (PbN2S, δ = 5318 ppm; PbS2O2, δ = 4100–4500 ppm).[8,24,26] Therefore, the observed 207Pb signal can be confidently assigned to the formation of a PbS3 coordination environment.
Figure 2.
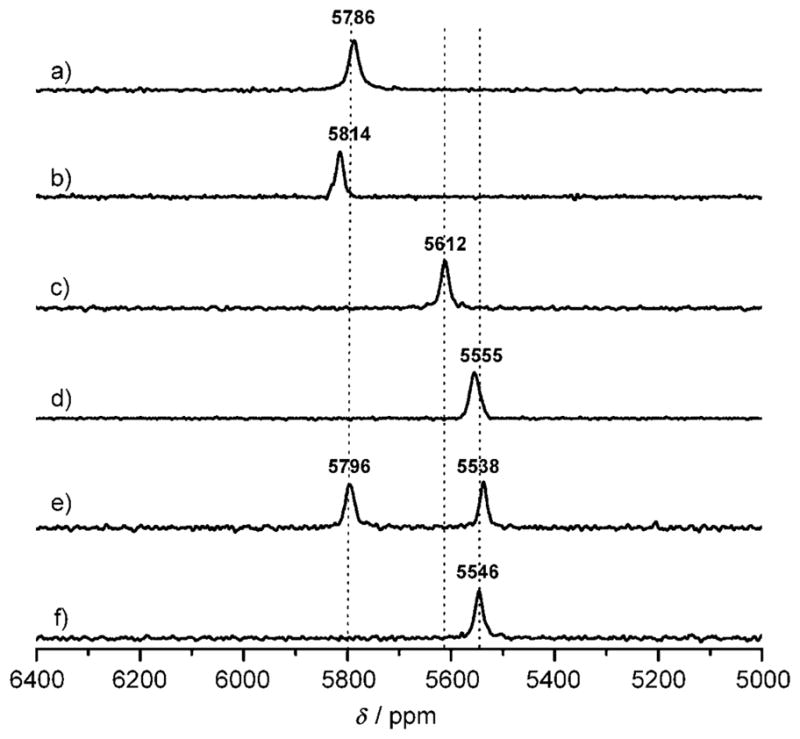
Natural-abundance 207Pb NMR spectra (104.435 MHz) of PbII-bound three-strand coiled-coil peptides (10–12 mM): a) Pb-(BabyL12C)3−, b) Pb(CSL12C)3−, c) Pb(CSL16C)3−, d) Pb(CSL12AL16C)3−, e) Pb2(GrandL12AL16L26C)32−, f) Pb(Grand-L12AL16L26C)3−. All spectra were recorded for 10–12 h using natural-abundance Pb(NO3)2, (207Pb = 22.6%), pH 7.35 ± 0.05, at 25°C.
An upfield chemical shift of approximately 200 ppm was observed when a lead-binding site was created in the a site peptide (Pb(CSL16C)3−; δ = 5612 ppm, w1/2 = 18 ppm). Furthermore, substitution of a sterically less-demanding amino acid residue above the a metal binding site leads to a 55–60 ppm further upfield shift (Pb(CSL12AL16C)3−; δ = 5555 ppm, w1/2 = 25 ppm). We conclude that 207Pb NMR spectroscopy is sufficiently sensitive to distinguish between two similar trigonal pyramidal PbS3 centers based on the a versus d substitution pattern of the peptide. Furthermore, the upfield shift in the Pb(CSL12AL16C)3− suggests that the additional space provided above the PbS3 plane by the alanine accommodates the bulky Pb2+ lone pair within the helical assembly better. A similar rationale can be applied to the longer Pb2(GrandL12AL16L26C)32−, in which the leucine layer above the a site is substituted by alanine and leucines at the 16th (a) and 26th (d) sites are replaced with cysteines, creating two Pb2+ binding sites. The d site has a 207Pb NMR signal at δ = 5796 ppm (Figure 2e; w1/2 = 17 ppm); this value is between the signal obtained for Pb(BabyL12C)3− and Pb(CSL12C)3−, but clearly in the region of d cysteine ligands. A 207Pb peak at 5538 ppm (w1/2 = 18 ppm) is assigned to the a site with a hole oriented towards the N terminus, which compares well with the value obtained for Pb(CSL12AL16C)3−. These data illustrate that 207Pb NMR is sufficiently sensitive to discriminate complexation of Pb2+ in these similar yet non-identical sites. Furthermore, the simultaneous observation of both peaks and the relatively narrow linewidths suggest that the Pb2+ ions are in slow exchange on the NMR timescale.
Interestingly, the addition of one equivalent of Pb(NO3)2 into GrandL12AL16L26C gives a 207Pb signal at the a site region only (δ = 5546 ppm, w1/2 = 19 ppm), indicating a selective binding of Pb2+ to the a site with a hole above (Figure 2f). This observation is in contrast to the previously reported a versus d preference for Pb2+ complexation. The inversion of selectivity is a consequence of the added space made available by substituting alanine for leucine. The steriochemically active lone pair of Pb2+ no longer clashes with the alkyl side chain of leucine and can now be accommodated within the generated cavity, leading to a higher thermodynamic stability of lead binding. It has been shown that Cd2+, which forms a mixture of three- (CdS3) and four-coordinate (CdS3(H2O)) structures with TRIL16C, becomes fully four-coordinate, using an exogenous water ligand, when space is made available above the metal by the same leucine to alanine substitution in TRIL12AL16C.[46]
These results provide experimental confirmation of the importance of the lone pair on the selectivity of Pb2+ for sulfur sites in proteins such as PbrR691 and ALAD (in which Pb2+ displaces Zn2+ from three cysteines and one exogeneous water rather than the five-coordinate zinc binding site with nitrogen and oxygen atoms as ligands). These data suggest that there will be a significant preference for Pb2+ to be sequestered into an environment that provides sufficient space to accommodate the large lone pair of this ion. Such a situation exists when Pb2+ displaces Zn2+ in ALAD.
Despite the fact that lead-substituted ALAD is strongly implicated in lead toxicity, to date there have been no examples of biomolecules or model compounds that have exhibited a 207Pb NMR spectrum for a PbS3 center in aqueous solution at physiological pH. Our ability to detect such a chromophore using natural-abundance isotope levels, to illustrate the sensitivity of the chemical shift range and to demonstrate how slight amino acid sequence changes affect lead binding to a protein are significant advances for understanding the biochemistry of human lead poisoning. Our data also indicate that Pb2+ exchange between homoleptic thiolate sites is slow on the NMR timescale. Most importantly, we have demonstrated that high-quality spectra do not require expensive enriched 207Pb, but can be obtained using natural-abundance lead salts. We hope that 207Pb NMR spectroscopy may now be useful to identify and characterize proteins associated with lead toxicity directly from human samples if a sufficiently concentrated sample can be obtained.
Experimental Section
Peptide synthesis and purification
All of the peptides were synthesized on an Applied Biosystems 433A peptide synthesizer by using standard Fmoc/tBu-based protection strategies on Rink Amide MBHA resin (0.25 mmol scale) with HBTU/HOBt/DIEPA coupling methods.[47] The peptides were then cleaved from the resin either using a mixture of 95% trifluoroacetic acid (TFA), 2.5% ethanedithiol, and 2.5% triisopropyl silane or a mixture of 90% TFA, 5% thioanisole, 3% ethanedithiol, and 2% anisole. The cleaved peptide solutions were filtered and then evaporated under a dry N2 flow to give a glassy film. The white film was washed with ice-cold diethyl ether (peroxide free) to obtain a crude peptide powder. The peptides were dissolved in 10% acetic acid, lyophilized, and subsequently purified by reverse-phase HPLC (Waters 600 with Vydac protein and peptide C-18 column; solvent A: 0.1% TFA in H2O; solvent B: 0.1% TFA in acetonitrile/H2O (9:1); linear gradient 20–80% of solvent B over 30 min; flow rate: 10 mL min−1). The identity and purity of the purified peptides was confirmed by electrospray mass spectrometry (Waters) in positive-ion mode and by analytical HPLC. The purity of peptides was more than 95%. All of the peptides studied herein were N-terminally acetylated and C-terminally amidated. A list of the peptides synthesized with their sequences is given in Table 1.
Natural abundance 207Pb NMR spectroscopy
NMR samples (10–12 mM) were prepared under a nitrogen atmosphere by dissolving of pure and dried peptide (70–80 mg) in D2O/H2O (15%, 400–500 μL; degassed). The peptide concentration was determined by Ellman’s test.[48] Calculated amounts of 250 mM Pb(NO3)2 (natural abundance) stock solution was added to the peptide solution and the pH was adjusted by the slow addition of a small aliquot of KOH/15% D2O until the pH reached 7.35 ± 0.05. All of the 207Pb NMR spectra were recorded at a frequency of 104.435 MHz on a Varian 500 MHz NMR spectrometer at room temperature (25°C) using 60° pulses, a 20 ms relaxation delay, and a 20 ms acquisition time. Initially, a large spectral width of 300 KHz was used to find the position of the peak. Once the peak position was found, the spectral window was reduced to about 166 KHz. However, the chemical shift difference was not observed when the spectral window was about 300 KHz (3000 ppm), 166 KHz (1500 ppm), or 50 KHz (500 ppm). A linear prediction was performed to remove the noise, and the real FID was determined before the data processing. After zero-filling, the data (128 K data points) were processed with an exponential line broadening of 200–250 Hz using the software MestRe-C.[49] The 207Pb NMR chemical shifts are reported downfield from tetramethyllead (δ = 0 ppm; toluene) using 1.0M Pb(NO3)2 salt (natural) as an external standard (δ = −2990 ppm, D2O, 25 °C; relative to PbMe4).
Footnotes
V.L.P. thanks the National Institute of Health for support of this research (R01 ES0 12236).
References
- 1.Lanphear BP. Science. 1998;281:1617–1618. doi: 10.1126/science.281.5383.1617. [DOI] [PubMed] [Google Scholar]
- 2.Simons TJB. Eur J Biochem. 1995;234:178–183. doi: 10.1111/j.1432-1033.1995.178_c.x. [DOI] [PubMed] [Google Scholar]
- 3.Zawia NH, Crumpton T, Brydie M, Reddy GR, Razmiafshari M. Neurotoxicology. 2000;21:1069–1080. [PubMed] [Google Scholar]
- 4.Simons TJB. Neurotoxicology. 1993;14:77–85. [PubMed] [Google Scholar]
- 5.Razmiafshari M, Zawia NH. Toxicol Appl Pharmacol. 2000;166:1–12. doi: 10.1006/taap.2000.8950. [DOI] [PubMed] [Google Scholar]
- 6.Zawia NH, Sharan R, Brydie M, Oyama T, Crumpton T. Dev Brain Res. 1999;107:291–298. doi: 10.1016/s0165-3806(98)00023-6. [DOI] [PubMed] [Google Scholar]
- 7.Erskine PT, Senior N, Awan S, Lambert R, Lewis G, Tickle IJ, Sarwar M, Spencer P, Thomas P, Warren MJ, Shoolingin-Jordan PM, Wood SP, Cooper JB. Nat Struct Biol. 1997;4:1025–1031. doi: 10.1038/nsb1297-1025. [DOI] [PubMed] [Google Scholar]
- 8.Andersen RJ, diTargiani RC, Hancock RD, Stern CL, Goldberg DP, Godwin HA. Inorg Chem. 2006;45:6574–6576. doi: 10.1021/ic060497z. [DOI] [PubMed] [Google Scholar]
- 9.Payne JC, ter Horst MA, Godwin HA. J Am Chem Soc. 1999;121:6850–6855. [Google Scholar]
- 10.Godwin HA. Curr Opin Chem Biol. 2001;5:223–227. doi: 10.1016/s1367-5931(00)00194-0. [DOI] [PubMed] [Google Scholar]
- 11.Magyar JS, Weng TC, Stern CM, Dye DF, Rous BW, Payne JC, Bridgewater BM, Mijovilovich A, Parkin G, Zaleski JM, Penner-Hahn JE, Godwin HA. J Am Chem Soc. 2005;127:9495–9505. doi: 10.1021/ja0424530. [DOI] [PubMed] [Google Scholar]
- 12.Bridgewater BM, Parkin G. J Am Chem Soc. 2000;122:7140–7141. [Google Scholar]
- 13.Chen PR, He C. Curr Opin Chem Biol. 2008;12:214–221. doi: 10.1016/j.cbpa.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 14.Armitage IM, Pajer RT, Uiterkamp AJMS, Chlebowski JF, Coleman JE. J Am Chem Soc. 1976;98:5710–5712. doi: 10.1021/ja00434a058. [DOI] [PubMed] [Google Scholar]
- 15.Armitage IM, Uiterkamp AJMS, Chlebowski JF, Coleman JE. J Magn Reson. 1978;29:375–392. [Google Scholar]
- 16.Forsen S, Johansson C, Linse S. Methods Enzymol. 1993;227:107–118. doi: 10.1016/0076-6879(93)27007-4. [DOI] [PubMed] [Google Scholar]
- 17.Ellis PD. Science. 1983;221:1141–1146. doi: 10.1126/science.221.4616.1141. [DOI] [PubMed] [Google Scholar]
- 18.Summers MF. Coord Chem Rev. 1988;86:43–134. [Google Scholar]
- 19.Dieckmann GR, McRorie DK, Tierney DL, Utschig LM, Singer CP, Ohalloran TV, PennerHahn JE, DeGrado WF, Pecoraro VL. J Am Chem Soc. 1997;119:6195–6196. [Google Scholar]
- 20.Matzapetakis M, Farrer BT, Weng TC, Hemmingsen L, Penner-Hahn JE, Pecoraro VL. J Am Chem Soc. 2002;124:8042–8054. doi: 10.1021/ja017520u. [DOI] [PubMed] [Google Scholar]
- 21.Utschig LM, Wright JG, Dieckmann G, Pecoraro VL, O’Halloran TV. Inorg Chem. 1995;34:2497–2498. [Google Scholar]
- 22.Wrackmeyer B, Horchler K. Annu Rep NMR Spectrosc. 1990;22:249–306. [Google Scholar]
- 23.Claudio ES, ter Horst MA, Forde CE, Stern CL, Zart MK, Godwin HA. Inorg Chem. 2000;39:1391–1397. doi: 10.1021/ic9909431. [DOI] [PubMed] [Google Scholar]
- 24.Rupprecht S, Franklin SJ, Raymond KN. Inorg Chim Acta. 1995;235:185–194. [Google Scholar]
- 25.Aramini JM, Hiraoki T, Yazawa M, Yuan T, Zhang MJ, Vogel HJ. J Biol Inorg Chem. 1996;1:39–48. [Google Scholar]
- 26.Rupprecht S, Langemann K, Lugger T, McCormick JM, Raymond KN. Inorg Chim Acta. 1996;243:79–90. [Google Scholar]
- 27.Pedrido R, Bermejo MR, Romero MJ, Vazquez M, Gonzalez-Noya AM, Maneiro M, Rodriguez MJ, Fernandez MI. Dalton Trans. 2005:572–579. doi: 10.1039/b416296j. [DOI] [PubMed] [Google Scholar]
- 28.Reger DL, Ding Y, Rheingold AL, Ostrander RL. Inorg Chem. 1994;33:4226–4230. [Google Scholar]
- 29.Claudio ES, Godwin HA, Magyar JS. Prog Inorg Chem. 2003;51:1–144. [Google Scholar]
- 30.Arsenault JJI, Dean PAW. Can J Chem. 1983;61:1516–1523. [Google Scholar]
- 31.Dean PW, Vittal JJ, Payne NC. Inorg Chem. 1984;23:4232–4236. [Google Scholar]
- 32.Christou G, Folting K, Huffman JC. Polyhedron. 1984;3:1247–1253. [Google Scholar]
- 33.Farrer BT, McClure CP, Penner-Hahn JE, Pecoraro VL. Inorg Chem. 2000;39:5422–5423. doi: 10.1021/ic0010149. [DOI] [PubMed] [Google Scholar]
- 34.Iranzo O, Ghosh D, Pecoraro VL. Inorg Chem. 2006;45:9959–9973. doi: 10.1021/ic061183e. [DOI] [PubMed] [Google Scholar]
- 35.Matzapetakis M, Ghosh D, Weng TC, Penner-Hahn JE, Pecoraro VL. J Biol Inorg Chem. 2006;11:876–890. doi: 10.1007/s00775-006-0140-7. [DOI] [PubMed] [Google Scholar]
- 36.Dieckmann GR, McRorie DK, Lear JD, Sharp KA, DeGrado WF, Pecoraro VL. J Mol Biol. 1998;280:897–912. doi: 10.1006/jmbi.1998.1891. [DOI] [PubMed] [Google Scholar]
- 37.Farrer BT, Pecoraro VL. Proc Natl Acad Sci USA. 2003;100:3760–3765. doi: 10.1073/pnas.0336055100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghosh D, Pecoraro VL. Inorg Chem. 2004;43:7902–7915. doi: 10.1021/ic048939z. [DOI] [PubMed] [Google Scholar]
- 39.Peacock AFA, Iranzo O, Pecoraro VL. Dalton Trans. 2009:2271–2280. doi: 10.1039/b818306f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matzapetakis M. University of Michigan. Ann Arbor: 2004. [Google Scholar]
- 41.Busenlehner LS, Cosper NJ, Scott RA, Rosen BP, Wong MD, Giedroc DP. Biochemistry. 2001;40:4426–4436. doi: 10.1021/bi010006g. [DOI] [PubMed] [Google Scholar]
- 42.Busenlehner LS, Weng TC, Penner-Hahn JE, Giedroc DP. J Mol Biol. 2002;319:685–701. doi: 10.1016/S0022-2836(02)00299-1. [DOI] [PubMed] [Google Scholar]
- 43.Utschig LM, Bryson JW, O’Halloran TV. Science. 1995;268:380–385. doi: 10.1126/science.7716541. [DOI] [PubMed] [Google Scholar]
- 44.Aramini JM, Krygsman PH, Vogel HJ. Biochemistry. 1994;33:3304–3311. doi: 10.1021/bi00177a022. [DOI] [PubMed] [Google Scholar]
- 45.Peacock AFA, Stuckey JA, Pecoraro VL. Angew Chem. 2009;121:7507–7510. [Google Scholar]; Angew Chem Int Ed. 2009;48:7371–7374. doi: 10.1002/anie.200902166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iranzo O, Jakusch T, Lee KH, Hemmingsen L, Pecoraro VL. Chem Eur J. 2009;15:3761–3772. doi: 10.1002/chem.200802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chan WC, White PD. Fmoc Solid-Phase Peptide Synthesis: A Practical Approach. Oxford University Press; New York: 2000. [Google Scholar]
- 48.Ellman GL. Arch Biochem Biophys. 1958;74:443–450. doi: 10.1016/0003-9861(58)90014-6. [DOI] [PubMed] [Google Scholar]
- 49.Cobas C, Cruces J, Sardina FJ. MestRe-C version 2.3. Universidad de Santiago de Compostela; Spain: 2000. [Google Scholar]