

Abstract
DNA double-strand break (DSB) repair occurs within chromatin and can be modulated by chromatin modifying enzymes. Here we identify the related human histone deacetylases HDAC1 and HDAC2 as two participants in the DNA-damage response (DDR). We show that acetylation of histone H3 lysine 56 (H3K56) is regulated by HDAC1/2, and that HDAC1/2 are rapidly recruited to DNA-damage sites to promote H3K56 hypo-acetylation. Furthermore, we establish that HDAC1/2-depleted cells are hypersensitive to DNA-damaging agents and exhibit sustained DNA-damage signaling, phenotypes that reflect defective DSB repair, particularly by the pathway of non-homologous end-joining (NHEJ). Collectively, these results demonstrate that HDAC1 and HDAC2 function in the DDR by promoting DSB repair and thus provide important insights into the radio-sensitizing effects of HDAC inhibitors that are being developed as cancer therapies.
Keywords: HDAC1, HDAC2, DNA damage, chromatin, non-homologous end-joining
Because DNA damage represents a formidable challenge to the integrity of genetic material, cells have evolved multifaceted systems, collectively termed the DNA-damage response (DDR), to detect, signal and repair various types of DNA damage1,2. DNA double-strand breaks (DSBs) represent one of the most challenging forms of DNA damage which, if left unrepaired, can trigger cellular death and can contribute to human diseases, including cancer1. In eukaryotes, DSBs are repaired by two main pathways: non-homologous end-joining (NHEJ), which operates throughout the cell cycle, and homologous recombination (HR) which is limited to S and G2 cell-cycle phases3. For NHEJ, the Ku70/Ku80 complex loads onto free DNA ends where it helps recruit the DDR protein kinase DNA-PK (DNA-dependent protein kinase)4, as well as other factors, including the nuclease Artemis and the ligase IV-XRCC4 complex, which are required for NHEJ to ensue5. NHEJ occurs rapidly within cells and mostly requires minimal processing of DNA ends. By contrast, HR requires extensive DNA end-resection to create stretches of single-stranded DNA (ssDNA) that bind factors such as RPA and RAD51 to promote the various steps in HR3. Since HR and NHEJ function together during certain phases of the cell cycle, mechanisms must exist to modulate repair-pathway-choice for each DSB. Although DNA end-resection, as well as the cell cycle dependent phosphorylations, regulates DSB repair pathway usage, it is tempting to speculate that the chromatin context of a DSB will also influence whether NHEJ or HR repairs it.
In regards to the above, it is becoming increasingly clear that, as well as exerting profound effects on transcription, chromatin structure also markedly influences DNA-damage recognition, signalling and repair6-8. Histones, the basic protein units of chromatin, are subjected to modifications such as acetylation, phosphorylation and ubiquitylation that can alter the properties of chromatin and thus influence DNA-based processes, including DNA damage responses9. For instance, activation of the apical DDR protein kinases ATM (ataxia telangiectasia mutated), ATR (ATM and Rad3 related) and DNA-PK leads to phosphorylation of the histone variant H2AX on chromatin flanking DSB sites10. This phospho-form of H2AX, termed gH2AX, is one of the earliest chromatin markers of DSBs and is critical for the accumulation of repair and signalling proteins, such as 53BP1, into foci at DNA-damage sites, thus promoting DSB signalling and repair6-8,11. Likewise, the recruitment of Ubiquitin E3 ligases RNF8 and RNF168 to DSB sites mediates histone H2A and H2AX ubiquitylation that is vital for effective execution of the DDR12-16. Histone acetylations, together with the enzymes mediating their addition and removal, have also been implicated in the DDR, although their modes of action are less well defined compared to histone phosphorylations and ubiquitylations17-19. Notably, by conducting a screen for DNA-damage-responsive histone modifications in human cells, we recently found that H3K9 and H3K56 acetylations (H3K9Ac and H3K56Ac) are negatively regulated by DNA damage20. Other studies, however, reported differential effects of DNA damage on H3K56Ac in mammalian cells21-23, thus highlighting the need for additional analysis of H3K56Ac and its functions in mammalian cells.
Histone acetylations are regulated by the concerted actions of histone acetyltransferases (HATs) and histone deacetylases (HDACs) that work by adding and removing acetyl groups from lysine residues. Human cells contain eighteen known HDACs that fall into four classes. Class I HDACs, which include HDAC1, HDAC2, HDAC3 and HDAC8, are most closely related to budding yeast Rpd324,25. These HDACs are ubiquitously expressed and are mainly localized within the nucleus, where they function in diverse processes, including transcription. Class II HDACs consist of HDAC4-7, HDAC9 and HDAC10. These HDACs are most closely rlated to yeast Hda1, are not ubiquitously expressed and localize chiefly to the cytoplasm, although they can become nuclear through their regulation by 14-3-3 proteins and phosphorylations25. Class III HDACs, also known as “Sirtuins”, are NAD-dependent and consist of SIRT1-7 that are closely related to the yeast Sir2 protein26. Sirtuins can be nuclear or cytoplasmic, and are involved in diverse processes including metabolism, transcription and the DDR. The HDAC Class IV consists of only one member, HDAC11, whose functions are as yet poorly understood. A key goal for studies on HDAC enzymes is to define their in vivo targets, as this can provide crucial insights into how these multi-tasking enzymes carry out their diverse biological functions26. Such work is also of medical relevance, given that broad specificity HDAC inhibitors are promising anti-cancer drugs that can selectively kill cancer cells and are known to sensitize cells to treatments that induce DNA damage24,27,28.
In this study, our previous observation of a decrease of H3K56Ac upon DNA damage prompted us to evaluate the role of HDACs in the DDR. Here, we describe how human HDAC1 and HDAC2 respond to DNA damage and mediate changes in histone acetylation, including H3K56, following DNA-damage induction. Furthermore, by defining the effects of impairing HDAC1/2 function, we establish that these enzymes serve as important components of the DDR by promoting DSB signalling and repair, principally through their requirement for effective NHEJ.
RESULTS
HDAC1 and HDAC2 localize to sites of DNA damage
We previously demonstrated that H3K56Ac levels are reduced by treatments that induce DNA damage, including the drug phleomycin that produces DSBs20. Although damage-dependent H3K56Ac loss could occur through multiple mechanisms, we reasoned that it might reflect the action of one or more HDAC. Consequently, we tested whether levels of H3K56Ac and their reduction upon DNA damage were affected by the Class I/II HDAC inhibitors Trichostatin A (TSA) and sodium butyrate (NaB), or by the Sirtuin inhibitor, nicotinamide. Notably, in cells that had not been treated with a DNA-damaging agent, TSA and NaB but not nicotinamide, resulted in increased H3K56Ac levels (Fig. 1a). This suggested that Class I/II HDACs regulate H3K56Ac levels under normal growth conditions. Furthermore, while DNA damage resulted in decreased H3K56Ac in cells treated with no inhibitors or with nicotinamide, for TSA- or NaB-treated cells, H3K56Ac levels were similar under both unchallenged and DNA-damaging conditions (Fig. 1a). Collectively, these data indicated that H3K56Ac levels are mainly controlled by Class I/II HDACs and that the activity of such HDACs is also needed for H3K56Ac loss upon DNA damage induction.
Figure 1.

HDAC1 and HDAC2 localize to, and H3K56Ac is reduced at, sites of DNA damage. (a) Treatment with the Class I/II HDAC inhibitors sodium butyrate (NaB) or Trichostatin A (TSA) increases H3K56Ac levels and blocks damage-dependent deacetylation of H3K56. U2OS cells were untreated or treated with HDAC inhibitors followed by DNA damage induction by phleomycin. Samples were analyzed by western blotting (WB) with the indicated antibodies; γH2AX detection denotes DNA damage. (b) The Class I HDACs, HDAC1 and HDAC2, localize to sites of laser-induced DNA damage. DNA damage was generated by laser micro-irradiation followed by immunofluorescence (IF, 5 min after damage) with the indicated antibodies. γH2AX marks the “laser-lines” containing damaged DNA. (c) H3K56Ac levels are reduced at DNA-damage sites in both Hela and U2OS cells. DNA damage was generated as in b followed by IF (2 h) for H3K56Ac and γH2AX. Boxes show zoomed sections of damaged areas. (d) H3K56Ac levels are reduced at site-specific DSBs. Site-specific DSBs were induced by addition of 4-OHT into U2OS cells containing AsiSI-ER. 4-OHT treated or untreated cells were analyzed by ChIP followed by qPCR. AsiSI sites (DSB1 and DSB2) were compared to locus lacking AsiSI sites (no DSB). For H3 acetylations, qPCR values were normalized to input and total H3. All values were then normalized to untreated samples for each respective antibody and genomic locus (n=3, error bars represent S.E.M.). (e) H3K56Ac loss, and HDAC1 and HDAC2 localization, at DNA-damage sites is blocked by HDAC inhibitors. Analysis was performed as in a and b.
Based on the above results, we speculated that the HDAC(s) responsible for H3K56 deacetylation might be activated by DNA damage and/or localized to DNA damage sites. Furthermore, we reasoned that Class I HDACs would be prime candidates for such factors, as these enzymes are mainly nuclear. Notably, when we screened all Class I HDACs for their ability to localize to sites of laser-induced DNA damage; we could detect the recruitment of endogenous HDAC1 and HDAC2, but not HDAC3 or HDAC8, to sites of laser micro-irradiation under these conditions and with commercially available antibodies (Fig. 1b; antibody specificities for HDAC1 and HDAC2 are verified in Supplementary Fig. 1). HDAC1/2 accumulation occurred within 5 minutes of DNA-damage induction and then gradually declined, being no longer visible after 30 minutes (Fig. 1b and data not shown). Consistent with HDAC1 and HDAC2 functioning at DNA damage sites, we also observed that staining for H3K56Ac was diminished at sites of DNA damage containing γH2AX (Fig. 1c and Supplementary Fig. 2a; compare the dotted lines representing average H3K56Ac intensities of lines 1 and 2 that represent damaged and undamaged regions, respectively). By contrast, other histone acetylations that we analyzed did not change detectably in response to laser micro-irradiation (Supplementary Figs. 2e). Furthermore, by using immunofluorescence (IF) to detect IR-induced foci (IRIF), we found that H3K56Ac exhibited punctate staining, the intensity of which was inversely correlated with that of γH2AX or 53BP1 (Supplementary Fig. 2b,c,d). Quantification of H3K56Ac intensities demonstrated that this modification was indeed diminished in damaged regions (IRIF) versus undamaged regions (Supplementary Fig. 2b,c).
H3K56Ac levels are reduced upon DNA damage
To analyze the behaviour of H3K56Ac at site-specific DNA breaks, as well as to complement our western blotting and IF data with another technique, we used a U2OS cell line expressing a 4-hydroxytamoxifen (4-OHT) inducible restriction enzyme (AsiSI-ER) that causes DSBs at specific genomic loci29 (see Supplementary Fig. 3 for confirmation of DNA damage induction by 4-OHT addition, which induced γH2AX and 53BP1 foci). By using chromatin immunoprecipitation (ChIP) analysis, we found that AsiSI-ER induction decreased H3K56Ac at DSB sites, while no reduction was observed at another site that did not contain an AsiSI sequence (Fig. 1d; note that the effects on H3K56Ac are specific because H3K14Ac did not decrease at either DSB site, while γH2AX was increased). Taken together, these results established that H3K56Ac levels are reduced at sites of DNA damage.
Since HDAC1 and HDAC2 localize to sites of laser micro-irradiation, and because we had observed H3K56Ac loss at such sites, we next determined whether HDAC inhibitors could block the localized decrease of H3K56Ac in damaged regions. Indeed, we found that Class I/II HDAC inhibitors suppressed H3K56 deacetylation at sites of laser micro-irradiation and also blocked the localization of HDAC1 and HDAC2 to DNA-damage regions (Fig. 1e). Taken together, these data suggested that HDAC1/2 regulate H3K56Ac and deacetylate H3K56 at sites of DNA damage through their localization to damaged DNA via an HDAC inhibitor-sensitive process.
In addition to being produced in response to exogenously applied agents, DNA damage is also generated when cells enter oncogene-induced senescence (OIS) or replicative senescence30-32. Notably, we found that human diploid fibroblast exhibiting OIS due to RAS over-expression32-34 displayed decreased H3K56Ac compared to control cells, as well as H3K56Ac exclusion from DNA-damage sites (Fig. 2a,b). Similarly, late-passage human diploid fibroblasts displaying replicative senescence exhibited lower H3K56Ac levels than earlier-passage cells (Fig. 2c.d and Supplementary Fig. 4a). Importantly, the observed decrease in H3K56Ac levels were not due to effects of DNA damage on cell-cycle distributions, as this histone mark is not cell cycle regulated20,35; (see also Supplementary Fig. 4b).
Figure 2.

H3K56Ac levels decrease during oncogene-induced and replicative senescence and H4K16Ac is a DNA-damage responsive histone mark. (a) H3K56Ac levels are reduced in 53BP1-positive, RAS-overexpressing OIS cells. Human BJ primary fibroblast cells untreated or undergoing OIS were analyzed by IF with the indicated antibodies. Note that H3K56Ac is excluded from 53BP1 foci. (b) H3K56Ac is reduced and H4K16Ac is increased in OIS cells as analyzed by WB from cells in a. H3K56Ac is reduced in replicative-senescent BJ cells (arrows) by IF (c) and WB (d). PD: population doublings. (e and f) H4K16Ac levels rapidly decrease (5 min) then increase (2 h) at sites of damage following laser-microirradiation. Cells were analyzed as in Fig. 1b.
During the course of the above studies, we found that H4K16Ac was reduced by replicative senescence but enhanced upon OIS. Furthermore, by carrying out laser-micro-irradiation studies, we found that H4K16Ac displayed a biphasic response at DNA-damage sites: being reduced at early time points, (Fig. 2e) but enhanced at later times (Fig. 2f). The kinetics of H4K16Ac loss was similar to that of H3K56Ac by both IRIF (Supplementary Fig. 2b) and at sites of laser micro-irradiation (Supplementary Fig. 4c). Our interpretation of these data is that, while both H3K56Ac and H4K16Ac signals are rapidly lost at DNA-damage sites, H4K16Ac levels then increase at damaged regions as a consequence of ongoing DNA repair. This would also provide an explanation for the differential effects seen for H4K16Ac in senescent cells because increased H4K16Ac could reflect ongoing DNA repair during OIS but not during replicative senescence. Together, the above findings thereby established that H3K56Ac levels are decreased by both exogenous and endogenously produced DNA damage, and indicated that H4K16Ac is also a DNA-damage responsive histone mark.
HDAC1 and HDAC2 deacetylate H3K56 and H4K16
As HDAC1 is implicated in cellular senescence36,37, and given the similar kinetics between HDAC1/2 recruitment to DNA damage sites and the loss of H3K56 and H4K16 acetylation at sites of damage, we tested whether HDAC1 and its close relative, HDAC2, regulated these histone marks. Indeed, siRNA-mediated depletion of HDAC1 and HDAC2 together, but not of either alone, caused H3K56 and H4K16 hyper-acetylation in U2OS cells (Fig. 3a; as shown in Supplementary Fig. 5a,b, HDAC1/2 depletion did not appreciably affect cell-cycle distributions). Additionally, HDAC1/2-depletion in HeLa cells also resulted in hyper-acetylation of H3K56Ac as detected by two different anti-H3K56Ac antibodies (Fig. 3b; also see below). By contrast, depletion of HDAC3, which we had found not to be detectably recruited to DNA-damage sites, did not yield H3K56Ac or H4K16Ac hyper-acetylation (Fig. 3c). Importantly, we carried out various experiments to verify that the observed effects reflected HDAC1/2 depletion rather than off-target effects. Thus, we found that induction of H3K56Ac and H4K16Ac was also observed when we used pair-wise combinations of various HDAC1- or HDAC2-targeting siRNA oligonucleotides (Supplementary Fig. 5c). Furthermore, the effects of HDAC1/2 depletion on H3K56Ac and H4K16Ac levels were circumvented when a siRNA-resistant HDAC1 derivative was employed (Fig. 4a). For this analysis, we created a cell line expressing a form of HDAC1 (siResA-HDAC1) that was resistant to HDAC1 siRNA A. Importantly, siResA-HDAC1 rescued the hyper-acetylation of H3K56 and H4K16 in cells depleted for endogenous HDAC1 by siRNA A but did not rescue H3K56 hyper-acetyation caused by another HDAC1 siRNA (B) that targets both endogenous HDAC1 and siResA-HDAC1. Such effects were also observed in IF experiments for H3K56Ac where cells expressing GFP-siResA-HDAC1 displayed lower H3K56Ac levels than those lacking HDAC1 (Fig. 4b; only one siRNA resistant construct was required because, as shown in Supplementary Fig. S5d, depletion of HDAC1 or HDAC2 alone did not result in H3K56 hyper-acetylation).
Figure 3.

HDAC1 and HDAC2, co-regulate H3K56Ac and H4K16Ac. (a) siRNA depletion of HDAC1/2 together, but not singularly, causes H3K56 and H4K16 hyper-acetylation in U2OS cells; siLuciferase (siLuc) used as control. After siRNA transfections, cells were analyzed by WB. (b) HDAC1/2 regulates H3K56Ac in HeLa cells. Experiments were performed as in Figure 1a. An increase in H3K56Ac in siHDAC1/2-depleted cells was confirmed with two antibodies against H3K56Ac (Upstate and Epitomics). (b) The down-regulation of the Class I HDAC, HDAC3, does not affect H3K56Ac or H4K16Ac levels. Cells were analyzed as in a.
Figure 4.

HDAC1 and HDAC2 specifically and directly target H3K56 and H4K16 (a) siResistant HDAC1 (siResA-GFP-HDAC1) rescues defects of H3K56 and H4K16 hyper-acetylation in siHDAC1/2 U2OS cells. (see methods for details; Endo = endogenous protein). (b) U2OS cells expressing siResA-GFP-HDAC1 rescues H3K56 hyper-acetylation in cells depleted for endogenous HDAC1/2. Cells were treated with the indicated siRNAs and analyzed by IF with a H3K56Ac antibody (Epitomics). (c) HDAC1/2-depleted U2OS cells exhibit H3K56 and H4K16 hyper-acetylation. Quantification of samples in a was performed with the LI-COR Odyssey infrared imaging system; ratios of signals in siHDAC1/2 versus siLuc are given and histone-modification levels were normalized to histone H3 or H4 levels. (d) Recombinant HDAC1 deacetylates H3K56 and H4K16 in vitro. (e) Depletion of SIRT1/2 does not increase H3K56Ac and H4K16Ac levels in HeLa cells. Experiments were performed as in a. siRNA sequences targeting SIRT1 and SIRT2 were obtained from a previous study22. Lower right panel confirms the documented cytoplasmic localization of SIRT2 and the efficient depletion of GFP-SIRT2 by fluorescence microscopy.
Additional analyses confirmed that H3K56Ac and H4K16Ac are HDAC1/2 targets because, of the acetylations we tested, these increased most in HDAC1/2-depleted cells (Fig. 4c). Furthermore, recombinant HDAC1 deacetylated these sites on calf histones in vitro, suggesting that H3K56Ac and H4K16Ac are likely direct HDAC1/2 targets (Fig. 4d). Consistent with this, treating cells with the Class I/III HDAC inhibitors TSA or NaB enhanced both H3K56Ac and H4K16Ac (Supplementary Fig. 5e). In yeast, H3K56Ac is deacetylated by Sirtuin proteins, a family of HDACs also implicated in H3K56Ac deacetylation in mammalian cells22,38,39. However, when we treated cells with the Sirtuin inhibitor nicotinamide or depleted cells of SIRT1 and SIRT2, little or no increase in H3K56Ac or H4K16Ac was observed (Fig. 4e and Supplementary Fig. 5f; note that analysis of p53 acetylation, a known Sirtuin target40,41, revealed that nicotinamide was effective in our assays). Nevertheless, we note that SIRT6-depletion has been shown to enhance H3K56Ac specifically at telomeres21 and that knockout of SIRT6 or depletion of HDAC1/2 in mouse cells results in increased H3K56Ac23,42. Regardless, our data revealed that H3K56Ac and H4K16Ac levels are mainly controlled by HDAC1 and HDAC2 in human cells and highlight how regulation of H3K56Ac may differ in different organisms and genomic locations.
HDAC1 and HDAC2 are required for the DDR, particularly through NHEJ
During the above work, we noted that, while HDAC1/2 depletion on its own triggered only nominal gH2AX production, it reproducibly enhanced the levels of gH2AX produced in cells upon phleomycin treatment (Fig. 3b; also see below). This observation, along with HDAC1 and HDAC2 localizing to sites of DNA damage, suggested functional roles for these HDACs in the DDR. Consistent with this idea, we found that HDAC1/2 depletion caused hypersensitivity to the DSB-inducing agents IR and phleomycin (Fig. 5a). While investigating the molecular basis for these phenotypes, we found that HDAC1/2 depletion or HDAC-inhibitor treatment led to higher amounts of gH2AX being produced in response to DNA damage than in control cells (Fig. 5b, Fig. 1a and Supplementary Fig. 6a-d; as shown in Supplementary Fig. 7a-c, these phenotypes were rescued when cells were complemented with a siRNA resistant HDAC1). Furthermore, DNA-damage induced phosphorylations of CHK1, CHK2 and p53 were also higher and more sustained in HDAC1/2 depleted cells than in control cells (Fig. 5b). HDAC1/2 inhibition also enhanced the amount of γH2AX at a site-specific DSB as detected by ChIP analysis (see below).
Figure 5.

HDAC1/2-depleted cells exhibit defective DNA-damage responses. (a) HDAC1/2-depleted cells exhibit hypersensitivity to IR and phleomycin; data are from triplicate experiments, and CtIP acts as a positive control; error bars, +/− S.D. (b) HDAC1/2-depleted cells exhibit increased DDR signaling as monitored by WB; samples were from untreated (U) cells or cells harvested at the indicated times after release from a 2 h phleomycin-treatment (*cross-reacting proteins). (c) HDAC inhibition following IR causes increased γH2AX. Immediately after IR exposure, cells were incubated in the absence (Untr) or presence of NaB (5 mM) or TSA (1.3 mM), and then were analyzed by IF 2 h or 8 h later. Undamaged cells were untreated or treated with HDAC inhibitors and analyzed at 8 h. (d) HDAC1/2 depletion causes DSB-repair deficiency. Neutral comet assays were performed on control (siLuc) and HDAC1/2 depleted cells; representative images are shown. Quantification of tail moments for cells untreated (Untr) or treated with phleomycin (e) or IR (f). Cells were DNA damaged (2 h Phleo, (60 μg ml−1) or IR, (20 Gy) followed by 1 h recovery (Rec).
Collectively, the above data suggested that HDAC1/2 depletion or inhibition does not affect the amount of DNA damage produced by DSB-inducing agents but, rather, impairs DNA repair. In accord with this idea, and arguing against indirect transcriptional effects caused by HDAC1/2 depletion, when we treated cells with DNA-damaging agents and added HDAC inhibitors immediately afterwards, this also resulted in gH2AX hyper-phosphorylation (Fig. 5c). Moreover, by using comet assays, we established that while the levels of endogenous and induced DSBs were similar in control and HDAC1/2 depleted cells, the repair of IR- and phleomycin-induced DSBs was markedly impaired by HDAC1/2 depletion (Fig. 5d-f). Furthermore, HDAC1/2-depleted cells exhibited defective DSB repair as revealed by a modest reduction in homologous recombination (HR) as measured by a I-Sce1 based HR assay, and a major defect in non-homologous end-joining (NHEJ) as measured by random-plasmid integration (Fig. 6a,b; as shown in Supplementary Fig. S7d, HDAC1/2-depleted cells expressed normal levels of core NHEJ factors, thus ruling out indirect transcriptional effects). In addition, HDAC1/2 depletion led to higher levels of DNA-damage induced auto-phosphorylation (S2056p) and ATM-mediated phosphorylation (T2609p) of the NHEJ factor DNA-PKcs (Fig. 6c). By contrast, HDAC1/2 depletion did not markedly affect DDR markers arising when cells were treated with camptothecin (Fig. 6d), which produces DSBs in S-phase that are repaired by HR43. These data therefore indicated that HDAC1 and HDAC2 promote proper DSB signalling and repair, predominately through their requirement for effective NHEJ.
Figure 6.
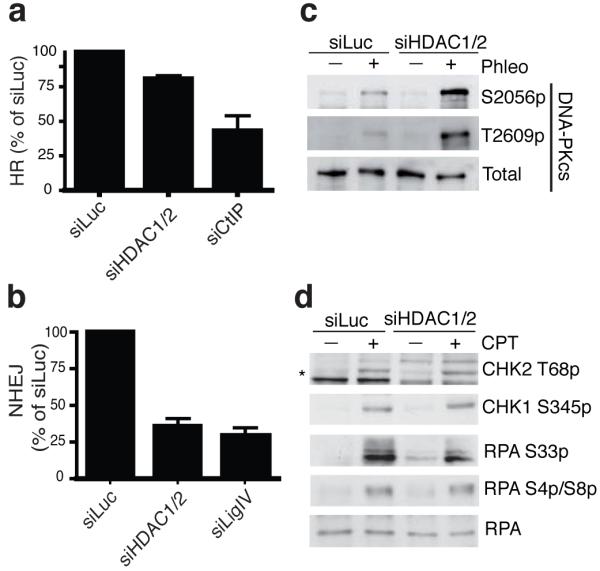
HDAC1 and HDAC2 promote efficient DNA repair, particularly through NHEJ. HDAC1/2 depleted cells are defective in HR (a) and NHEJ (b); graphs represent three experiments +/− S.D. CtIP and Ligase IV depletions are controls. (c) DNA-PKcs is hyper-phosphorylated upon DNA damage in HDAC1/2-depleted cells. Experiments were performed as in Fig. 5b. (d) DNA-damage induction resulting from camptothecin (CPT) does not hyper-activate DDR signaling in siHDAC1/2 cells. Cells were siRNA treated and processed as in b but treated with CPT (1 μM for 1 h; * denotes CHK2 T68p).
HDACs, including HDAC1 and HDAC2, influence NHEJ factor persistence at DSBs
Finally, we used live-cell imaging to determine whether HDAC1/2 affected the recruitment and/or persistence of the NHEJ factors Ku70 and Artemis at sites of DNA damage44,45. Thus, we found that green-fluorescent protein (GFP)-tagged Ku70 and Artemis were rapidly recruited to sites of laser-induced DNA damage (Fig. 7a,b; bright staining of Ku70-GFP under all conditions reflects nucleolar localization, as reported previously46). Notably, both Ku70 and Artemis displayed increased association with damaged regions in cells treated with HDAC inhibitors as compared to control cells (Fig. 7a,b; see Fig. 7c for quantitative graphs). Furthermore, HDAC1/2 depletion produced similar results (Fig. 7d). Because it is established that NHEJ factor persistence at DSB sites occurs under conditions of defective NHEJ44, these data were in accord with HDAC1/2 depletion rendering cells NHEJ defective. However, we do not think that these data solely reflect a NHEJ defect because the persistence of Artemis at sites of DNA damage was greater in HDAC1/2-depleted cells than in Ligase IV-depleted cells (Fig. 7d), despite Ligase IV depletion causing an equal, if not greater, NHEJ deficiency than HDAC1/2 depletion (Fig. 6b). In corroboration with these findings, by using ChIP analyses, we found that HDAC inhibitors caused the NHEJ factors Ku70/80 and XRCC4 to display enhanced associations with an AsiSI-ER induced site-specific DSB (Fig. 7e). Together, these results thereby established that HDAC1 and HDAC2 promote NHEJ, at least in part by regulating the proper assembly and/or disassembly of NHEJ factors from DSB sites.
Figure 7.

HDAC inhibition results in NHEJ factor persistence at sites of DNA damage. (a) and (b) Persistence of KU70 and Artemis is increased after HDAC inhibition at DSBs. Accumulation of KU70-GFP or ARTEMIS-GFP to sites of laser-microirradiation (white circle) from cells either in the absence or presence of the HDAC inhibitor NaB are shown. (c) The difference in average fluorescence intensity in the damaged versus an undamaged region is plotted in relationship to time for each condition as described in methods for KU70-GFP and ARTEMIS-GFP. Error bars represent S.E.M. (d) HDAC1 and HDAC2 depletion affects ARTEMIS persistence at DSBs. siRNA experiments were performed as in b and quantifications were done as in h. (e) HDAC inhibition results in enhanced NHEJ factor binding and γH2AX production at a site-specific DSB. U2OS-AsiSI-ER cells were untreated or treated with 5 mM NaB for 16 h. Cells were pre-treated for 1 h with 10 mM DNA-PKcs inhibitor followed by 2 h with 4-OHT, then analyzed by ChIP as in Fig. 1d with the indicated antibodies (n=2, error bars= S.E.M.).
DISCUSSION
Our data suggest a model in which HDAC1/2 mediated protein deacetylations, including those on H3K56 and H4K16, promote DSB repair, particularly NHEJ (Fig. 8). Such a model therefore has resonance with the situation in budding yeast, where mutation of H4K16 or deletion of the HDAC1/2 homolog Rpd3 renders cells NHEJ defective and deacetylation of H4K16 occurs at DSBs47,48. Furthermore, H3K56Ac in yeast is absent in G1 when NHEJ is most active and is highest in S-phase, when HR predominates, while H3K56 hyper-acetylation in G1 is detrimental to genome stability39,49. Although such findings might initially seem at odds with work showing that histone hyper-acetylation occurs at DNA-damage sites, it is noteworthy that such H4 acetylation increases were analyzed and detected only at late times after DSB-induction, and that HDAC inhibitors were found to enhance the loading of Rad51, a HR protein19. Consequently, we suggest that histone acetylation changes are likely to occur in a biphasic manner following DSB induction, with rapid deacetylation on sites such as H3K56 and H4K16 occurring to promote NHEJ (possibly by generating a less dynamic “repressed” chromatin state) being followed by histone acetylations that enhance HR (and potentially alternative NHEJ pathways) by making chromatin more “open”. Thus, as for transcription, HDACs and HATs may function in a coordinated manner in the DDR to promote both “open” and “repressed” chromatin states to facilitate distinct events50.
Figure 8.

Model for the role of HDAC1/2, as well as the inhibitory effects of Class I/II HDAC inhibitors, in the DDR. HDAC1 and HDAC2 participate in the DDR through their (i) recruitment to DNA damage, (ii) regulation of H3K56 and H4K16 acetylations and (iii) requirement for DNA repair, particularly through NHEJ. Class I/II HDAC inhibitors block the activity of HDAC1 and HDAC2 resulting in defects in the DDR, including hyper-acetylation of H3K56 and H4K16 as well as an impairment of NHEJ.
There are various ways in which HDAC1 and HDAC2 could influence NHEJ. One possibility is that transcription is inhibited at sites of DNA damage and rapid histone deacetylation by HDAC1/2 at DNA-damage sites acts to repress transcription, thus preventing transcription from interfering with repair processes. In this regard, we note that histone hypo-acetylation is generally associated with transcriptionally silent chromatin and that H3K56Ac and H4K16Ac are histone marks associated with transcriptional activation35,51. Consistent with HDAC1/2 being recruited to DNA damage sites as components of transcriptional co-repressor complexes (such as NuRD, Sin3 and CoREST), at sites of laser micro-irradiation, we have also observed the hypo-phosphorylation of RNA polymerase II Ser-5, as well as ubiquitylated histone H2A and SUMO1, three marks that are all associated with transcriptional repression (Supplementary Fig. 8; also see Refs14,52). An additional way that HDAC1 and HDAC2 could promote NHEJ is to affect the ability of NHEJ factors to bind to DSB sites and/or function effectively there. Notably, in vitro studies have shown that, while Ku can bind and slide freely on naked DNA, nucleosomes can act as a barrier to such sliding53. It could therefore be that HDAC1/2 act to alter chromatin into a state that promotes NHEJ, at least in part by restraining unproductive Ku sliding and, instead, maintaining Ku at DSB termini to mediate repair. In this regard, the enhanced accrual of Ku and Artemis at sites of DNA damage that we observe upon HDAC1/2 inactivation is not only due to impaired NHEJ but also reflects the NHEJ machinery unproductively spreading onto chromatin flanking the DSB sites. We can not rule out that in addition to deacetylating histones, HDAC1 and HDAC2 more directly influence DSB repair by targeting NHEJ factors themselves or other regulators, such as ATM that may be important for NHEJ in particular chromatin contexts54-56. Clearly, studies aimed at systematically identifying HDAC1 and HDAC2 substrates will be valuable in exploring such possibilities.
DNA-damage induction occurs during tumour evolution57,58 and HDAC1/2 are overexpressed in many cancers24, possibly to promote effective DNA repair by regulating histone acetylations, including H4K16Ac59. Furthermore, broad specificity HDAC inhibitors are promising anti-cancer drugs that sensitize cancer cells to DNA-damaging treatments24,27,28. Hypo-acetylated histones found in cancer cells can epigenetically silence tumor-suppressor genes (including p21), and HDAC inhibitors can reverse this silencing, leading to cell-cycle arrest24,60. However, the specific HDACs whose inhibition results in these effects, as well as their targets, have not yet been identified. Our data highlight the potential for HDAC1 and HDAC2, as well as their targets, H3K56Ac and H4K16Ac, to be involved – through their impact on NHEJ – in modulating a cancer cells’ susceptibility to HDAC inhibitors and DNA-damage based therapies. Finally, we speculate that, while broad-specificity HDAC inhibitors are used for cancer treatments and are being combined with radiotherapies in clinical trials28, specific co-inhibition of HDAC1/2 would be efficacious in such contexts.
Supplementary Material
ACKNOWLEDGEMENTS
We thank all members of the Jackson lab for help and support, P. Huertas, A. Kaidi, and B. Xhemalce for critical reading of the manuscript and B. Xhemalce for assistance with ChIP. We thank F. d’Adda di Fagagna for providing OIS cells, A. Meier for replicative senescent BJ cells and P. Frit for GFP-KU70. We thank NEB Biolabs for kindly providing AsiSI genomic DNA to G.L. K.M.M. is funded by a Wellcome Trust project grant (086861/Z/08/Z). J.V.T. is supported by a BBSRC Cooperative Award in Science and Engineering (CASE) studentship with KuDOS Pharmaceuticals. G.L research is funded by ARC, CNRS, and PRES-University of Toulouse grants. S.E.P. is funded by Human Frontier Science Program Organization. S.B. is funded by Cancer Research U.K. and an EMBO Long-Term Fellowship. Research in the S.P.J. laboratory is also supported by the European Community (EU Projects DNA Repair (LSHG-CT-2005-512113) and GENICA) and by core infrastructure provided by Cancer Research UK and the Wellcome Trust.
Appendix
Online Methods
Cell culture, reagents and treatments
Cells were maintained as described in Supplementary Methods. Phleomycin treatments were with 60 μg ml−1 for 2 h unless stated otherwise. Cells were exposed to IR as delivered by a Faxitron X-ray unit. Unless indicated otherwise, HDAC inhibitors were added for 16-24 h and final concentrations were: sodium butyrate (5 mM), TSA (1.3 mM) or nicotinamide (25 mM). Antibodies used in this study are listed in Supplementary Table S1. Details of HDAC1 complementation and analysis of NHEJ factors are described in Supplementary Methods.
Laser micro-irradiation
Laser micro-irradiation is described in detail in Supplementary Methods.
Protein extracts and Western blotting
For whole cell extracts, cells were washed once with PBS (phosphate buffered saline), collected by adding Laemmli buffer (4% (v/v) SDS, 20% (v/v) glycerol and 120 mM Tris, pH 6.8), boiled for 5 min at 95°C, sheared through a 23-gauge needle and boiled again before loading. Samples were resolved by SDS-PAGE and analyzed by standard western blotting techniques. Antigens were detected by standard chemiluminescence (ECL; Amersham) or quantified with a LI-COR Odyssey infrared imaging system (LI-COR Biosciences). Secondary antibodies used for ECL were goat anti-rabbit HRP (Perbio Science Ltd) and rabbit anti-mouse HRP (Dako Ltd). Secondary antibodies used for quantification were IRDye 680CW Donkey anti-rabbit or anti-mouse (LI-COR Biosciences).
siRNA transfection
RNA interference was performed as previously described20. Briefly, cells were subjected to two rounds of siRNA transfections with the indicated siRNAs using Qiagen HiPerFect (following manufacturer’s protocol) and after 72 h, cells were processed and analyzed as described for each experiment. siRNA sequences used in this study are listed in Supplementary online material. Western blot analyses with the indicated antibodies were performed to analyze depletion efficiencies for each siRNA. For human SIRT2, siRNA depletion was tested on GFP-SIRT2 (cloned into eGFP-C1) that was transiently transfected into U2OS cells followed by two rounds of siRNA with the indicated siRNAs against SIRT2.
Immunofluorescence analyses
Cells were grown on poly-L-lysine-treated coverslips. After indicated treatments, coverslips were washed once with PBS at room temperature (RT). Cells were pre-extracted by incubating coverslips in CSK buffer (10 mM PIPES pH 6.8, 100 NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA, 0.5% (v/v) Triton X-100) for 5 min on ice. Cells were washed once in cold PBS and fixed with 2% (v/v) paraformaldehyde (PFA) for 20 min at RT followed by three washes with PBS-T (PBS containing 0.1% (v/v) Tween-20) and subsequently blocked for 15 min at RT in blocking buffer (PBS containing 3% BSA). Primary antibodies were incubated for 1 h at RT in the same buffer. Cells were then washed three times in PBS-T before incubation in the dark with Alexafluor-conjugated secondary antibodies (Molecular Probes) in blocking buffer for 45 min at RT. Cells were again washed three times in PBS-T followed by a final wash in PBS. The coverslips were then mounted on slides in Vectashield containing DAPI (Vector laboratories). Cells were imaged with an inverted FV1000 confocal microscope (Olympus) or a Radiance confocal microscope (BioRad). Quantitative analyses of intensities versus distance for IF images were obtained with the line-intensity function by using Volocity software. For BrdU labelling combined with dual-immunofluorescence, cells were grown on coverslips and incubated with 10 mM BrdU for 20 min. Coverslips were pre-extracted and processed for IF for either H3K56Ac or gH2AX. DNA-damage analysis with BrdU is described in Supplementary Methods.
Chromatin Immunoprecipitation (ChIP) analysis
U2OS cells containing a stably integrated AsiSI-ER-PURO construct were grown in selective media (1 μg ml−1 puromycin). To induce the nuclear expression of AsiSI-ER, these cells were treated with 300 nM of 4-OHT (4-hydroxytamoxifen) for 4 h to induce site specific DSBs. Cells untreated or treated with 4-OHT and analyzed by ChIP as described in Supplementary Methods. To ChIP NHEJ factors, experiments were done essentially as described for Fig. 1F with the following exceptions. Cells were either untreated or treated with sodium butyrate (5 mM) for 16 h. Cells were then pre-treated with DNA-PKcs inhibitor (NU7441, 10 μM) for 1 h followed by 4-OHT (300 nM) for 2 h. Antibodies and primer sequences are given in Supplementary Methods.
In vitro HDAC assays
2.5 μg of recombinant HDAC1 (Millipore) were incubated with 2.5 μg of calf thymus histones in deacetylase buffer (10 mM Tris-HCl pH 8.0, 10 mM NaCl, 10% (v/v) glycerol) for 2 h at 37°C. 3X sample loading buffer was added to the protein mix and samples were analyzed by Western blotting with the indicated antibodies.
Cell survival assays
72 h post-siRNA transfection, U2OS cells were treated with IR (Faxitron X-ray machine) or phleomycin (for 2 h followed by washing). Cells were left to form colonies for 10-14 days at 37°C. Colonies were stained with 0.5% (v/v) crystal violet/20% (v/v) ethanol and counted. Results were normalized to plating efficiencies of untreated cells for each siRNA.
NHEJ Assays
Assays were performed as detailed in Supplementary Methods.
Homologous recombination (HR) assays
HR assays were performed as described in Supplementary Methods.
Neutral Comet assays
Comet assays were with the Single Cell Gel Electrophoresis Assay-kit (Trevigen) and performed as described in Supplementary Methods.
REFERENCES
- 1.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–8. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–45. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 3.Huertas P. DNA resection in eukaryotes: deciding how to fix the break. Nat Struct Mol Biol. 2010;17:11–6. doi: 10.1038/nsmb.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gottlieb TM, Jackson SP. The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell. 1993;72:131–42. doi: 10.1016/0092-8674(93)90057-w. [DOI] [PubMed] [Google Scholar]
- 5.Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J. 2009;417:639–50. doi: 10.1042/BJ20080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Downs JA, Nussenzweig MC, Nussenzweig A. Chromatin dynamics and the preservation of genetic information. Nature. 2007;447:951–8. doi: 10.1038/nature05980. [DOI] [PubMed] [Google Scholar]
- 7.Groth A, Rocha W, Verreault A, Almouzni G. Chromatin challenges during DNA replication and repair. Cell. 2007;128:721–33. doi: 10.1016/j.cell.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 8.Misteli T, Soutoglou E. The emerging role of nuclear architecture in DNA repair and genome maintenance. Nat Rev Mol Cell Biol. 2009;10:243–54. doi: 10.1038/nrm2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 10.Bonner WM, et al. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–67. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stucki M, Jackson SP. gammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair (Amst) 2006;5:534–43. doi: 10.1016/j.dnarep.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 12.Doil C, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136:435–46. doi: 10.1016/j.cell.2008.12.041. [DOI] [PubMed] [Google Scholar]
- 13.Huen MS, et al. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007;131:901–14. doi: 10.1016/j.cell.2007.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mailand N, et al. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007;131:887–900. doi: 10.1016/j.cell.2007.09.040. [DOI] [PubMed] [Google Scholar]
- 15.Kolas NK, et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007;318:1637–40. doi: 10.1126/science.1150034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stewart GS, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009;136:420–34. doi: 10.1016/j.cell.2008.12.042. [DOI] [PubMed] [Google Scholar]
- 17.Escargueil AE, Soares DG, Salvador M, Larsen AK, Henriques JA. What histone code for DNA repair? Mutat Res. 2008;658:259–70. doi: 10.1016/j.mrrev.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 18.Corpet A, Almouzni G. A histone code for the DNA damage response in mammalian cells? EMBO J. 2009;28:1828–30. doi: 10.1038/emboj.2009.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murr R, et al. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol. 2006;8:91–9. doi: 10.1038/ncb1343. [DOI] [PubMed] [Google Scholar]
- 20.Tjeertes JV, Miller KM, Jackson SP. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. Embo J. 2009;28:1878–89. doi: 10.1038/emboj.2009.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michishita E, et al. Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle. 2009;8:2664–6. doi: 10.4161/cc.8.16.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Das C, Lucia MS, Hansen KC, Tyler JK. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature. 2009;459:113–7. doi: 10.1038/nature07861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang B, Zwaans BM, Eckersdorff M, Lombard DB. The sirtuin SIRT6 deacetylates H3 K56Ac in vivo to promote genomic stability. Cell Cycle. 2009;8:2662–3. doi: 10.4161/cc.8.16.9329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26:5420–32. doi: 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- 25.Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008;9:206–18. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–95. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith KT, Workman JL. Histone deacetylase inhibitors: anticancer compounds. Int J Biochem Cell Biol. 2009;41:21–5. doi: 10.1016/j.biocel.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 28.Camphausen K, Tofilon PJ. Inhibition of histone deacetylation: a strategy for tumor radiosensitization. J Clin Oncol. 2007;25:4051–6. doi: 10.1200/JCO.2007.11.6202. [DOI] [PubMed] [Google Scholar]
- 29.Iacovoni JS, et al. High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 2010 doi: 10.1038/emboj.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.d’Adda di Fagagna F, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–8. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 31.d’Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer. 2008;8:512–22. doi: 10.1038/nrc2440. [DOI] [PubMed] [Google Scholar]
- 32.Bartkova J, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–7. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 33.Di Micco R, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–42. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 34.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 35.Xie W, et al. Histone h3 lysine 56 acetylation is linked to the core transcriptional network in human embryonic stem cells. Mol Cell. 2009;33:417–27. doi: 10.1016/j.molcel.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Willis-Martinez D, Richards HW, Timchenko NA, Medrano EE. Role of HDAC1 in senescence, aging, and cancer. Exp Gerontol. 2009 doi: 10.1016/j.exger.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bandyopadhyay D, et al. Down-regulation of p300/CBP histone acetyltransferase activates a senescence checkpoint in human melanocytes. Cancer Res. 2002;62:6231–9. [PubMed] [Google Scholar]
- 38.Yuan J, Pu M, Zhang Z, Lou Z. Histone H3-K56 acetylation is important for genomic stability in mammals. Cell Cycle. 2009;8:1747–53. doi: 10.4161/cc.8.11.8620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller KM, Maas NL, Toczyski DP. Taking it off: regulation of H3 K56 acetylation by Hst3 and Hst4. Cell Cycle. 2006;5:2561–5. doi: 10.4161/cc.5.22.3501. [DOI] [PubMed] [Google Scholar]
- 40.Luo J, et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–48. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 41.Cheng HL, et al. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A. 2003;100:10794–9. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dovey OM, Foster CT, Cowley SM. Histone deacetylase 1 (HDAC1), but not HDAC2, controls embryonic stem cell differentiation. Proc Natl Acad Sci U S A. 2010;107:8242–7. doi: 10.1073/pnas.1000478107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pommier Y, et al. Repair of and checkpoint response to topoisomerase I-mediated DNA damage. Mutat Res. 2003;532:173–203. doi: 10.1016/j.mrfmmm.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 44.Uematsu N, et al. Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J Cell Biol. 2007;177:219–29. doi: 10.1083/jcb.200608077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mari PO, et al. Dynamic assembly of end-joining complexes requires interaction between Ku70/80 and XRCC4. Proc Natl Acad Sci U S A. 2006;103:18597–602. doi: 10.1073/pnas.0609061103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Higashiura M, Shimizu Y, Tanimoto M, Morita T, Yagura T. Immunolocalization of Ku-proteins (p80/p70): localization of p70 to nucleoli and periphery of both interphase nuclei and metaphase chromosomes. Exp Cell Res. 1992;201:444–51. doi: 10.1016/0014-4827(92)90293-h. [DOI] [PubMed] [Google Scholar]
- 47.Bird AW, et al. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature. 2002;419:411–5. doi: 10.1038/nature01035. [DOI] [PubMed] [Google Scholar]
- 48.Jazayeri A, McAinsh AD, Jackson SP. Saccharomyces cerevisiae Sin3p facilitates DNA double-strand break repair. Proc Natl Acad Sci U S A. 2004;101:1644–9. doi: 10.1073/pnas.0304797101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xhemalce B, et al. Regulation of histone H3 lysine 56 acetylation in Schizosaccharomyces pombe. J Biol Chem. 2007;282:15040–7. doi: 10.1074/jbc.M701197200. [DOI] [PubMed] [Google Scholar]
- 50.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–19. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 51.Wang Z, et al. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–31. doi: 10.1016/j.cell.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Galanty Y, et al. Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature. 2009;462:935–9. doi: 10.1038/nature08657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roberts SA, Ramsden DA. Loading of the nonhomologous end joining factor, Ku, on protein-occluded DNA ends. J Biol Chem. 2007;282:10605–13. doi: 10.1074/jbc.M611125200. [DOI] [PubMed] [Google Scholar]
- 54.Kim GD, et al. Sensing of ionizing radiation-induced DNA damage by ATM through interaction with histone deacetylase. J Biol Chem. 1999;274:31127–30. doi: 10.1074/jbc.274.44.31127. [DOI] [PubMed] [Google Scholar]
- 55.Rimkus SA, et al. Mutations in String/CDC25 inhibit cell cycle re-entry and neurodegeneration in a Drosophila model of Ataxia telangiectasia. Genes Dev. 2008;22:1205–20. doi: 10.1101/gad.1639608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goodarzi AA, et al. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell. 2008;31:167–77. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 57.Bartkova J, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–70. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 58.Gorgoulis VG, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–13. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 59.Fraga MF, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 60.Ocker M, Schneider-Stock R. Histone deacetylase inhibitors: signalling towards p21cip1/waf1. Int J Biochem Cell Biol. 2007;39:1367–74. doi: 10.1016/j.biocel.2007.03.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.