

Abstract
A wealth of evidence supports increased NO (NO⋅) in asthma,
but its roles are unknown. To investigate how NO participates in
inflammatory airway events in asthma, we measured NO⋅ and
NO⋅ chemical reaction products [nitrite, nitrate,
S-nitrosothiols (SNO), and nitrotyrosine] before,
immediately and 48 h after bronchoscopic antigen (Ag) challenge of
the peripheral airways in atopic asthmatic individuals and nonatopic
healthy controls. Strikingly, NO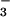 was the only
NO⋅ derivative to increase during the immediate Ag-induced
asthmatic response and continued to increase over 2-fold at 48 h
after Ag challenge in contrast to controls [P <
0.05]. NO
was the only
NO⋅ derivative to increase during the immediate Ag-induced
asthmatic response and continued to increase over 2-fold at 48 h
after Ag challenge in contrast to controls [P <
0.05]. NO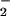 was not affected by Ag challenge at 10
min or 48 h after Ag challenge. Although SNO was not detectable in
asthmatic airways at baseline or immediately after Ag, SNO increased
during the late response to levels found in healthy controls. A model
of NO⋅ dynamics derived from the current findings predicts that
NO⋅ may have harmful effects through formation of peroxynitrite,
but also subserves an antioxidant role by consuming reactive oxygen
species during the immediate asthmatic response, whereas nitrosylation
during the late asthmatic response generates SNO, safe reservoirs for
removal of toxic NO⋅ derivatives.
was not affected by Ag challenge at 10
min or 48 h after Ag challenge. Although SNO was not detectable in
asthmatic airways at baseline or immediately after Ag, SNO increased
during the late response to levels found in healthy controls. A model
of NO⋅ dynamics derived from the current findings predicts that
NO⋅ may have harmful effects through formation of peroxynitrite,
but also subserves an antioxidant role by consuming reactive oxygen
species during the immediate asthmatic response, whereas nitrosylation
during the late asthmatic response generates SNO, safe reservoirs for
removal of toxic NO⋅ derivatives.
Keywords: reactive oxygen species, reactive nitrogen species, asthma, allergy
The exhaled air of asthmatic
individuals contains higher levels of NO⋅ than found in exhalate
of healthy nonsmoking individuals (1–7). The role of NO⋅ in
asthma is unclear, but studies suggest that NO⋅ relaxes
bronchial smooth muscle, leading to bronchodilatation, inhibits
proinflammatory signaling events (8, 9), or conversely contributes to
airway inflammation and injury through formation of toxic reactive
nitrogen species (RNS) (10). Ultimately, the functional role of
NO⋅, as any molecule, will depend on both its concentration and
association with other biomolecules and proteins (11). In this context,
NO⋅ is a highly reactive molecule, and exhaled NO⋅ likely
represents only a fraction of the total NO⋅ in the lung.
NO⋅ reacts with oxygen or reactive oxygen species (ROS) to form
oxidation products, such as NO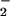 ,
NO
,
NO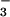 , and RNS (8, 10–13). NO⋅ or RNS may lead
to nitration of tyrosine residues in proteins or nitrosylation of
biologic constituents to form S-nitrosothiols (SNO); 100%
of NO⋅ reaction products are partitioned in the liquid phase of
the lung (8, 10, 11, 13). Notably, the temporal sequence of change in
NO⋅ and its downstream reaction products within airways during
an asthmatic attack is unknown.
, and RNS (8, 10–13). NO⋅ or RNS may lead
to nitration of tyrosine residues in proteins or nitrosylation of
biologic constituents to form S-nitrosothiols (SNO); 100%
of NO⋅ reaction products are partitioned in the liquid phase of
the lung (8, 10, 11, 13). Notably, the temporal sequence of change in
NO⋅ and its downstream reaction products within airways during
an asthmatic attack is unknown.
Airway antigen (Ag) challenge has been used in atopic individuals as an experimental model to study mechanisms/mediators that lead to asthmatic responses and airway inflammation (14–19). Exposure of asthmatic individuals to appropriate Ag results in both an immediate asthmatic response occurring within minutes and a similar but prolonged late response after many hours. The immediate response has been associated with release of bronchoconstrictor mediators and ROS, and the late response with thickening of the airway mucosa by edema and inflammatory cell influx (14–19). To investigate the functional role of NO⋅ in the asthmatic response, we measured NO⋅ generation and downstream product formation (nitrite, nitrate, SNO, and nitrotyrosine) before, immediately and 48 h after bronchoscopic Ag challenge of the peripheral airways in atopic asthmatic individuals and nonatopic healthy controls. The results reveal clear differences in NO⋅ chemical reactions in the early and late responses to Ag challenge in asthmatic airways, which support distinct functions for NO⋅ in asthmatic inflammatory events during the immediate and late phase of an asthma attack.
Methods
Study Population.
All individuals were screened by history, physical examination, spirometry, methacholine provocation to establish reactivity, and allergy prick skin testing to a standard panel of aeroallergens. All subjects were nonsmokers. Healthy controls had no cardiopulmonary symptoms, normal baseline spirometry, negative methacholine challenge, and were nonatopic. Inclusion criteria for atopic asthmatic individuals were positive skin testing for atopy, a forced expiratory volume in 1 sec (FEV1) ≥60% predicted, no asthma exacerbation within the previous month, no inhaled antiinflammatory agents for ≥2 weeks, and no oral corticosteroids for ≥4 weeks. Asthma severity and temporal course in volunteers included mild intermittent and mild persistent asthma (20). Individuals were studied outside of the specific allergen season. Samples examined in this study were obtained as part of a separate clinical investigation (14). Detailed clinical characteristics of individuals in this study including atopic status, pulmonary functions, nonspecific airway reactivity to methacholine, and specific airway reactivity to Ag were recently described in a separate report (14). The study was approved by the Cleveland Clinic Institutional Review Board, and all individuals gave informed written consent.
Segmental Ag Challenge.
Asthmatics with ≥2 positive skin tests underwent whole-lung bronchoprovocation with a relevant Ag. Escalating doses of Ag were aerosolized to determine the provocative dose that caused a 20% decline in FEV1. A minimum of 4 weeks after the whole-lung Ag aerosol challenge, individuals underwent segmental Ag challenge during bronchoscopy. For segmental Ag challenge, the dose of Ag instilled into each segment was 10% of the whole-lung Ag dose (typically 1–50 protein nitrogen (PN) units/segment) diluted in 10 ml of sterile saline (14). Controls also underwent a lung segmental Ag challenge with 100 PN units ragweed Ag per segment. Baseline NO⋅ levels were measured in orally exhaled gases before bronchoscopy and in the gases of the lower airway during a breath hold, as described below. Baseline bronchoalveolar lavage (BAL) was obtained from the lingula. Ag was instilled into two lung subsegments of the right middle lobe via the bronchoscope. The bronchoscope was maintained in the airway. Ten minutes after Ag challenge, NO⋅ was measured in gases sampled from the right middle lobe bronchus during a breath hold, and BAL was obtained from one of the Ag challenged right middle lobe subsegments. Individuals returned 48 h later for measurement of NO⋅ in orally exhaled gases, and a second bronchoscopy to measure intrapulmonary NO⋅ and obtain BAL from the second Ag challenged subsegment of the right middle lobe.
BAL Fluid.
To recover lung epithelial lining fluid, 150 ml of sterile 0.9% saline warmed to 37°C was instilled in 50-ml aliquots and immediately aspirated back through the suction channel of the bronchoscope (4, 11). The recovered BAL fluid was immediately centrifuged at 400 × g for 10 min. The cell pellet was removed and the supernatant immediately frozen at −80°C for determination of NO⋅ reaction products including nitrite, nitrate, and SNO.
Intrapulmonary NO⋅ levels.
The bronchoscope was advanced into the lung, and real-time NO⋅ measurements in the lower airway gases were obtained at a rate of 20 samplings per second by using a Teflon tube inserted through the working channel of the bronchoscope and connected to a chemiluminescence analyzer for detection of nitric oxide (NOA 280, Sievers, Boulder, CO), as previously described (11). Individuals were instructed to breath-hold during NO⋅ measures. Breath holding was confirmed by monitoring chest wall movement and airflow at the mouth and nose. Intrapulmonary NO⋅ levels were determined in individuals breathing room air and while breathing air with no NO⋅ (zero air) when ambient levels were >20 parts per billion.
Exhaled NO⋅ at the Mouth.
NO⋅ in gases exhaled at the mouth was measured in asthmatic and healthy control individuals by using a chemiluminescent analyzer (NOA 280, Sievers) by collecting a single exhaled breath from total lung capacity against a resistance of 10 cm H2O, after a 15-second breath hold at total lung capacity.
NO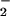 , NO
, NO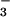 , and
S-nitrosothiols in BAL.
, and
S-nitrosothiols in BAL.
Nitrate and nitrite in BAL fluid were measured by chemiluminescence as
previously described (11). Briefly, NO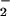 and
NO
and
NO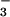 were converted to NO⋅ by a saturated
solution of VCl3 in 0.8 M HCl, and the NO⋅
detected by using the NOA 280, and interpolation by using standard
authentic NO
were converted to NO⋅ by a saturated
solution of VCl3 in 0.8 M HCl, and the NO⋅
detected by using the NOA 280, and interpolation by using standard
authentic NO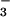 and NO
and NO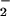 .
NO
.
NO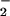 alone was determined by using a 1% wt/vol
solution of KI in glacial acetic acid to convert NO
alone was determined by using a 1% wt/vol
solution of KI in glacial acetic acid to convert NO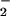 to NO. NO
to NO. NO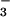 was subsequently calculated as the
difference between total [NO
was subsequently calculated as the
difference between total [NO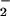 and
NO
and
NO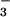 ] and NO
] and NO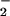 . Levels of
NO
. Levels of
NO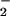 or NO
or NO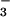 were expressed as
micromolar levels in BAL fluid.
were expressed as
micromolar levels in BAL fluid.
SNO were assayed as NO⋅ by chemiluminescence (NOA 280) after reduction by using a saturated solution of CuCl and 1 mM cysteine in a purge vessel by using a continuous helium stream in 10 mM PBS, pH 7.0, at 50°C, as previously described (13, 21). This assay is specific and linear to 5 nM (21).
Immunohistochemical Detection of Nitrotyrosine.
Endobronchial biopsies were taken from the lingula and at 48 h after segmental Ag challenge from the right middle lobe of asthmatic and control individuals for immunostaining. Tissues were fixed in 10% buffered formalin, embedded in paraffin, and 5-mm sections were placed on charged slides for immunohistochemistry. The slides were incubated at 37°C with 0.01 mg/ml protease K for 15 min. After wash with PBS containing 0.5 mM levamisole (Sigma), the tissue was treated with 1% BSA in PBS to block nonspecific binding, then incubated for 2 h with the primary polyclonal antibody directed against nitrotyrosine (Upstate Biotechnology, Lake Placid, NY) (1:150 diluted in 1% BSA/PBS). After wash with PBS/0.5 mM levamisole, tissue was incubated with a biotin-conjugated secondary antibody (Dako) for 10 min. Because peroxidase-dependent immunohistochemistry may give rise to artifactual protein nitration in tissue sections (10), an alkaline phosphatase method was used to detect nitrotyrosine in tissue sections. Washing was followed by another 10-min incubation with alkaline phosphatase-labeled streptavidin (Dako). Immunostaining was visualized with an alkaline phosphate substrate solution containing naphtol AS-MX phosphatase, Fast red, and levamisole in Tris buffer (pH 8.2) (Dako) and counterstained with the nuclear stain, hematoxylin (Sigma). Specificity of staining was confirmed by immunoabsorption of nitrotyrosine antibody with 5 mM nitrotyrosine (Sigma) before incubation with tissue sections or with PBS instead of the primary antibody.
Statistical Analyses.
Summaries of the clinical characteristics at individual time points and their differences from baseline are reported as mean ± standard error. We used paired t tests within the healthy control and asthmatic groups to compare the mean levels of clinical characteristics. Asthmatics and controls were compared with respect to mean levels at individual time points (baseline, 10 m, 48 h) and with respect to differences from baseline by using t tests for independent samples. The normality assumption of the t tests was assessed visually. The nonparametric Mann–Whitney test was used as an alternative to the t test for independent samples when nonnormality was demonstrated. statistical analysis system (sas) Ver. 6.12 (SAS Institute, Cary, NC) and s-plus Ver. 3.4 (Mathsoft, Seattle, WA) were used to perform the statistical tests and visual assessments.
Results
Characterization of Study Population.
Eight mild intermittent asthmatics [2 male, 6 female, age 37 ± 7 (SD), range 27–47 yr] and six healthy nonasthmatic nonatopic controls [3 male, 3 female, age 42 ± 9 (SD), range 26–53 yr] participated in the study. Bronchoscopy and segmental Ag challenge were well tolerated without complications. Clinical characteristics of these individuals have been previously reported (14). Because sample obtained from some subjects was limiting, not all studies could be performed on every individual. The n for experiments in which all individuals could not be evaluated is stated in the text.
NO⋅ in Exhaled and Intrapulmonary Gases.
NO⋅ at baseline and 48 h after Ag challenge have been reported in part previously (14). In all individuals, NO⋅ achieved a steady-state plateau that was quantitated (Fig. 1). Baseline intrapulmonary NO⋅ in asthmatic airways tended to be higher than controls (Fig. 1 and Table 1). NO⋅ at 10 min after Ag challenge was similar to baseline levels, with only a small tendency to decreased NO⋅ in asthmatic airways (Table 1). In contrast, intrapulmonary NO⋅ at 48 h after Ag challenge increased 2-fold in asthma but not in controls (Table 1). Exhaled NO⋅ clearly reflected the changes in intrapulmonary NO⋅, although exhaled NO⋅ underestimated intrapulmonary NO⋅ in asthma (Table 1). Baseline NO⋅ levels in asthmatics in this study were not significantly higher than healthy controls. Measures of exhaled NO in a larger population of asthmatic individuals (n = 60) revealed that NO⋅ levels vary widely in asthmatics, with many asthmatics having NO⋅ in the range of healthy controls (range 4–49 parts per billion). Variability in NO was independent of age (ANOVA P = 0.427) (data not shown) but may reflect asthma severity (2–5, 23).
Figure 1.

An asthmatic individual with higher NO⋅ plateau in intrapulmonary gases than a healthy control individual at Baseline before Ag challenge. At 10 min after Ag challenge (Ag 10 min), NO⋅ plateau levels are similar to baseline levels, but by 48 h after Ag challenge (Ag 48 h), the NO⋅ plateau in the asthmatic airway gases is much higher than plateau levels at baseline. In contrast, the nonatopic healthy control had no increase in plateau NO⋅ in intrapulmonary gases. Each point represents a single NO⋅ determination in gases continually sampled from the lower airway at bronchoscopy at intervals of 0.05 sec.
Table 1.
Effect of segmental antigen challenge on NO in exhaled and lower airway gases
| Baseline, 0
min
|
Ag, 10 min
|
Ag, 48 h
|
|||
|---|---|---|---|---|---|
| Exhaled | Lower airway | Lower airway | Exhaled | Lower airway | |
| NO, ppb | |||||
| Asthma | 9 ± 1 | 14 ± 5 | 10 ± 2 | 15 ± 5 | 26 ± 7 |
| Control | 7 ± 1 | 5 ± 2 | 6 ± 2 | 7 ± 1 | 6 ± 3 |
| P* | 0.2 | 0.1 | 0.3 | 0.05 | 0.03 |
T test comparison of asthma vs. control.
ppb, parts per billion.
NO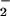 and NO
and NO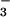 in BAL Fluid.
in BAL Fluid.
Total NO⋅ reaction products in BAL fluid tended to be higher in
asthma at baseline [baseline NO⋅ products (μM): asthma 3
± 1, controls 1.7 ± 0.3, P = 0.06]. NO⋅
reaction products increased in asthmatics but not controls, after Ag
challenge [NO⋅ products (μM): Ag 10 min, asthma 4.4 ±
0.7, controls 1.7 ± 0.5, P = 0.01; Ag 48 h,
asthma 6 ± 1, controls 1.6 ± 0.4, P =
0.02]. The major NO⋅ reaction product in the healthy control
was NO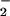 , whereas NO
, whereas NO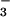 was
predominant in the asthmatic BAL fluid (Fig.
2). Levels of NO
was
predominant in the asthmatic BAL fluid (Fig.
2). Levels of NO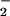 were
remarkably similar in asthmatics and controls, with no changes during
Ag challenge [NO
were
remarkably similar in asthmatics and controls, with no changes during
Ag challenge [NO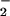 (μM): baseline, asthma 1.3
± 0.1, controls 1.4 ± 0.2, P = 0.9; Ag 10 min,
asthma 1.4 ± 0.2, controls 1.9 ± 0.4, P =
0.3; Ag 48 h, asthma 1.4 ± 0.2, controls 1.3 ± 0.1,
P = 0.9] (Fig. 2). In contrast, NO
(μM): baseline, asthma 1.3
± 0.1, controls 1.4 ± 0.2, P = 0.9; Ag 10 min,
asthma 1.4 ± 0.2, controls 1.9 ± 0.4, P =
0.3; Ag 48 h, asthma 1.4 ± 0.2, controls 1.3 ± 0.1,
P = 0.9] (Fig. 2). In contrast, NO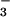 in asthmatic BAL fluid increased at 10 min of Ag challenge with further
increases at 48 h, whereas control NO
in asthmatic BAL fluid increased at 10 min of Ag challenge with further
increases at 48 h, whereas control NO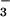 did not
change [NO
did not
change [NO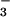 (μM): Baseline, asthma 1.9 ±
0.7, controls 0.5 ± 0.2, P = 0.08; Ag 10 min,
asthma 3.1 ± 0.7, controls 0.4 ± 0.2, P =
0.008; Ag 48 h, asthma 4.4 ± 1.4, controls 0.5 ± 0.4,
P = 0.02] (Fig. 2).
(μM): Baseline, asthma 1.9 ±
0.7, controls 0.5 ± 0.2, P = 0.08; Ag 10 min,
asthma 3.1 ± 0.7, controls 0.4 ± 0.2, P =
0.008; Ag 48 h, asthma 4.4 ± 1.4, controls 0.5 ± 0.4,
P = 0.02] (Fig. 2).
Figure 2.

NO metabolites in BAL fluid from asthmatic and control airways before
(0 min), and 10 min or 48 h after segmental Ag challenge.
NO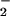 in asthmatic or control BAL fluid did not change
significantly with Ag challenge from baseline (all comparisons.
P > 0.5). NO
in asthmatic or control BAL fluid did not change
significantly with Ag challenge from baseline (all comparisons.
P > 0.5). NO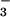 increases at 10
min and 48 h after Ag challenge in asthmatics, whereas
NO
increases at 10
min and 48 h after Ag challenge in asthmatics, whereas
NO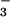 did not change in control airways. SNO were
undetectable in the asthmatic airway at baseline and 10 min after Ag
challenge but increased at 48 h after Ag challenge.
did not change in control airways. SNO were
undetectable in the asthmatic airway at baseline and 10 min after Ag
challenge but increased at 48 h after Ag challenge.
Levels of SNO.
In contrast to healthy controls, SNO were undetectable in BAL fluid from asthmatics at baseline or 10 min after Ag challenge (Fig. 2). However, 48 h after Ag challenge, SNO in asthmatic BAL fluid increased and was similar to control levels [SNO (nM): baseline, asthma 0 ± 0, control 10 ± 6, P = 0.1; Ag 10 min, asthma 0 ± 0, control 7 ± 3, P = 0.06; Ag 48 h, asthma 12 ± 5, control 12 ± 6, P = 0.9 (all time points, asthma n = 4, control n = 3)] (Fig. 2).
Nitrotyrosine in Endobronchial Biopsies.
Biopsies of asthmatic airways demonstrated thickened basement membranes, epithelial shedding, and increased numbers of goblet cells at baseline. As previously shown (4, 22, 23), immunoreactivity for nitrotyrosine was positive in epithelial cells of the asthmatic airways as compared with healthy control epithelial cells (Fig. 3). At 48 h after Ag challenge, loss of epithelial cells was noted, with areas of denuded basement membrane. Furthermore, prominent influx of eosinophils was noted in Ag-challenged segments. Epithelial cells and eosinophils stained positive for nitrotyrosine. Because of marked loss of epithelial cells with Ag challenge, quantitation of nitrotyrosine at 48 h Ag challenge was not possible. Semiquantitative assessment of the basal cells in epithelial biopsies at baseline revealed a tendency to more intense staining for nitrotyrosine in asthmatic, as compared with control epithelium.
Figure 3.

Nitrotyrosine immunostaining of asthmatic and healthy control bronchial mucosa. (A) Asthmatic bronchial mucosa has marked increase in goblet cells and thickened basement membrane. The epithelial cells stain prominently for nitrotyrosine (red staining); red, arrow; g, goblet cell. (B) At 48 h after Ag challenge, epithelial cells are sloughing from the thickened basement membrane (bm), but the cells remaining show positivity for nitrotyrosine. (C) Healthy control bronchial mucosa shows typical pseudostratified columnar epithelium, with red staining present in apical portions of cells (×40; hematoxylin counterstaining).
Discussion
The dynamics of NO⋅ metabolism during an asthmatic response
reveal multiple and sequential reactions and suggest a multifunctional
role for NO⋅ in the airway. In comparison to healthy controls,
mild well-controlled atopic asthmatics tend to have increased NO,
NO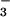 , and nitrotyrosine (22, 23), but undetectable SNO
in the lower airways. Within 10 min of Ag-induced asthmatic response,
NO
, and nitrotyrosine (22, 23), but undetectable SNO
in the lower airways. Within 10 min of Ag-induced asthmatic response,
NO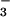 increases markedly in all asthmatics, whereas
NO
increases markedly in all asthmatics, whereas
NO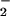 or SNO do not change, and NO⋅ tends to
decrease. By 48 h after Ag-induced asthmatic response, NO⋅,
SNO and NO
or SNO do not change, and NO⋅ tends to
decrease. By 48 h after Ag-induced asthmatic response, NO⋅,
SNO and NO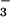 are strikingly increased. Despite notable
changes in asthmatic airways, healthy control individuals have no
changes in levels of NO⋅ or NO⋅ reaction products. Thus,
these changes are distinct to atopic asthmatic individuals. How can
these changes be explained? Recent work has revealed that the chemistry
in the lower airway of asthmatic individuals is oxidizing and most
likely acidic, as opposed to the neutral and overall reducing
conditions in the normal airway (19, 25). On the basis of the known
chemistry of NO, we can begin to construct a model of the events in the
asthmatic airway.
are strikingly increased. Despite notable
changes in asthmatic airways, healthy control individuals have no
changes in levels of NO⋅ or NO⋅ reaction products. Thus,
these changes are distinct to atopic asthmatic individuals. How can
these changes be explained? Recent work has revealed that the chemistry
in the lower airway of asthmatic individuals is oxidizing and most
likely acidic, as opposed to the neutral and overall reducing
conditions in the normal airway (19, 25). On the basis of the known
chemistry of NO, we can begin to construct a model of the events in the
asthmatic airway.
A kinetic scheme based on our findings and current understanding of
NO⋅ reactions is presented in Fig.
4. Although NO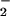 and
NO
and
NO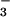 are both detected in the lung,
NO
are both detected in the lung,
NO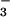 in the asthmatic lung is higher than in controls
and further increases by over 50% within minutes of an asthmatic
reaction. Several pathways may account for the enhanced formation of
NO
in the asthmatic lung is higher than in controls
and further increases by over 50% within minutes of an asthmatic
reaction. Several pathways may account for the enhanced formation of
NO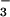 in the asthmatic airway. First, in the presence
of the rich supply of oxyhemoglobin in the lung (represented as a
combination of two resonance forms
FeII–O2 and
FeIII–O
in the asthmatic airway. First, in the presence
of the rich supply of oxyhemoglobin in the lung (represented as a
combination of two resonance forms
FeII–O2 and
FeIII–O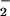 ), reaction of
NO⋅ with oxyhemoglobin results in NO
), reaction of
NO⋅ with oxyhemoglobin results in NO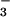 and
FeIII (reaction A, Fig. 4) (26):
and
FeIII (reaction A, Fig. 4) (26):
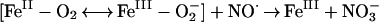 |
A |
Although oxyhemoglobin concentration is the same in
well-controlled asthmatic and healthy control lungs, enhanced formation
of NO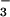 in asthmatic lungs at baseline may result from
the increased NO⋅ generated in asthma (4).
in asthmatic lungs at baseline may result from
the increased NO⋅ generated in asthma (4).
Figure 4.

Model of NO⋅ reactions in the lung during an Ag-induced asthmatic response.
The lack of any significant increase of NO⋅ during the
immediate asthmatic response indicates that pathway A is not the
etiology of the rapid rise in NO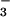 within minutes of
Ag challenge. Indeed, the lack of immediate increase of intrapulmonary
NO⋅, but rather a tendency to decrease, suggests that NO⋅
is being consumed by a more rapid reaction. Given the premise that the
amount of oxyhemoglobin did not change in the asthmatic lung (oxygen
saturation > 98% in all individuals), the increased formation of
NO
within minutes of
Ag challenge. Indeed, the lack of immediate increase of intrapulmonary
NO⋅, but rather a tendency to decrease, suggests that NO⋅
is being consumed by a more rapid reaction. Given the premise that the
amount of oxyhemoglobin did not change in the asthmatic lung (oxygen
saturation > 98% in all individuals), the increased formation of
NO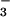 likely results from rapid generation of
peroxynitrite (ONOO−) via reaction of superoxide and
NO⋅ [reaction B, Fig. 4] (27, 28). Indeed, spontaneous asthma
attacks and models of experimental Ag challenge are both associated
with immediate release of superoxide (O
likely results from rapid generation of
peroxynitrite (ONOO−) via reaction of superoxide and
NO⋅ [reaction B, Fig. 4] (27, 28). Indeed, spontaneous asthma
attacks and models of experimental Ag challenge are both associated
with immediate release of superoxide (O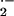 ),
which persists throughout the late asthmatic response (15–18). As
early as 10 min after local instillation of Ag into airways of atopic
individuals, O
),
which persists throughout the late asthmatic response (15–18). As
early as 10 min after local instillation of Ag into airways of atopic
individuals, O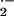 is generated by airspace cells,
with micromolar levels of superoxide at sites of Ag challenge (15–18).
The oxidative environment in the airway during asthmatic response is
confirmed by a decrease in reduced glutathione and increase in oxidized
glutathione at 10 min after Ag challenge (19). Notably, NO⋅
undergoes a direct bimolecular reaction with O
is generated by airspace cells,
with micromolar levels of superoxide at sites of Ag challenge (15–18).
The oxidative environment in the airway during asthmatic response is
confirmed by a decrease in reduced glutathione and increase in oxidized
glutathione at 10 min after Ag challenge (19). Notably, NO⋅
undergoes a direct bimolecular reaction with O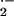 ,
yielding ONOO− at almost diffusion limited rates (reaction
B, k = 6.7 − 19 × 109
⋅ M−1 ⋅ sec−1)
(Fig. 4) (28, 31, 32). The rate constant is over 3.5 times faster than
the dismutation of O
,
yielding ONOO− at almost diffusion limited rates (reaction
B, k = 6.7 − 19 × 109
⋅ M−1 ⋅ sec−1)
(Fig. 4) (28, 31, 32). The rate constant is over 3.5 times faster than
the dismutation of O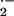 by superoxide dismutases
(SOD). Taken together with the rapid loss of SOD activity during the
immediate Ag-induced asthmatic response (19), reaction B is
overwhelmingly favored in the asthmatic airway.
by superoxide dismutases
(SOD). Taken together with the rapid loss of SOD activity during the
immediate Ag-induced asthmatic response (19), reaction B is
overwhelmingly favored in the asthmatic airway.
After NO⋅ reaction with superoxide and generation of
ONOO−, stopped flow experiments demonstrate peroxynitrite
decay depends on both CO2 and pH (reactions C and
D, Fig. 4) (29–31). At physiologic CO2 and pH,
the CO2-catalyzed decomposition of
ONOO− (reaction C) is faster than the proton-catalyzed
decomposition/isomerization to NO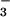 (reaction
D) (28, 29, 31). CO2 levels are relatively high
in the lung (1.76 mM), and the reaction rate with ONOO− is
one of the fastest for ONOO− that likely occurs in
biologic systems (reaction C, rate constant of 3 − 6 ×
104 M−1
s−1) (29, 31, 33). ONOO− and
CO2 lead to nitrosoperoxycarbonate adduct
(ONOOCO
(reaction
D) (28, 29, 31). CO2 levels are relatively high
in the lung (1.76 mM), and the reaction rate with ONOO− is
one of the fastest for ONOO− that likely occurs in
biologic systems (reaction C, rate constant of 3 − 6 ×
104 M−1
s−1) (29, 31, 33). ONOO− and
CO2 lead to nitrosoperoxycarbonate adduct
(ONOOCO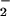 ), which has a lifetime of <3 ms (28, 31).
On the basis of kinetics, ONOOCO
), which has a lifetime of <3 ms (28, 31).
On the basis of kinetics, ONOOCO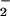 has enhanced
nitrating capabilities as compared with ONOO− or ONOOH,
such that nonenzymatic nitration of tyrosines becomes a physiologically
significant process in cells (29, 31). As in this study, previous
studies have shown increased immunoreactivity for nitrotyrosine in
airway epithelial cells of stable asthmatics as compared with healthy
controls (4, 22, 23). In the absence of target molecules, however,
decomposition of ONOOCO
has enhanced
nitrating capabilities as compared with ONOO− or ONOOH,
such that nonenzymatic nitration of tyrosines becomes a physiologically
significant process in cells (29, 31). As in this study, previous
studies have shown increased immunoreactivity for nitrotyrosine in
airway epithelial cells of stable asthmatics as compared with healthy
controls (4, 22, 23). In the absence of target molecules, however,
decomposition of ONOOCO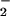 to NO
to NO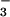 occurs rapidly with regeneration of CO2.
occurs rapidly with regeneration of CO2.
Protonation of ONOO− (reaction D) is markedly enhanced by
acidification (27, 28, 31), and increased acidity occurs in asthmatic
airway during an attack (25). The H+ in the
asthmatic airway may reach levels of 6.3 μM, as opposed to
H+ levels in the healthy airway of 22 nM (25).
Thus, enhanced NO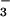 formation in asthmatic response is
likely because of increased ROS and acidity in the asthmatic airway.
formation in asthmatic response is
likely because of increased ROS and acidity in the asthmatic airway.
Importantly, NO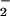 is not predicted to increase in the
reaction scheme in Fig. 4. The third order reaction of NO⋅ and
O2 is relatively very slow in comparison to the
rapid consumption of NO⋅ by superoxide (27). Depending on the
reactions available for NO⋅, some fraction of the NO⋅
in vivo reacts with molecular oxygen to yield
NO
is not predicted to increase in the
reaction scheme in Fig. 4. The third order reaction of NO⋅ and
O2 is relatively very slow in comparison to the
rapid consumption of NO⋅ by superoxide (27). Depending on the
reactions available for NO⋅, some fraction of the NO⋅
in vivo reacts with molecular oxygen to yield
NO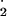 , and subsequently NO
, and subsequently NO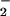 .
Although NO
.
Although NO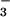 can be produced from dimerization of
NO
can be produced from dimerization of
NO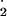 to yield
N2O4 followed by
nitrosation of water to give NO
to yield
N2O4 followed by
nitrosation of water to give NO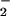 and
NO
and
NO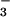 , this is an unlikely route in biological systems,
because NO
, this is an unlikely route in biological systems,
because NO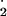 is formed slowly at NO⋅
concentrations found in vivo (27).
is formed slowly at NO⋅
concentrations found in vivo (27).
Although studies have shown that humans with severe asthma have low
airway SNO (25), this is, to our knowledge, the first report that SNO
is undetectable in airways of mild well-controlled asthmatic
individuals. Low levels of SNO in asthma have been previously
attributed to increased catabolic processes (34). Recently, two soluble
protein fractions have been identified from homogenates of rodent lung
that specifically break down SNO and prevent airway smooth muscle
relaxation in vitro (34). Furthermore, increased catabolic
activity has been identified in rodent lung homogenates after ovalbumin
sensitization. Although enzymatic processes for SNO catabolism exist in
neutrophils (34), neutrophils constitute <1% of cells in asthmatic
and control lungs at baseline, and do not increase significantly at 10
min after allergen challenge (24). Thus, neutrophils are a less likely
mechanism for low SNO in the asthmatic lung. Nevertheless, these
findings indicate that SNO catabolism is accelerated in mild stable
asthma as well as in severe asthma. Interestingly, SNO increases to
“normal” during the late asthmatic response. SNO may be formed in
the lung by organic or inorganic reactions. NO⋅, present at high
levels in asthmatic airways (4), may produce SNO under conditions in
which glutathione levels are high, as found in the airway (8, 16, 34).
Specifically, ONOO− may be scavenged by high thiol
concentrations producing a source of SNO formation by NO⋅
synthases (NOS) (35). Reactions between oxygen, superoxide and NO may
also form nitrosating species in the oxidative environment of the
asthmatic airway. Nitrosylation of thiols has also been proposed to
occur via reaction with ONOOCO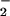 or by formation of
thiyl radicals (13, 27–29, 31). Oxygen-dependent pathways for SNO
formation through thiol autooxidation or NO
or by formation of
thiyl radicals (13, 27–29, 31). Oxygen-dependent pathways for SNO
formation through thiol autooxidation or NO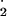 formation may also be relevant in the oxidizing environment of the
asthmatic lung (13). However, SNO increase during the late asthmatic
response, and not in the immediate response, implies a mechanism of
formation that may depend on an influx of inflammatory cells
and/or enzymatic processes, e.g., peroxidases (24). Overall,
these findings support that the ongoing inflammation up-regulates SNO
catabolism in mild asthma, but that SNO formation overwhelms catabolism
in the late asthmatic response, perhaps through enzymatic acceleration
of reactions or acidification, which favor SNO synthesis and stability.
formation may also be relevant in the oxidizing environment of the
asthmatic lung (13). However, SNO increase during the late asthmatic
response, and not in the immediate response, implies a mechanism of
formation that may depend on an influx of inflammatory cells
and/or enzymatic processes, e.g., peroxidases (24). Overall,
these findings support that the ongoing inflammation up-regulates SNO
catabolism in mild asthma, but that SNO formation overwhelms catabolism
in the late asthmatic response, perhaps through enzymatic acceleration
of reactions or acidification, which favor SNO synthesis and stability.
NO⋅ increases only during the late asthmatic response (6, 7, 14). NO⋅ is endogenously produced in the lung by NOS 1–3 (4, 36–38). Of the three enzymes, NOS2 expression is regulated primarily at the transcriptional level by cytokines (4, 36). Specifically, up-regulation of NOS2 gene expression in airway epithelial cells occurs in a delayed fashion at 8–24 h after exposure to interferon-γ in vitro (36, 37). The increase of NO⋅ during the late asthmatic response is consistent with a mechanism that may include up-regulation of NOS2 gene expression by cytokines generated by cells recruited into Ag-challenged airways.
Our current findings and model suggest several relevant biologic
functions for NO⋅ in the airway. NO⋅ rapidly consumes
cytotoxic ROS produced during the immediate asthmatic response. The
reaction product ONOO−/ONOOH is far less reactive
than superoxide and leads to the accumulation of the innocuous product
NO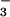 . This immediate detoxification role for
NO⋅ is supported by animal models of asthma in which inhibition
of NO⋅ synthesis leads to worsening toxic reaction to an Ag
challenge (6, 39, 40). Nitrosylation reactions predominate during the
late asthmatic response with accumulation of SNO, which have been
proposed as safe reservoirs for removal of toxic NO⋅ derivatives
(13). Although NO⋅ may have some harmful effects in the airways,
collectively the current findings reveal a temporal sequence of
NO⋅ participation in asthmatic airway chemical events, which
suggests that it may also serve a protective role in the asthmatic
response.
. This immediate detoxification role for
NO⋅ is supported by animal models of asthma in which inhibition
of NO⋅ synthesis leads to worsening toxic reaction to an Ag
challenge (6, 39, 40). Nitrosylation reactions predominate during the
late asthmatic response with accumulation of SNO, which have been
proposed as safe reservoirs for removal of toxic NO⋅ derivatives
(13). Although NO⋅ may have some harmful effects in the airways,
collectively the current findings reveal a temporal sequence of
NO⋅ participation in asthmatic airway chemical events, which
suggests that it may also serve a protective role in the asthmatic
response.
Acknowledgments
We thank D. Laskowski and R. Piccin for technical support, D. J. Stuehr for critical review of the manuscript, and L. Ignarro for helpful discussions. This work was supported by National Institutes of Health Grants HL60917 and BL59337.
Abbreviations
- Ag
antigen
- BAL
bronchoalveolar lavage
- NOS
NO synthase
- SNO
S-nitrosothiols
- ROS
reactive oxygen species
References
- 1.Persson M G, Zetterstrom O, Agrenius V, Ihre E, Gustafsson L E. Lancet. 1994;343:146–147. doi: 10.1016/s0140-6736(94)90935-0. [DOI] [PubMed] [Google Scholar]
- 2.Kharitonov S A, Yates D, Robbins R A, Logan-Sinclair R, Shinebourne E A, Barnes P J. Lancet. 1994;343:133–135. doi: 10.1016/s0140-6736(94)90931-8. [DOI] [PubMed] [Google Scholar]
- 3.Massaro A, Mehta S, Lilly C, Kobzik L, Reilly J, Drazen J. Am J Respir Crit Care Med. 1996;153:1510–1514. doi: 10.1164/ajrccm.153.5.8630594. [DOI] [PubMed] [Google Scholar]
- 4.Guo F H, Comhair S A A, Zheng S, Dweik R A, Eissa N T, Thomassen M J, Calhoun W, Erzurum S C. J Immunol. 2000;164:5970–5980. doi: 10.4049/jimmunol.164.11.5970. [DOI] [PubMed] [Google Scholar]
- 5.Silkoff P, Sylvester J, Zamel N, Permutt S. Am J Respir Crit Care Med. 2000;161:1218–1228. doi: 10.1164/ajrccm.161.4.9903111. [DOI] [PubMed] [Google Scholar]
- 6.Mehta S, Lilly C, Rollenhagen J, Haley K, Asano K, Drazen J. Am J Physiol. 1997;272:L124–L131. doi: 10.1152/ajplung.1997.272.1.L124. [DOI] [PubMed] [Google Scholar]
- 7.Kharitonov S, O'Connor B, Evans D, Barnes P. Am J Respir Crit Care Med. 1995;151:1894–1899. doi: 10.1164/ajrccm.151.6.7767537. [DOI] [PubMed] [Google Scholar]
- 8.Sanders S P. Am J Respir Cell Mol Biol. 1999;21:147–149. doi: 10.1165/ajrcmb.21.2.f158. [DOI] [PubMed] [Google Scholar]
- 9.Raychaudhuri B, Dweik R, Connors M J, Buhrow L T, Malur A, Drazba J, Erzurum S C, Kavuru M S, Thomassen M J. Am J Respir Cell Mol Biol. 1999;21:311–316. doi: 10.1165/ajrcmb.21.3.3611. [DOI] [PubMed] [Google Scholar]
- 10.van der Vliet A, Elserich J P, Shigenaga M K, Cross C E. Am J Respir Crit Care Med. 1999;160:1–9. doi: 10.1164/ajrccm.160.1.9807044. [DOI] [PubMed] [Google Scholar]
- 11.Dweik R A, Laskowski D, Abu-Soud H M, Kaneko F T, Hutte R, Stuehr D J, Erzurum S C. J Clin Invest. 1998;101:660–666. doi: 10.1172/JCI1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wink D A, Hanbauer I, Grisham M B, Laval F, Nims R W, Laval J, Cook J, Pacelli R, Liebmann J, Krishna M, et al. Curr Top Cell Regul. 1996;34:159–187. doi: 10.1016/s0070-2137(96)80006-9. [DOI] [PubMed] [Google Scholar]
- 13.Gaston B, Sears S, Woods J, Hunt J, Ponaman M, McMahon T, Stamler J S. Lancet. 1998;351:1317–1319. doi: 10.1016/S0140-6736(97)07485-0. [DOI] [PubMed] [Google Scholar]
- 14.Thomassen M J, Raychaudhuri B, Dweik R A, Farver C, Buhrow L T, Malur A, Hammel J, Erzurum S C, Kavuru M S. J Allergy Clin Immunol. 1999;104:1174–1182. doi: 10.1016/s0091-6749(99)70010-2. [DOI] [PubMed] [Google Scholar]
- 15.Calhoun W J, Reed H E, Moest D R, Stevens C A. Am Rev Respir Dis. 1992;145:317–325. doi: 10.1164/ajrccm/145.2_Pt_1.317. [DOI] [PubMed] [Google Scholar]
- 16.Liu M C, Hubbard W C, Proud D, Stealey B, Galli S, Gagey-Sobotka A, Bleeker E, Lichtrenstein L. Am Rev Respir Dis. 1991;144:51–58. doi: 10.1164/ajrccm/144.1.51. [DOI] [PubMed] [Google Scholar]
- 17.Jourjour N, Calhoun W, Kelley E A, Gleich G, Schwartz L, Busse W. Am J Respir Crit Care Med. 1997;155:1515–1521. doi: 10.1164/ajrccm.155.5.9154851. [DOI] [PubMed] [Google Scholar]
- 18.Sanders S, Zweier J, Harrison S, Trush M, Rembish S, Liu M. Am J Respir Crit Care Med. 1995;151:1725–1733. doi: 10.1164/ajrccm.151.6.7767513. [DOI] [PubMed] [Google Scholar]
- 19.Comhair S A A, Bhathena P R, Dweik R A, Kavuru M, Erzurum S C. Lancet. 2000;355:624. doi: 10.1016/S0140-6736(99)04736-4. [DOI] [PubMed] [Google Scholar]
- 20.Guidelines for the Diagnosis and the Management of Asthma, Expert Panel Report II (1997) (National Asthma Education and Prevention Program, Bethesda, MD), National Institutes of Health Publication no. 97–4051.
- 21.Fang K, Ragsdale N V, Carey R M, Macdonald T, Gaston B. Biochem Biophys Res Commun. 1998;252:535–540. doi: 10.1006/bbrc.1998.9688. [DOI] [PubMed] [Google Scholar]
- 22.Kaminsky D, Mitchell J, Carroll N, James A, Soultanakis R, Janssen Y. J Allergy Clin Immunol. 1999;104:747–754. doi: 10.1016/s0091-6749(99)70283-6. [DOI] [PubMed] [Google Scholar]
- 23.Saleh D, Ernst P, Lim S, Barnes P J, Giaid A. FASEB J. 1998;12:929–937. [PubMed] [Google Scholar]
- 24.Wu W, Samoszuk M, Comhair S, Thomassen M J, Farver C, Dweik R A, Kavuru M, Erzurum S C, Hazen S L. J Clin Invest. 2000;105:1455–1463. doi: 10.1172/JCI9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hunt J, Fang K, Malik R, Snyder A, Malhotra N, Platts-Mills T, Gaston B. Am J, Respir Crit Care Med. 2000;161:694–699. doi: 10.1164/ajrccm.161.3.9911005. [DOI] [PubMed] [Google Scholar]
- 26.Fukuto J M. In: Nitric Oxide: Biochemistry, Molecular Biology, and Therapeutic Implications. Ignarro L, Murad F, editors. New York: Academic; 1995. pp. 1–15. [Google Scholar]
- 27.Crow J P, Beckman J S. In: Nitric Oxide: Biochemistry, Molecular Biology, and Therapeutic Implications. Ignarro L, Murad F, editors. New York: Academic; 1995. pp. 17–43. [Google Scholar]
- 28.Freeman B A, White C R, Gutierrez H, Paler-Martinez A, Trapey M M, Rubbo H. In: Nitric Oxide: Biochemistry, Molecular Biology, and Therapeutic Implications. Ignarro L, Murad F, editors. New York: Academic; 1995. pp. 45–69. [Google Scholar]
- 29.Radi R, Denicola A, Freeman B. Methods Enzymol. 1999;301:353–367. doi: 10.1016/s0076-6879(99)01099-x. [DOI] [PubMed] [Google Scholar]
- 30.Padmaja S, Kissner R, Bounds P, Koppenol W. Helv Chim Acta. 1998;7:1201–1206. [Google Scholar]
- 31.Koppenol W. In: Metal Ions in Biological Systems. Sigel A, Sigel H, editors. New York: Dekker; 1999. pp. 597–619. [PubMed] [Google Scholar]
- 32.Rochelle L, Fischer B, Adler K. Free Radic Biol Med. 1998;24:863–868. doi: 10.1016/s0891-5849(97)00375-4. [DOI] [PubMed] [Google Scholar]
- 33.Pryor W A, Lemercier J N, Zhang H, Uppu R M, Squadrito G L. Free Radic Biol Med. 1997;23:331–338. doi: 10.1016/s0891-5849(97)00121-4. [DOI] [PubMed] [Google Scholar]
- 34.Fang K, Johns R, Macdonald T, Kinter M, Gaston B. Am J Physiol Lung Cell Mol Physiol. 2000;23:L716–L721. doi: 10.1152/ajplung.2000.279.4.L716. [DOI] [PubMed] [Google Scholar]
- 35.Schmidt HHHW, Hofman H, Schindler U, Shutenko Z S, Cunningham D D, Feeelisch M. Proc Natl Acad Sci USA. 1996;93:14492–14497. doi: 10.1073/pnas.93.25.14492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo F H, Uetani K, Haque J, Williams B R G, Dweik R A, Thunnissen F B J M, Calhoun W, Erzurum S C. J Clin Invest. 1997;100:829–838. doi: 10.1172/JCI119598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uetani K, Der S D, Zamanian-Daryoush M, de La Motte C, Lieberman B Y, Williams B R, Erzurum S C. J Immunol. 2000;165:988–996. doi: 10.4049/jimmunol.165.2.988. [DOI] [PubMed] [Google Scholar]
- 38.De Sanctis G T, MacLean J A, Hamada K, Mehta S, Scott J A, Jiao A, Yandava C N, Kobzik L, Wolyniec W W, Fabian A J, et al. J Exp Med. 1999;189:1621–1630. doi: 10.1084/jem.189.10.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schuiling M, Meurs H, Zuidhof A B, Venema N, Zaagsma J. Am J Respir Crit Care Med. 1998;158:1442–1449. doi: 10.1164/ajrccm.158.5.9803027. [DOI] [PubMed] [Google Scholar]
- 40.Ricciardolo F L, Geppetti P, Mistretta A, Nadel J A, Sapienza M A, Bellofiore S, Di Maria G U. Lancet. 1996;348:374–377. doi: 10.1016/s0140-6736(96)04450-9. [DOI] [PubMed] [Google Scholar]