

Abstract
We present a novel cellular microarray assay using soluble peptide-loaded HLA A2-Ig dimer complexes that optimizes the avidity of peptide-HLA binding by preserving the molecular flexibility of the dimer complex, while attaining much higher concentrations of the complex relative to cognate T-cell receptors. A seminal advance in assay development is made by separating the molecular T-cell receptor recognition event from the binding interactions that lead to antigen-specific cell capture on the microarray. This advance enables the quantitative determination of antigen-specific frequencies in heterogeneous T-cell populations without enumerating the number of cells captured on the microarray. The specificity of cell capture, sensitivity to low antigen-specific frequencies, and quantitation of antigenic T-cell specificities are established using CD8 T-cell populations with prepared antigen-specific CTL frequencies, and heterogeneous T cells isolated from peripheral blood. The results demonstrate several advantages for high-throughput broad-based, quantitative assessments of low-frequency antigen specificities. The assay enables the use of cellular microarrays to determine the stability and flux of antigen-specific T-cell responses within and across populations.
Introduction
Cytotoxic T lymphocyte (CTL) mediated adaptive immune response plays a crucial role in the control of infectious, autoimmune, and cancerous diseases. The core molecular event that triggers intracellular signaling and subsequent T-cell effector functions is the interaction of T-cell receptors (TCRs) on the surface of CD8 T cells with an antigen-presenting cell surface that displays a peptide antigen 8-10 amino acids in length, presented by class I major histocompatibility complexes (human leukocyte antigen, HLA, in humans). Studying T-cell antigen specificities by identifying T-cell peptide antigens is therefore fundamentally important to disease control and therapies [i], peptide antigen-based vaccine design [ii], and understanding the underlying mechanisms of immune responses; e.g., immunodominance [iii]. However, the low frequencies of antigen-specific CTL and the concomitant diversity of CTL antigen specificities [iv], as well as limitations on sample sizes in clinical settings, require methods for assessing antigen specifies that are sensitive to antigen-specific CTL in populations consisting largely of irrelevant cells, and that enable high-throughput analyses of multiple specificities simultaneously on a single heterogeneous cell population.
With the development of HLA multimers [v,vi], cellular microarrays based on HLAs have shown potential as a fast, cost-effective, and high-throughput method for qualitatively characterizing T-cell antigen specificities [vii,viii,ix,x] and cytokine secretion in heterogeneous T-cell populations [xi]. The quantitative application of HLA-based cellular microarrays, however, remains elusive in large part due to the large variability in the molecular nature of proteins, exacerbated by the lack of control over many protein-specific physical, chemical, and biological processes associated with microarray fabrication and with the sensitivity of the binding assay. For protein microarrays, in general, preserving protein structure and function, and the accessibility of binding sites on the capture molecule are critical for performance. Proteins are prone to unfold and aggregate under chemical, physical or mechanical stresses associated with printing nanoliter quantities of protein solutions, as well as with immobilization [xii]. Elements that can influence protein-substrate interactions leading to unfolding and instability include surface properties of the microarray substrate, immobilization chemistry, and printing method [xiii,xiv,xv,xvi,xvii]. The challenges are particularly formidable for HLA microarrays because of the intrinsically labile, multidomain structure of the HLA complex. The relatively weak binding affinities that characterize peptide-mediated HLA-TCR interactions [xviii,xix] also place stringent demands on the accessibility of complementary binding sites on peptide-loaded HLA (pHLA) multimers.
Here, we describe a novel cellular microarray assay that addresses these challenges, and importantly, enables the quantitation of antigenic T-cell specificities. In this new assay, T cells are incubated with peptide-loaded HLA-Ig dimers in solution, and the antigen-specific CTL are subsequently captured on the microarray by binding to an anti-Ig antibody, as illustrated in Figure 1. The frequency of antigen-specific CTL in the population is quantified by the number of antigen-specific CTL captured on the microarray relative to the number of T cells captured by binding to anti-CD3 antibodies also printed on the microarray. The approach takes advantage of the high stability of immunoglobulins by printing the anti-Ig antibody instead of the labile pHLA-Ig complex, and also optimizes the avidity of pHLA binding to the TCRs by preserving the flexibility of the pHLA-Ig complex in solution. Much higher concentrations of the soluble dimer complex can be obtained in solution relative to surface-bound dimers, which also enhances antigen-specific binding to the TCRs. The ability to form microclusters of TCR-bound pHLA-Ig dimers on the T-cell surface may also contribute to the efficiency of capturing T cells on the microarray through the accumulation of ligands in the cell contact area and by increasing the rate of receptor-ligand complex formation [xx]. Most importantly, the use of anti-CD3 antibodies to capture CTL from the T-cell population as a whole, irrespective of antigen specificity enables the quantitation of antigen-specific frequencies as the ratio of fluorescence intensities of labeled cells on anti-Ig antibody spots relative to the anti-CD3 antibody spots, without enumerating the actual number of cells captured on the microarray. Thus, the new assay can provide quantitative assessments of antigen-specific T-cell responses over time for specific donors, and among donors within large, diverse populations.
Figure 1.

Schematic depiction of the cellular microarray assay in which soluble peptide-loaded HLA A2-Ig dimers are incubated with T cells in solution. The antigen-specific sub-population of cells are identified by the surface-bound pHLA A2-Ig dimers and subsequently captured on the microarray by binding to printed anti-Igλ antibodies that recognize the immunoglobulin (Ig) portion of the dimer.
Experimental Section
Anti-Igλ microarray fabrication
Anti-mouse Igλ antibody microarrays were fabricated by printing a solution of 0.5 mg/ml polyclonal anti-mouse Igλ antibody (Southern Biotech, AL), and separately, a solution of 0.5 mg/ml anti-human CD3 antibody (BD Biosciences, San Diego CA) on polyacrylamide film-coated glass slides - Nexterion Slide H (Schott, Elmsford NY) - using a PerkinElmer Piezorray noncontact microarrayer (PerkinElmer, Waltham MA). Bovine serum albumin (BSA, Sigma Aldrich, St Louis, MO) at 0.5 mg/ml (final concentration) was added to the printing solutions to maintain regular spot-to-spot morphology, produce uniform intra-spot antibody distributions, and minimize antibody loss during printing (Figure S1). The solutions were typically printed in pre-programmed patterns of 5×5 subarrays of spots, separated by a spot center-to-center distance of 1 mm. The individual spots consisted of about 10 nl of solution, and were 500 μm in diameter. For the experiments evaluating microarray sensitivity, 10×10 subarrays of spots were printed to facilitate the analysis of the statistical distribution of spot intensities. The individual spots consisted of ~ 0.33 nl of solution and were 150 μm in diameter with a spot center-to-center distance of 400 μm. The printed slides can be stored sealed at 4°C for more than a month before use without measurable loss of activity. Prior to an experiment, microarray slides were incubated in a humid chamber (relative humidity of 75%) for 1 hr at room temperature (24°C) to facilitate the immobilization of arrayed proteins within the gel substrate. After washing with 0.05% (v/v) Tween 20 in 1× PBS solution (PBST) followed by PBS solution, the microarray slides were blocked in blocking buffer (50 mM sodium borate containing 50 mM ethanolamine at pH 8.0) at room temperature for 1 hr. The slides were then washed again with 0.05% (v/v) PBST and PBS solutions.
Cellular microarray assays
Cells were first labeled with CFSE (carboxy-fluorescein diacetate, succinimidyl ester; Invitrogen, Carlsbad CA) according to manufacturer’s protocol, and then incubated with unloaded or peptide-loaded dimer at 4°C for 45 min. For every 1×105 cells, 1 μl of 0.5 mg/ml dimer was used. The cells were then centrifuged with 300 μl PBS solution at 700 rcf for 5 min to remove unbound dimer, and resuspended in PBS solution to a final concentration of ~1–2×106 cells/ml. The cells were then contacted with the microarray surface by pipetting 50 μl of this suspension onto an area of 6.8 mm × 6.8 mm to cover a 5×5 subarray of 500 μm spots or 10×10 subarray of 150 μm spots. Each of the 14 subarrays on the glass slide was separated from the other subarrays by a silicone gasket (Grace Bio Labs, Bend OR).
For microarray sensitivity experiments, cell suspensions containing known frequencies of antigen-specific CTLs ranging from 10% to 0%, obtained by serially mixing CFSE-labeled aAPC-enriched antigen-specific CTLs with unlabeled CD8 cell-depleted autologous PBMCs, were incubated with peptide-loaded dimer molecules and applied to anti-Igλ microarrays. The same frequencies of cells, but pre-incubated with unloaded dimer, were included as controls. The frequency of undiluted aAPC-enriched antigen-specific CTLs was kept at a moderate level of 10% before fluorescence labeling and mixing with unlabeled PBMCs, so that ultimately the CFSE-labeled cells in each cell suspension contained not only antigen-specific CTLs but also a pool of CD8 cells expressing TCRs of diverse specificities. After 1 hr incubation in the dark at room temperature, the microarray slide was then dip-washed in PBS solution to remove any unbound cells prior to scanning.
HLA A2-Ig microarray fabrication and assays
HLA A2-Ig dimer microarrays were fabricated by printing solutions of 0.5 mg/ml peptide-loaded dimer molecules and unloaded dimer molecules (negative control) onto a Nexterion Slide H, in 5×5 subarrays with 1 mm spot center-to-center distance. Individual spots consisted of ~10 nl of solution and were 500 μm in diameter. In some experiments, slides were incubated with a solution of goat anti-mouse IgG antibody (BD Biosciences, 100 μl of 0.5 mg/ml and spread by a 24 mm × 60 mm glass coverslip) for 1 hr at room temperature prior to printing, to improve the accessibility of peptide-dimer molecules on the substrate surface [viii]. After protein immobilization and blocking (with blocking buffer), antigen-specific CTLs, labeled with CFSE, were contacted with the microarray surface at a concentration of ~1–2×106/ml. Following incubation for 1 hr at room temperature in the dark, the microarray slide was dip-washed in PBS solution before scanning.
Cell-free HLA A2-Ig microarray assays
To assess the functionality of printed HLA A2-Ig molecules on the HLA A2-Ig dimer microarray, a cell-free antibody binding assay was carried out using two fluorescently labeled antibodies: PE-anti-HLA (clone W6/32, Dako North America, Carpinteria, CA) and FITC-anti-Igλ (Southern Biotech). W6/32 is a conformation-sensitive monoclonal antibody that reacts with the alpha-2 and alpha-3 domains of HLA class I molecules [xxi,xxii]. Studies have shown that W6/32 binding to HLA blocks or inhibits TCR activation or proliferation [xxiii,xxiv]. By incubating the HLA A2-Ig microarray with solutions containing W6/32 and anti-Igλ antibodies and measuring antibody binding, the structural integrity of the printed HLA A2-Ig molecules can be inferred. Briefly, spots were printed with 0.1 mg/ml unloaded HLA A2-Ig molecules in PBS solution, in 0.33% DMSO/PBS solution, or loaded with 66.7 μg/ml CMVpp65 in 0.33% DMSO/PBS solution. Alternatively, spots were printed with 0.43 mg/ml unloaded HLA A2-Ig dimer in PBS solution or loaded with 276 μg/ml CMVpp65 in 1.4% DMSO/PBS solution. Each sample was printed into 5×5 subarrays of spots, each consisting of 0.33 nanoliter of solution (150 μm in diameter). After protein immobilization and blocking, the microarray was incubated with PE-W6/32 or FITC-anti-Igλ antibody at a concentration of 20 μg/ml for 1 hr at room temperature in the dark. The slides were then washed with 0.05% PBST, followed by PBS solution before scanning.
Flow cytometry measurements
The frequencies of antigen-specific CTLs in aAPC-enriched populations or in donor CD8 cells were measured by flow cytometry using a FACSCalibur flow cytometer (BD Biosciences, San Jose CA). The procedure for dimer staining is described in [vi]. Briefly, ~150,000 cells in 100 μl of PBS solution were first incubated with 1 μl of unloaded or peptide-loaded HLA A2-Ig dimer at 4°C for 45 min. After washing with 500 μl of PBS solution at 700 rcf for 5 min, the cells were incubated with 2 μl of 0.1 mg/ml PE-conjugated goat anti-mouse IgG1 antibody (Invitrogen, Carlsbad CA) at 4°C for 10 min. After a second washing with 500 μl of PBS solution, the cells were incubated with 2 μl of 0.1 mg/ml FITC-conjugated anti-human CD8 monoclonal antibody (Sigma Aldrich, St Louis MO) at 4°C for 10 min. The cells were then washed a third time with 500 μl of the buffer solution, and fixed in 300 μl of 4% (w/v) paraformaldehyde before FACS analysis. FACS data reduction was carried out using FlowJo software (TreeStar, San Carlos CA).
Image acquisition and data analysis
Microarray images were acquired using a ProScanArray fluorescence scanner (PerkinElmer, Waltham MA). The microarray slides were scanned at 5 μm resolution. Image quantification and data analysis were performed using ScanArray Express 3.0 (PerkinElmer, Waltham MA) and JMP 6.0 (SAS, Cary NC) software. The data regression and plotting were performed using Sigma plot 10.0 (Systat Software, San Jose CA).
Preparation of peptide-HLA A2-Ig complexes
High purity (~95%) peptides CMVpp65 (NLVPMVATV), M1.58 (GILGFVFTL), PA.46 (FMYSDFHFI), and NA.75 (SLCPIRGWAI) were purchased from Genscript (Piscataway NJ). The peptides were reconstituted to 2 mg/ml in PBS solution containing 10 or 20% (v/v) DMSO (10% for CMVpp65, PA.46 and NA.75, and 20% for M1.58). Peptide-HLA A2-Ig dimer complexes were prepared by passive loading [xxv]. Briefly, unloaded HLA A2-Ig dimer (0.5 mg/ml, BD Pharmingen, San Diego CA) was incubated with the specific peptide of interest in 160:1 peptide:dimer molar ratio at 4°C or room temperature for up to 20 days. Subsequent separation of excess peptide was not performed.
Preparation of cell populations
Enriched populations of antigen-specific CTLs were obtained by repetitive stimulation with artificial antigen presenting cells (aAPCs), as described in [xxxi,xxvi]. Briefly, CD8 T lymphocytes were separated from peripheral blood of HLA-A2.1 healthy donors (phenotyped by FACS using FITC-labeled anti-HLA-A2.1 antibody) using a negative CD8 isolation kit (Miltenyi Biotec, Auburn CA). The resulting population consisted of >90% CD8 T cells, as determined by staining with FITC-labeled anti-human CD8 antibody in FACS. This cell population was stimulated with peptide-loaded aAPCs in a 96-well plate for 7 to 9 days over a period of up to 3 to 4 weeks. The cells were then collected, and an aliquot taken to measure the frequency of antigen-specific cells; the remaining cells were frozen in fetal bovine serum (FBS, Atlanta Biologicals, Lawrenceville GA) containing 10% DMSO, and stored at −80°C. Prior to use, the cells were thawed following standard procedures.
CD8-depleted autologous peripheral blood mononuclear cells (PBMCs) were isolated from peripheral blood using a positive CD8 isolation kit (Miltenyi Biotec, Auburn CA). CD8 cells from donor A8 were isolated from peripheral blood using a negative CD8 isolation kit (Miltenyi Biotec, Auburn CA).
Results and Discussion
In fabricating protein microarrays, rapid evaporation can occur when printing nanoliter quantities of protein solutions. Rapid evaporation, in turn, can produce abrupt changes in the solution conditions, exposing the protein solute to entirely different environments and compromising the functional integrity of the microarray [xv]. An example is given in Figure 2, which shows the effect of adding small amounts of dimethyl sulfoxide (DMSO) to the printing solution on the pHLA A2-Ig complexes printed on the microarray. DMSO is typically used to enhance the solubility of hydrophobic peptides in preparing peptide-loaded HLA complexes [xxvii,xxviii]. DMSO is also known to induce protein unfolding in aqueous solution at high concentrations on the order of 40% (v/v) or more [xxix]. We found that adding only 0.33% (v/v) DMSO to a printing solution containing the pHLA A2-Ig complex (Figure 2) reduces PE-labeled W6/32 antibody bound to the printed complex by more than 90%, independent of peptide loading with a specific peptide of interest – in this case, CMVpp65 – or with endogenous peptides resident in the “unloaded dimer” [vi]. A similar effect is observed for FITC-labeled anti-Igλ antibody binding to the CMVpp65-loaded dimer (Figure 2). The mean spot fluorescence intensity in this case is reduced by nearly half when the dimer complex is printed from solutions containing 1.4% (v/v) DMSO compared to solutions without DMSO.
Figure 2.

Effect of dimethyl sulfoxide (DMSO) on the binding of anti-HLA (clone W6/32) and anti-Ig antibodies binding to microarray spots printed with pHLA A2-Ig dimers. From left to right: PE-labeled W6/32 (20 μg/ml) contacted with spots printed with 0.1mg/ml unloaded pHLA A2-Ig dimer in PBS solution, in 0.33% (v/v) DMSO/PBS solution, and with 66.7μg/ml CMVpp65-loaded pHLA A2-Ig dimer in 0.33% (v/v) DMSO/PBS solution, and FITC-labeled anti-Igλ (20 μg/ml) contacted with spots printed with 0.43mg/ml unloaded pHLA A2-Ig dimer in PBS solution and with 276 μg/ml CMVpp65-loaded pHLA A2-Ig dimer in 1.4% (v/v) DMSO/PBS solution. Mean spot fluorescence intensities are normalized to the mean spot fluorescence intensities for the unloaded dimer in PBS solution for each antibody staining, with the percentages shown above the bars corresponding to these relative mean spots intensities. Insets: scanned fluorescence images of subarrays for the two printing solutions designated by the arrows.
Since monoclonal W6/32 antibody recognizes the folded HLA domain of the complex [xxx], while the anti-Igλ antibody recognizes the folded Ig domain of this complex, these results suggest that the presence of DMSO at dilute concentrations in the printing solution leads to the destabilization of the complex as a whole, rather than acting locally, for example, to render the antigen-recognition interface inaccessible. This deleterious effect is not observed when printing much larger (microliter) volumes of solution containing DMSO at dilute concentrations; thus implicating the printing/rapid evaporation of nanoliter solution volumes that leads to the loss of functionality.
We also considered the loss of functionality of the pHLA A2-Ig complex due to reaction with the amine-reactive NHS groups on the substrate by first pre-coating the entire slide with anti-Igλ antibody, then blocking with excess ethanolamine to quench unreacted NHS groups before printing the pHLA A2-Ig complex. We still observed very low levels of W6/32 antibody binding to the printed complex, and thus concluded that this reaction does not contribute significantly to the lability of the pHLA A2-Ig complex.
The strategy we adopt here circumvents printing the labile HLA molecule altogether, but takes advantage of the unique structure of this complex by printing instead the more robust and stable anti-Igλ antibody, which binds to the Ig domain of the complex, and enables printing a single solution on each spot across the entire microarray. Moreover, the TCR-pHLA molecular interactions that define T-cell antigen specificities take place in solution, rather than at the cell-microarray substrate interface, allowing for further control and optimization of the assay. Both factors contribute to the reliable quantitative determination of antigen-specific frequencies.
Quantifying antigen-specific cell capture on the pHLA A2-Ig microarray
We first examined the ability of this microarray assay to capture T cells specific for three peptide antigens: CMVpp65 from cytomegalovirus, and M1.58 and PA.46 from influenza A virus [xxxi]. Figure 3a depicts 5×5 subarrays of 25 anti-Igλ antibody spots with cells captured from these enriched populations when the cell suspension in each case was incubated with the cognate peptide-loaded HLA A2-Ig complex. Also shown is a 5×5 subarray of 25 anti-Igλ antibody spots contacted with the CMVpp65-enriched cell population incubated with the unloaded dimer (negative control). Essentially no cell binding to the anti-Igλ antibody spots is observed in this case. Similar results were obtained for the M1.58-enriched and PA.46-enriched cell populations when incubated with the unloaded dimer (images not shown).
Figure 3.

Comparison of pHLA A2-Ig dimer microarray and FACS assays of the frequency of antigen-specific CTLs in enriched T-cell populations. (a) Representative images of cell capture on 5 × 5 subarrays of 25 anti-Igλ spots and anti-CD3 spots. From left to right: CMVpp65-enriched CTLs incubated with the unloaded dimer, CMVpp65-enriched CTLs incubated with CMVpp65-loaded dimer, M1.58-enriched CTLs incubated with M1.58-loaded dimer, PA.46-enriched CTLs incubated with PA.46-loaded dimer, and CMVpp65-enriched CTLs on anti-CD3 spots. (b) Mean spot fluorescence intensities normalized by the mean fluorescence intensities for binding to the anti-CD3 spots on the microarray compared to the frequency of antigen-specific CTLs determined by FACS. For the microarray assay, the mean spot intensity for the unloaded dimer was subtracted before normalizing. For the FACS assay, the frequency measured for the unloaded dimer was subtracted beforehand. Error bars for the microarray assay represent standard errors for the 25 spot replicates. Dimer loading conditions: 15 days at 4°C for CMVpp65, 11 days at 24°C for M1.58, and 16 days at 24°C for PA.46. Individual spot are printed from 10 nl of solution and are 500μm in diameter. Spot center-to-center distance is ~ 1mm. Cell seeding conditions for each subarray: 50μl of ~0.8 × 106/ml total cells.
A 5×5 subarray of anti-CD3 antibody spots used for an internal calibration of the assay is also shown in Fig. 3a. Normalizing the mean intensity of the anti-Igλ antibody spots against the mean intensity of anti-CD3 antibody spots takes into account experimental variations in cell labeling across individual cell populations or from one experiment to another, as well as differences in capture efficiencies from slide to slide. Importantly, this normalization allows a quantitative assessment of the frequency of antigen-specific CTL in the total T-cell population. As illustrated in Figure 3b, the frequencies derived from the microarray assays using the enriched CTL populations are essentially equivalent to those obtained by flow-activated cell sorting (FACS; see Figure S2 for plots). The enriched CMVpp65-specific CTL frequency measured by FACS is 71.5% compared to 67.9±4.2% measured in the microarray assay. Similar agreement is obtained for the enriched PA.46-specific CTL frequency: 69.5% by FACS compared to 67.0±4.0% from the microarray assay; and for the enriched M1.58-specific CTL frequency: 13.2% by FACS compared to 14.6±1.3% from the microarray assay, which shows the quantitative accuracy of the microarray assay for these enriched cell populations.
Quantitative detection of low-frequency CTL
The ability to detect and quantify antigen-specific CTL at low frequencies on the order of 0.01% or less is critical to characterizing broad-based immune responses [xxxii]. To evaluate the sensitivity of the microarray assay to low-frequency CTL, we measured CMVpp65-specific T-cell capture from suspensions of CMVpp65-specific CTL in CD8 T cell-depleted autologous PBMCs. Assays were carried out for five different concentrations of CMVpp65-specific T cells: 0%, 0.01%, 0.1%, 1% and 10% of the total cell number. The cell suspension containing only CD8 T cell-depleted autologous PBMCs quantifies the background fluorescence intensity from endogenous fluorophores in the anti-Igλ antibodies and BSA printed on the microarray. Parallel assays in which each cell suspension is incubated with the unloaded dimer measure non-specific cell capture.
As shown in Figure 4, the mean spot intensities for cell suspensions incubated with the unloaded dimer are not significantly different from the background fluorescence intensity for the lower CMVpp65-specific CTL frequencies. In contrast, some non-specific cell capture is evident at CTL frequencies of 1% and 10% where the mean spot intensities are significantly greater than the background intensity. The quantitative impact of non-specific cell capture on the antigen-specific CTL frequency is, however, negligible. For example, the extent to which the mean spot intensity exceeds the background intensity for cells captured from the 1% CMVpp65-specific cell suspension incubated with the unloaded dimer is comparable to that for cells captured from the 0.01% cell suspension incubated with the CMVpp65-loaded dimer, and likewise for the 10% cell suspension incubated with the unloaded dimer relative to the 0.1% cell suspension incubated with the CMVpp65-loaded. Thus, the correction for non-specific cell capture is approximately two orders of magnitude less than the actual value of the antigen-specific CTL frequency at these higher frequencies.
Figure 4.

Quantified mean spot fluorescence intensities of cells captured from suspensions containing different concentrations of CMVpp65-specific CTLs incubated with either the CMVpp65-loaded dimer or the unloaded dimer. A CMVpp65-enriched cell population containing ~10% CMVpp65-specific CTL, as determined by FACS, was first labeled with CFSE, and incubated with either CMVpp65-loaded pHLA A2-Ig dimer (loading at 24°C for 2 days) or the unloaded dimer, and then serially diluted in unlabeled autologous CD8 T-cell-depleted PBMCs before contacting with the microarray. Concentrations of CMVpp65-specific CTL in the different suspensions were: 0% (100% CD8 T-cell-depleted PBMCs), 0.01%, 0.1%, 1% and 10% (undiluted) of the total cell number. Each suspension is contacted with a 10 × 10 subarray of 100 anti-Igλ antibody spots. Individual spots are printed with ~ 333pl of anti-Igλ antibody/BSA solution, and are 150μm in diameter. Cell seeding conditions: 50μl of 2 × 106/ml total cells on an area of 6.8 × 6.8 mm2. Error bars in the graph represent the standard deviation across the 100 spot replicates. The standard deviation for 0% frequency of CMVpp65-specific cells incubated with the CMVpp65-dimer is 10.0 (mean spot intensity = 60.7) compared to the standard deviation for 0% frequency of CMVpp65-specific cells incubated with unloaded dimer of 6.5 (mean spot intensity = 62.1). The horizontal dashed and dotted line designate the mean spot intensities corresponding to cells captured from the 0% CMVpp65-specific cell suspension incubated with the CMVpp65-loaded pHLA A2-Ig dimer and the unloaded dimer, respectively. Statistical differences between mean spot intensities for CMVpp65-specific and non-specific cell capture at each CTL frequency computed based on the student-t distribution produced the following p-values: p<0.0001 at 10%; p=0.0129 at 1%; p=0.1209 at 0.1%; and p=0.2175 at 0.01%.
The mean spot intensities in Figure 4 corresponding to average numbers of CMVpp65-specific T cells captured on each spot decrease monotonically as the frequency of those cells decreases from 10% to 0.01%, with the mean spot intensity for the lowest frequency still greater than the background fluorescence intensity. Nonetheless, the mean spot intensities for CMVpp65-specific T-cell capture from the 0.1% and 0.01% cell suspension are not statistically different than those corresponding to non-specific cell capture from these suspensions (p-values are given in Figure 4 caption), indicating a lower detection limit that is greater than 0.1%, or 100 CMVpp65-specific CTL in a total population of 100,000 CD8 T cells, the cell seeding condition used in these assays. Moreover, at the lowest antigen-specific CTL frequency of 0.01%, most of the microarray spots will not display captured antigen-specific T cells. This situation is seen in Figure 4 where the lower limit of the error bar for the mean spot intensity corresponding to CMVpp65-specific cells captured from the 0.01% cell suspension extends below the background fluorescence intensity. Clearly assays for low-frequency CTL can produce sparsely populated microarrays, especially when the total cell population is small, and this must be taken into account in order to obtain an accurate quantification of antigen-specific CTL frequencies.
To this end, we account for the uneven distribution of cells captured across the spot replicates by considering the variation in spot fluorescence intensities in the microarray assay. Figure 5a shows that variability for M1.58-specific T cells captured from suspensions of CFSE-labeled, M1.58-specific CTL in CD8 T cell-depleted autologous PBMCs. The absence of cells captured on a spot is defined to be the highest spot intensity observed over the entire distribution of spot intensities at 0% M1.58-specific CTL in CD8 T cell-depleted autologous PBMCs incubated with the M1.58-loaded dimer. This cutoff intensity is nearly five standard deviations above the mean spot intensity, and establishes a stringent criterion for cell capture at the higher (non-zero) frequencies. In addition, this cutoff intensity is more than two standard deviations above the mean spot intensity for non-specific cell capture from the undiluted, M1.58-enriched cell suspension incubated with the unloaded dimer – the negative control for M1.58-specific T cell capture in Figure 3 – and thus, also indirectly accounts for non-specific cell capture from this cell suspension at the low CTL frequencies considered. It is important to note that non-specific CD8+ T cells are present at a frequency nine times higher than the frequency of M1.58-specific CD8+ cells since the M1.58-enriched cell suspension contains only 10% M1.58-specific T cells.
Figure 5.

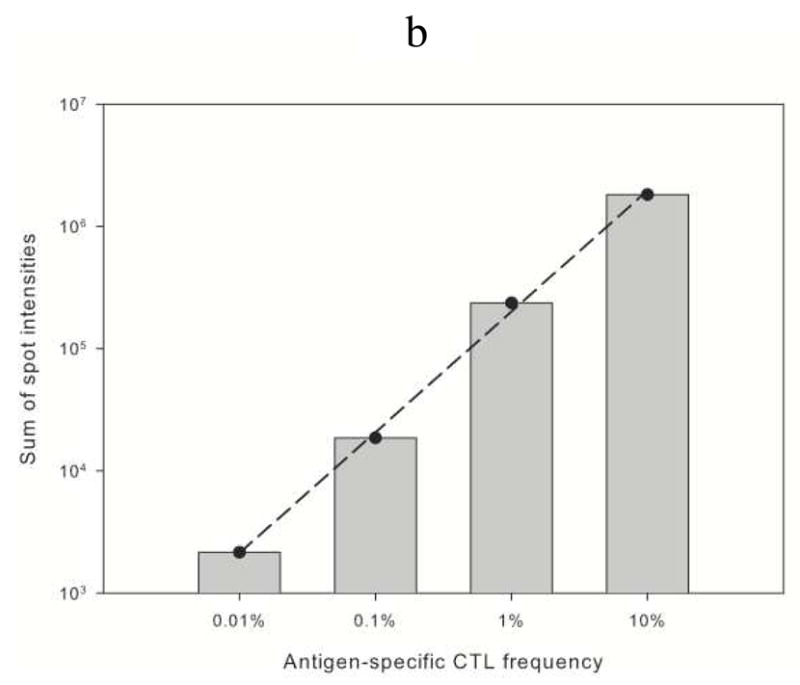
Individual spot intensities corresponding to CFSE-labeled M1.58-specific T-cell capture on a 10 × 10 subarray of 100 anti-Igλ antibody spots as a function of M1.58-specific CTL frequency. (a) Spot intensities corresponding to cells captured on subarrays contacted with cell suspensions containing CFSE-labeled M1.58-specific CTL at frequencies of 0% (100% CD8 cell-depleted autologous PBMCs), 0.01%, 0.1%, 1%, and 10%. Individual spot intensities are shown as open circles, vertically aligned for each frequency. Fluorescence intensities of spots contacted with the 0% M1.58-specific CTL suspension define the cutoff intensity for spots with captured cells. Spots with intensities below the cutoff are considered to have no captured cells and are shown as black circles, whereas spots with intensities above the cutoff are considered to have captured cells and are shown as red circles. For each frequency, the number to the left of the vertically aligned circles designates the number of spots with captured cells. (b) Sum of spot fluorescence intensities corresponding to the red circles in (a) as a function of the frequency of M1.58-specific CTL in the suspension. The linear dependence is log10[intensity] = 5.304+0.9886°log10[% freq.] with R2=0.998. Experimental conditions are the same as in Figure 4.
At 10% M1.58-specific CTL, we find that all 100 spot replicates have fluorescence intensities above this cutoff intensity indicating that captured cells are detected on each spot, although the variability in spot intensities is substantial. Considering a total population of 100,000 cells in the suspension, as many as 10,000 M1.58-specific CTL are captured, or 100 cells captured per spot on average. As the frequency of M1.58-specific CTL in the suspension decreases, captured cells are detected on fewer spots. At the lowest frequency of 0.01% M1.58-specific CTL, only two spots have fluorescence intensities higher than the cutoff, indicating that at least two cells have been captured out of a total 10 M1.58-specific CTL in the suspension. This lower bound sets the minimum efficiency for antigen-specific cell capture at 20%, which is much greater than the efficiency based on the fractional area of all the spots of 3.8%. The result implies that the cells sample the microarray surface more efficiently than expected from the quiescent settling of cells from the suspension. If the 10 M1.58-specific cells were randomly distributed over the 100 spots, the probability of capturing all of them on just two spots is approximately (1/50)8 or ~ 10−14. The random probability of capturing three M1.58-specific cells on just two of the 100 spots is about 2%, and it is ~ 10−4 for four cells on two of 100 spots. We conclude, therefore, that only two or three M1.58-specific cells were likely captured in this assay; thus establishing that the sensitivity of 0.01% antigen-specific CTL frequency corresponds to detecting the capture of two or three antigen-specific T cells.
An important implication of the large variability in spot intensities in Figure 5a is that mean spot intensities will not provide an accurate quantitative measure of low-frequency antigen-specific CTL, even after taking into account the unoccupied spots on sparsely populated microarrays. An alternative measure is the sum of spot intensities for only those spots displaying captured cells. Using this measure, a linear dependence over four orders of magnitude (R2=0.998) is obtained by plotting the logarithm of this sum as a function of the logarithm of the frequency of M1.58-specific CTL (Figure 5b). Our finding that the linear dependence holds even when the target cells are sparse in number compared to the number of microarray spots is especially significant for clinical and research applications where small volumes and consequently small numbers of target cells are dictated by limitations on sample size.
Profiling influenza-specific CTL in peripheral blood
We also evaluated the microarray assay for simultaneously detecting and quantifying antigen-specific CD8+ CTL frequencies in more complex cell populations from peripheral blood by comparing the assay with flow cytometry for three influenza A epitopes: the immunodominant M1.58 peptide, and the subdominant NA.75 and PA.46 peptides [xxxiii]. For the microarray assays, the spots displaying captured cells were identified by accounting for the variability in spot fluorescence intensities, as described above, and normalizing by the sum of spot intensities for all anti-CD3 antibody spots. Non-specific cell capture on the microarray was taken into account as described above and found to be less than the number of cells detected by FACS that bind the unloaded dimer.
The results in Figure 6 show that multiple antigen-specific CTL can be detected simultaneously and reliably for a variety of antigen-specific responses, including responses to subdominant as well as immunodominant epitopes. The M1.58-specific and PA.46-specific CTL frequencies were found to be 3.1% and 2.1%, respectively, in microarray assay compared to 1.5% and 0.67%, respectively, determined by FACS dimer staining. A significantly lower frequency of NA.75-specific CTL was obtained in both assays: 0.19% in the microarray assay compared to 0.07% determined by FACS dimer staining. The consistently higher frequencies derived from the microarray assays presumably reflect the high efficiency of cell capture on the microarray coupled with a higher level of non-specific binding of the unloaded dimer in the FACS analysis, which together predict comparatively higher antigen-specific CTL frequencies in the cell suspensions in contact with the microarray.
Figure 6.

Detection of M1.58-, NA.75-, and PA.46-specific CTL in CD8+ CTL isolated from peripheral blood of A2-positive human donor A8 by pHLA A2-Ig cellular microarrays and FACS. For each assay, the level of non-specific binding to the unloaded dimer was subtracted before plotting. Antigen-specific CTL frequencies in the microarray assay are quantified as the sum of spot intensities for only those spots displaying bound cells, normalized by the sum of anti-CD3 spot intensities. Individual spots of the 5 × 5 subarray were printed using 333pl of anti-Igλ antibody/BSA solution, and are 500 μm in diameter with a spot center-to-center distance of 1 mm. Cell seeding conditions: 50μl of ~2 × 106/ml total cells.
Conclusions
The HLA-based cellular microarray assay presented here offers several advantages for broad-based quantitative assessments of low-frequency antigen specificities in complex T-cell populations. These advantages stem from a number of notable advances in assay development, the central component of which is to separate the core TCR-pHLA molecular recognition event from the binding interactions that lead to capture of antigen-specific cells. Indeed, uncoupling these two steps allows for the optimization and control of each one separately. Significant enhancements in both the functionality and accessibility of the HLA complexes in binding cognate TCRs are achieved by incubating the pHLA-Ig dimers with the T cells in solution. Much higher concentrations of the pHLA complex are also possible in solution, and the concentration can be controlled to maximize TCR cross-linking for high TCR-pHLA avidity [xxxiv]. By replacing the relatively low affinity TCR-pHLA binding interactions with much higher affinity binding interactions between the pHLA-Ig complex and anti-Igλ antibody at the cell-microarray substrate interface, the cell capture efficiency is essentially uniform across the entire microarray. More importantly, antigen-specific cell capture can be calibrated against a comparable antibody-antigen binding interaction that characterizes cell capture from the CTL population as a whole. These advances yield a robust assay that is linear over four decades of antigen-specific CTL frequencies extending to frequencies as low as 0.01%.
These advantages are offset to an extent by the limited ability to multiplex the assay to quantify multiple antigen specificities simultaneously, since the current microarray assay uses only one unlabeled anti-Igλ antibody to capture antigen-specific cells. It may be feasible, however, to screen a single population of CD8 T cells for multiple antigen specificities if multiple capture molecules are used to distinguish the different pHLA-Ig dimer complexes of interest – e.g., by conjugating nucleic acid oligomers with different, predetermined sequences specific to different pHLA complexes, and capturing antigen-specific T cells by selective binding to complementary nucleic acid sequences printed on the microarray [x]. Combinatorial approaches using multiple pHLA complexes, each labeled with a unique combination of distinct fluorochromes, have also been applied to identify antigen-specific CTL on the basis of multi-color coding, thus enabling the simultaneous detection of multiple antigen specificities in a single cell population [xxxv,xxxvi,xxxvii]. Successful incorporation of these technologies into the current microarray assay, however, will likely involve tradeoffs in specificity and sensitivity that have yet to be evaluated in order to define the extent to which multiplexing can be achieved with the quantitation of low-frequency antigen specificities that has been demonstrated here.
With the current microarray design, we established a detection sensitivity of 0.01% antigen-specific CTL with sample sizes on the order of 100,000 CD8 T cells, which corresponds to the capture and detection of only two or three cells out of the 10 antigen-specific cells in this sample. Detection sensitivities higher than 0.01% antigen-specific CTL are also attainable by taking advantage of the scalability of cell capture in the microarray assay. Thus, increasing sample size, enhancing cell capture efficiency, or a combination of these two strategies would lead to higher sensitivities. Antigen-specific CTL frequencies lower than 0.01% can be detected, for example, using the existing microarray configuration and assay by simply increasing the sample size while maintaining the current efficiency of cell capture. The capture efficiency can also be improved; e.g., by increasing the spot surface area, enhancing contact between the cell suspension and the microarray substrate using microfluidics, or augmenting the intrinsic mobility of the cells on the substrate by patterning adhesion molecules on the microarray. Considering the practical limit on sensitivity to be the detection of two or three antigen-specific T cells captured on the microarray, a sensitivity of 0.001% antigen-specific CTL is feasible for sample sizes as small as 200,000 or 300,000 T cells.
Another possibility for improving sensitivity is to combine the detection of captured cells with a complementary functional assay of T-cell activation. The current microarray configuration and assay are ideally suited for such measurements. Specifically, T-cell activation in response to antigen-specific stimulation is enhanced by incubating the pHLA-Ig dimers with the T cells in solution, which allows for the accumulation of laterally mobile TCR-pHLA complexes on the cell surface and the formation of stable microclusters in the cell-substrate contact area [xxxviii]. Sustained T-cell activation on the microarray facilitates a complementary functional characterization through the detection of cytokines secreted by the captured cells. Indeed, multivalent binding of the bivalent pHLA-Ig dimer in combination with the bivalent anti-Igλ antibody will further stabilize TCR cross-linking and clustering, thereby inducing downstream signal transductions and subsequent responses, which would amplify T-cell activation to enhance their functional characterization.
The potential for high-throughput, broad-based assessments of diverse T-cell populations that this microarray assay offers would allow entirely different questions to be asked in the search for new vaccines or therapeutics. Specifically, the search paradigm would shift from identifying individual target molecules/antigens to one encompassing the simultaneous characterization of correlated responses to multiple targets. The search would therefore take place at the systems level with a focus on an “effective immune response,” – i.e., the ability to control disease. Moreover, this search would not be limited to a single, “one-size-fits-all” formulation, but could be tailored to the history of infections and vaccinations within specific human populations, or even at the level of individuals. The novel cellular microarray assay presented here thus opens up the exciting possibility of designing new vaccines or therapeutics that take advantage of both the complexity and the diversity of the human immune response.
Supplementary Material
Figure S1. Effect of print additives on human recombinant IFNγ (rhIFNγ) detection. Anti-IFNγ at a concentration of 0.5mg/ml was printed with 9 different additives, with no additive as a control. Each sample was printed at a replicate of 8 spots. Individual spots contain 333 picoliters of printing solution. A sandwich assay was conducted by contacting the array with 20ng/ml rhIFNγ followed by 20μg/ml PE-Cy7-anti-IFNγ. The mean spot intensity ± standard error for each sample across the spot replicates is shown to the left of the slide image and the identity of the spots in terms of the additives are shown to the right of the image.
Figure S2. FACS detection and determination of the frequency of antigen-specific CTLs enriched and expanded with aAPCs. For each aAPC-enriched population, FACS plots on the top panel show cells stained with dimer loaded with the inducing peptide. That is, from left to right: CMVpp65-enriched cells stained with CMVpp65-(loaded) dimer, M1.58-enriched cells stained with M1.58- dimer, and PA.46-enriched cells stained with PA.46-dimer. Cells stained with unloaded dimer were included as negative control, as shown in the bottom panel. The appearance of two CMVpp65-enriched dimer populations results from working with short-term T-cell cultures that contain a mixture of antigen-specific T-cell clones with different CMVpp65-dimer binding affinities. Furthermore, CTL with very high affinities can start to down-regulate CD8, as well as their TCRs specifically, when dimer or tetramer staining is carried out at room temperature.
Acknowledgments
This work was supported by NIH grant AI077097.
References
- i.Frahm N, et al. Control of human immunodeficiency virus replication by cytotoxic T lymphocytes targeting subdominant epitopes. Nature Immunology. 2006;7:173–178. doi: 10.1038/ni1281. [DOI] [PubMed] [Google Scholar]
- ii.Sette A, Peters B. Immune epitope mapping in the post-genomic era: lessons for vaccine development. Current Opinion in Immunology. 2007;19:106–110. doi: 10.1016/j.coi.2006.11.002. [DOI] [PubMed] [Google Scholar]
- iii.Yewdell JTW, Bennink JR. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annual Review of Immunology. 1999;17:51–88. doi: 10.1146/annurev.immunol.17.1.51. [DOI] [PubMed] [Google Scholar]
- iv.Arstila TP, et al. A direct estimate of the human alpha beta T cell receptor diversity. Science. 1999;286(5441):958–961. doi: 10.1126/science.286.5441.958. [DOI] [PubMed] [Google Scholar]
- v.Altman JD, et al. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274(5284):94–96. doi: 10.1126/science.274.5284.94. [DOI] [PubMed] [Google Scholar]
- vi.Greten TF, et al. Direct visualization of antigen-specific T cells: HTLV-1 Tax11–19-specific CD8(+) T cells are activated in peripheral blood and accumulate in cerebrospinal fluid from HAM/TSP patients. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(13):7568–7573. doi: 10.1073/pnas.95.13.7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vii.Chen DS, et al. Marked differences in human melanoma antigen-specific T cell responsiveness after vaccination using a functional microarray. PloS Medicine. 2005;2(10):1018–1030. doi: 10.1371/journal.pmed.0020265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- viii.Deviren G, et al. Detection of antigen-specific T cells on p/MHC microarrays. J Mol Recognit. 2007;20(1):32–38. doi: 10.1002/jmr.805. [DOI] [PubMed] [Google Scholar]
- ix.Soen Y, et al. Detection and characterization of cellular immune responses using peptide-MHC microarrays. PloS Biology. 2003;1(3):429–438. doi: 10.1371/journal.pbio.0000065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- x.Kwong GA, et al. Modular Nucleic Acid Assembled p/MHC Microarrays for Multiplexed Sorting of Antigen-Specific T Cells. Journal of the American Chemical Society. 2009;131(28):9695–9703. doi: 10.1021/ja9006707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xi.Stone JD, Walter J, Demkowicz E, Stern LJ. HLA-restricted epitope identification and detection of functional T cell responses by using MHC-peptide and costimualtory microarrays. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:3744–3749. doi: 10.1073/pnas.0407019102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xii.Nishioka GM, Markey AA, Holloway CK. Protein damage in drop-on-demand printers. Journal of the American Chemical Society. 2004;126(50):16320–16321. doi: 10.1021/ja044539z. [DOI] [PubMed] [Google Scholar]
- xiii.Zhu H, Snyder M. Protein chip technology. Current Opinion in Chemical Biology. 2003;7:55–63. doi: 10.1016/s1367-5931(02)00005-4. [DOI] [PubMed] [Google Scholar]
- xiv.Delehanty JB, Ligler FS. Method for printing functional protein microarrays. BioTechniques. 2003;34:380–385. doi: 10.2144/03342mt02. [DOI] [PubMed] [Google Scholar]
- xv.Wu P, Grainger DW. Comparison of hydroxylated print additives on antibody microarray performance. Journal of Proteome Research. 2006;5(11):2956–2965. doi: 10.1021/pr060217d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xvi.Liu YS, et al. Optimization of printing buffer for protein microarrays based on aldehyde-modified glass slides. Frontiers in Bioscience. 2007;12:3768–3773. doi: 10.2741/2350. [DOI] [PubMed] [Google Scholar]
- xvii.Ajikumar PK, et al. Carboxyl-terminated dendrimer-coated bioactive interface for protein microarray: High-sensitivity detection of antigen in complex biological samples. Langmuir. 2007;23(10):5670–5677. doi: 10.1021/la063717u. [DOI] [PubMed] [Google Scholar]
- xviii.Davis MM, et al. Ligand recognition by alpha beta T cell receptors. Annual Review of Immunology. 1998;16:523–544. doi: 10.1146/annurev.immunol.16.1.523. [DOI] [PubMed] [Google Scholar]
- xix.Armstrong KM, et al. Thermodynamics of T-cell receptor-peptide/MHC interactions: progress and opportunities. J Mol Recognit. 2008;21:275–287. doi: 10.1002/jmr.896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xx.Chan PY, et al. Influence of Receptor Lateral Mobility on Adhesion Strengthening between Membranes Containing Lfa-3 and Cd2. Journal of Cell Biology. 1991;115(1):245–255. doi: 10.1083/jcb.115.1.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxi.Parham P, Barnstable CJ, Bodmer WF. Use of a Monoclonal Antibody (W6-32) in Structural Studies of Hla-a,B,C Antigens. Journal of Immunology. 1979;123(1):342–349. [PubMed] [Google Scholar]
- xxii.Tanabe M, et al. Structural and Functional-Analysis of Monomorphic Determinants Recognized by Monoclonal-Antibodies Reacting with the Hla Class-I Alpha-3 Domain. Journal of Immunology. 1992;148(10):3202–3209. [PubMed] [Google Scholar]
- xxiii.Ware R, et al. Human Cd8(+) T-Lymphocyte Clones Specific for T-Cell Receptor V-Beta Families Expressed on Autologous Cd4(+) T-Cells. Immunity. 1995;2(2):177–184. doi: 10.1016/s1074-7613(95)80066-2. [DOI] [PubMed] [Google Scholar]
- xxiv.Genestier L, et al. Fas-independent apoptosis of activated T cells induced by antibodies to the HLA class I alpha 1 domain. Blood. 1997;90(9):3629–3639. [PubMed] [Google Scholar]
- xxv.Schneck JP, et al. Monitoring antigen-specific T cells using MHC-Ig dimers. In: Coligan JE, et al., editors. Short protocols in immunology: a compendium of methods from current protocols in immunology. Wiley; 2005. pp. 15-2–15-11. [Google Scholar]
- xxvi.Ndhlovu ZM, et al. Dynamic regulation of functionally distinct virus-specific T cells. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:3669–3674. doi: 10.1073/pnas.0915168107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxvii.Buchli R, et al. Real-time measurement of in vitro peptide binding to soluble HLA-A*0201 by fluorescence polarization. Biochemistry. 2004;43(46):14852–14863. doi: 10.1021/bi048580q. [DOI] [PubMed] [Google Scholar]
- xxviii.Rodenko B, et al. Generation of peptide-MHC class I complexes through UV-mediated ligand exchange. Nature Protocols. 2006;1(3):1120–1132. doi: 10.1038/nprot.2006.121. [DOI] [PubMed] [Google Scholar]
- xxix.Arakawa T, Kita Y, Timasheff SN. Protein precipitation and denaturation by dimethyl sulfoxide. Biophysical Chemistry. 2007;131(1–3):62–70. doi: 10.1016/j.bpc.2007.09.004. [DOI] [PubMed] [Google Scholar]
- xxx.Parnham P, et al. Use of a monoclonal antibody (W6/32) in structural studies of HLA-a,b,c antigens. Journal of Immunology. 1979;123(1):342–349. [PubMed] [Google Scholar]
- xxxi.Oelke M, et al. Ex vivo induction and expansion of antigen-specific cytotoxic T cells by HLA-Ig-coated artificial antigen-presenting cells. Nature Medicine. 2003;9(5):619–624. doi: 10.1038/nm869. [DOI] [PubMed] [Google Scholar]
- xxxii.Yewdell JW. Confronting complexity: Real-world immunodominance in antiviral CD8(+) T cell responses. Immunity. 2006;25(4):533–543. doi: 10.1016/j.immuni.2006.09.005. [DOI] [PubMed] [Google Scholar]
- xxxiii.Gianfrani C, et al. Human memory CTL response specific for influenza A virus is broad and multispecific. Human Immunology. 2000;61(5):438–452. doi: 10.1016/s0198-8859(00)00105-1. [DOI] [PubMed] [Google Scholar]
- xxxiv.Fahmy TM, et al. Probing T cell membrane organization using dimeric MHC-Ig complexes. J Immunol Methods. 2002;268:93–106. doi: 10.1016/s0022-1759(02)00203-x. [DOI] [PubMed] [Google Scholar]
- xxxv.Kedziershka K, et al. Finding multiple needles in one immune haystack. Nature Methods. 2009;6(7):489–490. doi: 10.1038/nmeth0709-489. [DOI] [PubMed] [Google Scholar]
- xxxvi.Newell EW, et al. Simultaneous detection of many T-cell specificities using combinatorial tetramer staining. Nature Methods. 2009;6(7):497–499. doi: 10.1038/nmeth.1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxxvii.Hadrup SR, et al. Parallel detection of antigen-specific T-cell responses by multidimensional encoding of MHC multimers. Nature Methods. 2009;6(7):520–526. doi: 10.1038/nmeth.1345. [DOI] [PubMed] [Google Scholar]
- xxxviii.Grakoui A, et al. The immunological synapse: A molecular machine controlling T cell activation. Science. 1999;285(5425):221–227. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Effect of print additives on human recombinant IFNγ (rhIFNγ) detection. Anti-IFNγ at a concentration of 0.5mg/ml was printed with 9 different additives, with no additive as a control. Each sample was printed at a replicate of 8 spots. Individual spots contain 333 picoliters of printing solution. A sandwich assay was conducted by contacting the array with 20ng/ml rhIFNγ followed by 20μg/ml PE-Cy7-anti-IFNγ. The mean spot intensity ± standard error for each sample across the spot replicates is shown to the left of the slide image and the identity of the spots in terms of the additives are shown to the right of the image.
Figure S2. FACS detection and determination of the frequency of antigen-specific CTLs enriched and expanded with aAPCs. For each aAPC-enriched population, FACS plots on the top panel show cells stained with dimer loaded with the inducing peptide. That is, from left to right: CMVpp65-enriched cells stained with CMVpp65-(loaded) dimer, M1.58-enriched cells stained with M1.58- dimer, and PA.46-enriched cells stained with PA.46-dimer. Cells stained with unloaded dimer were included as negative control, as shown in the bottom panel. The appearance of two CMVpp65-enriched dimer populations results from working with short-term T-cell cultures that contain a mixture of antigen-specific T-cell clones with different CMVpp65-dimer binding affinities. Furthermore, CTL with very high affinities can start to down-regulate CD8, as well as their TCRs specifically, when dimer or tetramer staining is carried out at room temperature.