

Abstract
Paragangliomas (PGLs) are rare chromaffin cell tumors that can often be cured by resection. Although described for the first time in 1886 1, the diagnosis of PGL remains a challenge, because patients do not present with characteristic signs and symptoms. If untreated, PGL can have a devastating outcome due to myocardial infarction, severe hypertension, stroke and/or arrhytmia caused by catecholamine excess. Even after proper diagnosis, the risk of metastatic disease remains. In recent years the opinion that metastatic disease is rare in PGL had to be revised, particularly in patients presenting with extra-adrenal PGL, with a PGL exceeding a size of 5 cm and/or carrying an SDHB germline mutation (especially for children and adolescents). In up to 10 % of patients, metastases are already present at diagnosis of PGL.
Measurement of plasma and urinary metanephrine levels has long been used effectively in the diagnosis of PGL. Recently, a dopaminergic phenotype (excess dopamine, L-3,4-dihydroxyphenylalanine and or methoxytyramine) was recognized as a good indicator for metastatic disease. Vast progress in targeted PET imaging (e.g. 18F-FDA, 18F-FDOPA, 18F-FDG) now allows for reliable early detection of metastatic disease. However, once metastatses are present, treatment options are limited. Survival of patients with metastatic PGL is variable. Depending on the study population the overall 5 year survival is 35–60 %, 2.
Here we review recent advances involving findings about the genetic background, the molecular pathogenesis, new diagnostic indicators, pathologic markers and emerging treatment options for metastatic PGL.
Definition
Following the definition of the world health organization, paragangliomas (PGLs) are chromaffin cell tumors developing from the sympathetic and parasympathetic ganglia throughout the abdomen and head and neck area. A PGL arising from the adrenal gland is called pheochromocytoma (PHEO). In addition, here we will distinguish between sympathetic extra-adrenal PGL (eaPGL) and parasympathetic PGL from the head and neck area (HNP). In general PGLs are well curable, however once a patient presents with metastases, treatment options are limited and rarely curative.
Metastatic disease due to PGL can only be diagnosed based on the presence of chromaffin tumors in locations where chromaffin cells are not usually present. Thus, the widely used term ‘malignancy’, that is spreading of a primary tumor by tissue and/or vascular invasion and/or metastazising, does not necessarily apply to PGL. While tissue and/or vascular invasion are sometimes observed in PGLs these observations don’t correlate well with the severity of the disease and a patient’s prognosis. Thus, in contrast to other malignancies, the development of metastases cannot be predicted or evaluated by high vascularization and mitotic rate, or vascular and/or tissue invasion. Despite huge efforts, currently no gene or protein has been identified as a definite marker or predictor for metastatic disease and no reliable cure has been developed yet.
Rule of 10 % Overcome
PGL has long been considered as the disease of 10 % (10 % metastatic, 10 % familial, 10 % recurring, 10 % extra-adrenal, 10 % occurring in children). However, improved diagnostic techniques showed that the rule of 10 % does not accurately characterize PGL. Overall, 0–36 % of PGL patients develop metastatic disease, depending on the type of tumor 3 (table 1). The percentage of PGL with family history has been revised to around 30 % 4, 5. Extra-adrenal tumors have been reported in 15–20 % of patients 6.
Table 1.
Predisposition to malignancy with respect to hereditary background.
Genetic Predispositions
Up to 30 % of PGL appear to present in a hereditary manner 4, 5. To date, 8 different germline mutations are associated with PGL (NF1: van Recklinghausen Neurofibromatosis Type 1, RET: Multiple Endrocrine Neoplasia Type 2, VHL: von Hippel-Lindau disease, SDH-B, -C, -D: familial PGL syndrome 4, 3, 1, SDH5 7 (also referred to as SDHAF1): familial PGL syndrome 2, TMEM127: familial PGL, respectively). However, SDH5/SDHAF1 mutation seems to be rare and should only be suspected in very young patients with HNP and family history thereof in absence of SDHB, C and D mutation 8. TMEM127 germline mutation seems to occur in up to 30 % of patients with other mutations predisposing to PGL. All PGL patients presenting with TMEM127 germline mutation had PHEO. However, during follow up of 4–16 years none of the patients with TMEM127 germline mutation developed metastases or recurrence 9. In the majority of PGL the underlying dysfunction remains unknown and they are referred to as sporadic.
In search of additional genes that may be involved in the development of PGL, a somatic heterozygous mutation of isocitrate dehydrogenase has been reported in one patient with HNP, but was not found again in a large cohort of apparently sporadic PGL 10. In addition, prolyl hydroxylase 2 (PHD2/EGLN1) mutation has been reported in a PGL patient who also presented with erythrocytosis 11. So far, this mutation has not been confirmed, neither for other patients with PGL nor erythrocytosis.
Correlation of patient’s presentation with their genetic background revealed a distinct manner of representation with respect to metastatic potential, catecholamine expression, tumor location 12 and ultra-structural appearance (unpublished observations) (Table 1). Patients with NF1, MEN and VHL related PGL rarely develop metastases (0–11 %) 6, 13, 14, while patients with SDHB germline mutation develop metastases in 50–97 % 6, 13-16 (table 1). The metastatic potential of PGL that developed due to germline mutation of SDH5 and TMEM127 is not yet clear, but suspected to be low.
Metastatic Paraganglioma: Does Location Play a Role?
PGL tissue from different locations appears similar microscopically. However, the prognosis is quite different for different tumor locations.
Patients with PHEO rarely develop metastases. Also, metastases secondary to HNP seem to be infrequent 17. However, development of metastatic disease in patients with eaPGL and/or multiple PGL has been reported frequently 18. Patients with SDHB derived PGL are more prone to developing metastatic disease. However, the primary tumor location most often found in patients with metastatic disease remains eaPGL 16, 19. This may imply an effect of the tumor environment on the development of metastatic disease.
The majority of PGL related metastases occur in local and distant lymphatic nodes, bones, liver and lungs 20, 21. Thus, PGL cells seem to spread via the lymphatic as well as the hematogenic route. Patients with long bone metastases have a more promising prognosis with up to 20 years of survival after detection of metastases, while patients with soft tissue metastases usually die much sooner (own unpublished observations). The cause for the unfavorable outcome of organ lesions remains to be established.
Metastatic Paraganglioma in Children
PGL in children is rare, thus studies including a representative cohort of pediatric patients are sparse and often contradictory. Available studies were recently reviewed by Havekes et al. 22.
Pediatric PGL appeared to be related to germline mutation frequently, even in the absence of family history (30 %23, 39 % 24, 59 % 25, up to 80 % (own unpublished observations). Mutation of the VHL gene has been reported as predominant in pediatric patients 24, 26. However, in a large group of children and adolescents with PGL (n=41), we observed a 54 % predominance for an SDHB mutation (own unpublished observations).
The primary PGLs in pediatric patients have been reported most frequently to be extra-adrenal (50 %), followed by PHEO (40 %) and HNP (10 %) 23. These numbers mainly agree with our findings (54 %, 36 % and 1 %, respectively). As suggested by the high incidence of extra-adrenal primary tumors and SDHB mutation in children, the rate of the development of metastases seems to be higher than in adult patients (47% 23, 68% (own unpublished observations), 5 out of 7 patients 27). In contrast, an Indian study including 11 pediatric patients almost always found the primary tumor to be PHEO 28. No case of metastatic disease was reported for this cohort.
Positive family history or genetic testing, eaPGL, and a tumor diameter > 5cm have been shown to correlate with the development of metastatic disease in children 23.
Post surgical treatment with 131I-MIBG seems to be beneficial in pediatric patients 27. Thus, if detected early, the prognosis for pediatric patients with metastatic PGL may be more promising than for adult patients.
Insights into the Pathogenesis of Metastatic Paraganglioma
Cell culture experiments suggest that apoptosis resistance in absence of nerve growth factor (NGF) is common to several hereditary forms of PGL 29. Thus, familial PGL may develop due to impaired culling of neural crest cells during the development of chromaffin tissues. NGF is an essential survival factor for developing neuroendocrine cells. Later in development, NGF becomes scarce, leading to neuronal culling. Survival of cultured cells, with impaired NF1, RET or VHL gene expression, was connected to JunB mediated apoptosis resistance in absence of NGF. For SDHx dysfunction, apoptosis resistance was also shown, but here it seems to be caused further downstream in the same pathway. Accumulation of succinate, as appears in presence of SDHx mutations, inhibits prolyl hydroxylase (PHD) function. This can stabilize hypoxia inducible factor α (HIFα), promoting pseudo-hypoxic conditions 30, 31. On the other hand, particularly inhibition of PHD3, also called EglN3, has been shown to hamper NGF related survival 29. These results promote a common pathway for the development of PGL, but have not yet been proven in human tumor tissue.
Based on a gene expression profiling study, PGL of different genetic backgrounds could be separated into two groups. VHL and SDHx mutation derived tumors shared up-regulation of hypoxia-, angiogenesis- and oxidoreductase imbalance related genes. MEN and NF1 derived tumors clustered separately 32, 33. A more recent study confirmed this expression based clustering into different types of PGL 34.
Hypoxia is common to many tumors. However, it is not yet clear if a (pseudo-) hypoxic state with according up-regulation of angiogenesis and cell proliferation genes is sufficient for tumor development 35, 36. In addition, while common to SDHx and some VHL related PGL, HIFα stabilization has not been found in von Hippel-Lindau 2C related PGL 37. Thus, HIF stabilization alone, while potentially crucial to SDHx and some VHL related tumors, is not a common feature in all SDHx and VHL related PGL.
Interestingly, (pseudo-) hypoxia was shown to inhibit SDHB protein expression. Accordingly, SDHB protein is absent or low in a number of VHL derived tumors 38.
Although tumors derived based on mutation in the VHL and SDHB genes share pseudo-hypoxic features, tumor location, symptomatic manifestation, and metastatic potential are very distinct 15, 16, 39. SDHB derived PGL typically present in a more aggressive manner with mainly extra-adrenal primary tumors, occurring at an earlier age with a high likelihood for the development of metastases. Thus SDHB mutations have been suggested as marker of poor prognosis 40.
It has been proposed, that the route of hypoxia development in VHL and SDHx derived tumors differs and thus results in up-regulation of distinct target genes 41. It has been reported that in SDHx mutation derived tumors HIF1α expression is more often increased, while in VHL mutation derived PGL HIF2α stabilization seems to be predominant. However, Favier et al. reported that HIF2α is more highly expressed in SDHx and VHL derived tumors, while absent in MEN and NF1 related tumors 34.
Another currently debated hypothesis to explain more aggressive behavior of SDHx related tumors are elevated levels of reactive oxygen species (ROS). In addition to succinate accumulation in SDHB and D mutation related PGL, complex II dysfunction has been observed 15, 42 and may lead to inappropriate electron transfer and increased ROS production. Mutation of the SDHC gene in model systems was shown to result in high levels of ROS, which lead to an increased frequency of mitochondrial and nuclear gene mutations, promoting tumor development and perhaps an aggressive phenotype 43, 44. In a yeast model, mutation of the SDHB-equivalent lead to increased ROS production. However, no mutagenic DNA damage was detected 45. However, SDHB silencing in cultured cells showed ambiguous results with respect to ROS production 46, 47. It remains to be examined if ROS production differs between VHL and SDHx derived tumors, because the down-regulation of SDHB protein in VHL derived PGL 38 may also result in increased ROS production.
Dysfunction of complex II in SDHx as well as VHL derived tumors may promote aerobic glycolysis as the main source of energy (34; own unpublished data). Accordingly, lactic acidosis, which can be a result of decoupled glycolysis and oxidative phosphorylation, has been described in some patients with PHEO 48.
The development of metastatic disease on the molecular level remains to be elucidated for PGL. Suppressed expression of p16INK4A and p14ARF has been reported to promote the development of metastases in Pten knock–out mice 49. In human PGL samples, promoter CpG methylation of the p16INK4A gene alone as well as promoter methylation of at least 3 of the following genes RASSF1A, NORE1A, p16INK4A, RARB, DCR2, CDH1 and APC (i.e. CpG island methylator phenotype (CIMP)) but not p14ARF has been shown to correlate with aggressive tumor behavior 50, 51. However, the same patient population was used in both studies and out of 6 primary metastatic tumors evaluated, only 3 were positive for p16INK4A hypermethylation or CIMP. All reported samples that had tested positive for SDHB mutation showed p16INK4A hypermethylation and CIMP, including 1 primary benign tumor. Thus, hypermethylation seems to primarily correlate with SDHB mutation, which has been suggested previously as a predictor for metastatic potential 40. Nevertheless, it may be of great interest to evaluate the molecular connection between SDHB mutation and a changed DNA methylation pattern. The consequences on the molecular biology of cells that carry these characteristic DNA methylations may help to explain why SDHB related PGL present in a more aggressive manner compared to other PGL.
Mutations of the SDHB gene as well as an extra-adrenal tumor environment seem to promote the development of metastases. However, a clear molecular biologic explanation for the more aggressive phenotype has not yet been found.
Markers for Malignant Paraganglioma
Currently there are no reliable markers for metastatic disease in PGL. The single way to diagnose malignancy is the presence of metastases. Thus, patients with PGL have to be followed-up on ultimately, because metastatic disease or recurrence can appear even after decades free of disease.
Many pathologic markers of malignancy used in other tumors were evaluated for PGL, but to date none could be sufficiently confirmed as a diagnostic or prognostic tool (for a summary see 2, 52–54). Amongst them were tissue invasion, high vascularization, and staining for specific proteins (e.g. Ki67, hTERT 55), characteristics that correlate with metastatic disease in other cancers. However, most of them proved to be not predictive of metastases or are controversial in PGL 54. In a recent study that includes 11 tumors of patients that presented with PGL related metastases, Ki67 staining has been shown to have a sensitivity and specificity of 100 % 56. However, this will have to be evaluated in a larger cohort of patients. In 2002 Thompson introduced the ‘pheochromocytoma of the adrenal gland scaled score’ (PASS), which is a scoring system based on 17 distinct morphologic features of PHEO to distinguish benign from primary metastatic tumors 57. However, considerable inter- and intra-observer variation suggest that the PASS system is of limited diagnostic and predictive value 58.
Recently, absence of SDHB protein in PGL specimens was suggested as predictive of metastatic disease 59. Absence of SDHB staining was found in some sporadic metastatic lesions in addition to others with known SDHB mutations. However, absence or low levels of SDHB protein were previously reported in VHL related PGL, which rarely metastasize 38. Thus, the predictive value of metastatic disease based on low SDHB protein levels may be limited to a distinct group of PGL.
Stathmin has been shown to be a good predictive marker in endocrine tumors. In PGL tissue, staining for stathmin was more intense in primary metastatic tumors. However, its stand-alone value as a marker of malignancy is limited, because primary non-metastatic tumors also show stathmin expression. Differentiation between weak and more intense staining may be difficult when examining individual tumors 60.
The most promising diagnostic/predictive marker for metastatic PGL so far seems to be the expression of the HIF 2α regulated SNAIL alone 61, or in combination with its target twist 62. Positive staining for one or both in primary tumors correlates well with the presence of distant metastases. However, these findings have to be carefully evaluated for the different types of PGL.
To date, SDHB mutation (particularly with a tumor size >5cm) and/or highly elevated levels of normetanephrine (NMN), dopamine (DA), L-3,4-dihydroxyphenylalanine (DOPA), and/or methoxytyramine are the best indicators for the development or presence of metastatic disease (Eisenhofer 2005, JCEM, Timmers?!, Eisenhofer et al. unpublished observation).
Diagnosis of Metastatic Paraganglioma
Signs and symptoms of patients bearing metastatic disease cannot be distinguished from those of patients with solitary or multiple PGL. In some cases, symptoms related to tumor burden of metastases are present. Most often patients with metastatic as well as non-metastatic PGL suffer from hypertension caused by the tumor’s hypersecretion of catecholamines. As described below, the secretion profile of a tumor can present valuable clues about tumor location and the possibility of multiple lesions or metastases.
Presence of metastases can be ruled out by imaging studies. However, a possible later development of metastases has to be monitored continuously. Considerations about the imaging approach of choice should be based on the genetic background and biochemical phenotype of each patient, as described below.
Biochemical Phenotype of Metastatic Paraganglioma
Currently, the diagnostic gold standard for PGL is an elevated plasma and/or urine metanephrine level. When a PGL is present, catecholamine levels can also be elevated in plasma and urine, but due to their lower stability they show a lower sensitivity as a diagnostic tool than metanephrines 63. For reliable results it is recommended to collect samples of relaxed patients. For plasma collection, patients should rest in the supine position without a pillow for at least 20 minutes after insertion of the cannula 64.
PGL can be differentiated based on their biochemistry in secreting (adrenergic, noradrenergic, or mixed phenotype) and non-secreting (biochemically silent) tumors. The latter most commonly arise from parasympathetic tissue in the head and neck area 65, 66. Knowledge of the biochemical phenotype is an important diagnostic indicator for tumor localization and possible underlying mutation as well as the presence of metastases (summarized in table 1 and 2).
Table 2.
Presentation, imaging and suggested treatment in PGL by hereditary backgrounds.
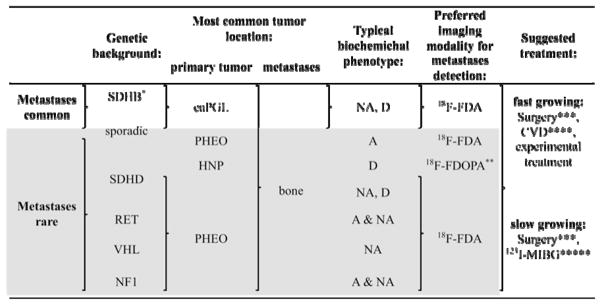 |
Abbreviations: adrenergic (A) (elevated epinephrine (Epi) and/or metanephrine (MN)); noradrenergic (NA) (elevated norepinephrine (NE) and/or normetanephrine (NMN)); dopaminerg (D) (elevated dopamine (DA); L-3,4-dihydroxyphenylalanine (DOPA) and/or methoxytyramine); adrenal PGL (PHEO); extra-adrenal PGL (eaPGL); head and neck PGL (HNP).
Risk of metastatic disease particularly high in children and adolescents.
Mainly studied on primary HNP but potentially similarly effective for metastases.
Complete resection of metastases is rarely possible, but resection can have a good palliative effect (avoidance of further organ or bone destruction by tumor growth as well as reduction of catecholamine levels which leads to a decrease in related signs and symptoms).
own unpublished observations suggest that (at least) patients with SDHB related fast growing tumors initially respond well. Physicians are recommended not to stop CVD, because resumption of therapy almost always results in chemotherapy resistant tumors (if toxicity occurs, longer intervals between cycles or a reduced dosage can be used).
High doses may be effective 111, however, additional studies and long-term observations are needed.
Metastases are most common in eaPGL, thus a noradrenergic phenotype (elevated norepinephrine (NE) or NMN) is often present in patients with metastatic disease. Metastases frequently appear dedifferentiated, potentially lacking the later enzymes of catecholamine synthesis. Thus, elevated methoxytyramine, DA, and/or DOPA levels can indicate metastatic disease (Eisenhofer et al. unpublished observations). Particularly in patients with SDHB mutation, eaPGL, and a primary tumor size of >5 cm metastatic disease is likely. Nevertheless, elevated DA, DOPA or methoxytyramine levels may also occur in solitary HNP 65, 67. A combination of elevated epinephrine (EPI)/metanephrine (MN) with NE/NMN may be caused by a solitary adrenal PHEO or multifocal adrenal and extra adrenal PGL or metastases. Metastatic disease in patients with an adrenergic phenotype is extremely rare.
Current Approaches to Detect Metastatic Paraganglioma
Following detection of elevated catecholamines and/or their metabolites or if PGL is otherwise suspected, confirmatory diagnosis and/or localization of PGL and possible metastases is necessary. Tumors can often be localized by computer tomography (CT) and/or magnetic resonance imaging (MRI). Non-specific functional imaging modalities, such as 18F-FDG positrom emission tomography (PET) and octreotide scintigraphy are also applied. However, only functional imaging techniques using tracers that are specifically internalized by catecholamine metabolism related mechanisms may confirm a tumor as PGL (e.g. 123/131I-MIBG, 18F-FDA, 18F-FDOPA1, 11C-Epinephrine, 11C-Hydroxyephedrine). Excellent in depth considerations which imaging modality is indicated under particular circumstances are presented elsewhere (e.g. 20, 69). Here the focus will be on imaging of metastatic lesions in PGL.
Use of functional imaging compounds that target specific receptors generally expressed in PGL - namely the NE and somatostatin (ST) 70 transporters - can lead to false negative results in metastatic lesions. Expression of the NE 71, 72 as well as the ST transporter 73, 74 was shown to be reduced or absent in PGL related metastases. It is believed that cellular dedifferentiation in metastatic lesions is the cause for this. Decreased sensitivity and specificity of targeted functional imaging agents (i.e. the ‘flipflop’ phenomenon) is especially pronounced in PGL related to SDHB mutation. Thus, the preferred imaging modalities differ for SDHB and non-SDHB related metastatic disease 75, 76. In SDHB related metastatic disease, 18F-FDG PET is preferred. 18F-FDG enters a cell through the glucose transporter (GLUT1). GLUT1 expression and 18F-FDG uptake is elevated in highly metabolically active tissue, such as metastases with increased glycolytic energy production. Glycolytic activity appears to be increased in SDHx mutation related PGL, possibly due to mitochondrial dysfunction (own unpublished results). Despite its capability to reliably detect SDHB related metastases, an 18F-FDG PET positive signal is not specific for PGL related lesions. When the genetic background is unknown and in non-SDHB related PGL, specific imaging of metastases should be attempted via 18F-FDA or 18F-FDOPA PET. Non-SDHx related bone metastases have been shown to be most reliably and specifically detected with 18F-FDA, while SDHB related bone metastases could be more efficiently detected by non-specific scintigraphy and 18F-FDG PET 77.
To date, the most widely used functional imaging modality for PGL is 123I-MIBG imaging. While useful in evaluating a patient as potential candidate for treatment with 131I-MIBG, sensitivity and specificity in the detection of metastases is inferior to 18F-FDA and 18F-FDOPA PET in patients with non-SDHB related disease and 18F-FDG in patients with SDHB related PGL. Overall, 18F-FDA PET remains the functional imaging approach of choice to detect PGL related metastases 20, 69, 78. Functional PET images of a patient with apparently sporadic disease, presenting with extensive bone metastases 16 years after initial diagnosis of a right adrenal PHEO are shown in Fig.1.
Fig.1.
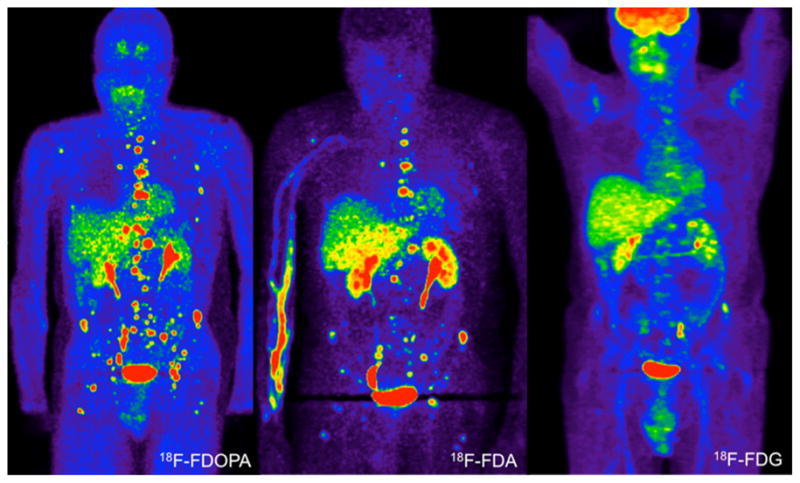
Anterior reprojected functional PET images of a patient with extensive bone metastases, detected 16 years after diagnosis of a right adrenal pheochromocytoma. The scans were performed on three consecutive days. Genetic testing for SDHB, C, and D germline mutations or deletion were negative.
Management and Treatment of Malignant Paraganglioma
Once presence of metastases is verified, there is no reliable cure. Treatment options are: 1) mechanical removal by excision or ablation, 2) targeted pharmaceutics or 3) chemotherapy. Recently, current treatment options were excellently summarized 79–81.
Surgical Approaches
Treatment options include palliative tumor resection, to reduce tumor burden and excess catecholamine related symptoms. Although in benign cases, a laparoscopic approach is preferred, in metastatic PGL this is controversial. Laparoscopy is not advisable when local invasion and/or large tumors are present or organ resection is required 82.
In patients with painful bone metastases and inoperable hepatic lesions, radiofrequency ablation (RFA) and cryoablation may be of benefit. However, before any tumor manipulation, patients have to be medicated with α– and β–blockers and catecholamine synthesis inhibitors for 7–21 days, to prevent hypertensive crisis during tumor manipulation (phenoxybenzamine, atenolol and metyrosine, respectively) 83. Following pretreatment, RFA has shown to be manageable in patients with secreting PGL 84. More recently, complete ablation of 6 out of 7 metastatic lesions (4 hepatic, 3 bone and 1 ischiatic) in 6 patients suffering from PGL has been presented 85.
Successful cryoablation on a hepatic lesion of a patient with non-secreting metastatic PGL has been reported 86.
Chemotherapy
The most widely used chemotherapeutic regimen is the CVD regimen (cyclophosphamide 750 mg/m2 body surface on day 1, vincristine 1.4 mg/m2 body surface on day 1, and dacarbazine 600 mg/m2 body surface on day 1 and 2, repeated every 21 days), developed by Keiser et al. 1985 87. However, benefits of CVD therapy for metastatic pheochromocytoma appears to be short-term and do not include an increase in patient survival. Particularly in women and patients with primary PHEO, CVD is counter-indicated 88. However, in a patient with SDHB mutation related disease, great results were achieved under the CVD regimen 89. Thus, CVD therapy may be considered in patients with fast growing tumors due to SDHB mutation, in patients where tumor shrinkage prior to resection is required or in patients whose symptoms related to catecholamine excess cannot be managed otherwise 90.
A recent case study has reported on a patient with metastatic PGL who has been orally treated with temozolomide. The patient received 250 mg temozolomide daily for 5 days, repeated every 28 days. After 21 weeks of treatment, the patient was free of symptoms, with a significant decrease of plasma catecholamine and metanephrine levels. The primary tumor and liver metastases shrank and allowed tumor resection. The authors conclude that treatment of metastatic PGL with temozolomide may be of benefit prior to tumor resection 91.
Temozolomide has also been used in combination with the targeted drug thalidomide for the treatment of metastatic PGL. In a phase II study including 3 patients with metastatic PGL, patients have been treated with 150 mg/m2 temozolomide for 7 days every other week and 50–400 mg/m2 daily. One out of 3 patients showed a positive response to the regimen, however cytotoxic effect that caused subjects to end this treatment regimen were observed in patients with other endocrine malignancies 92.
Targeted Therapy
In addition to tumor removal, treatment with targeted radiotherapeutics can be beneficial. The most frequently used radiotherapy is treatment with 131I-MIBG. This treatment option was developed as early as 1984 93. Feasibility of this treatment for each patient needs to be verified by positive 123/131I-MIBG imaging. Regimens of high or low doses were performed (>600 mCi and 100–500 mCi, respectively). Symptomatic response has been reported in as much as 67–98 % of patients, while objective tumor shrinkage/stable disease or decreased secretion was observed in 27–47 % and 45–67 % of the patients, respectively 94–97. Comparable results with a lower rate of adverse reactions could be achieved in a low dose regimen; hormonal and symptomatic relive for 50 % of patients have been reported 98.
In an attempt to increase the curative effect of 131I-MIBG treatment, recently Ultratrace Iobenguane I-131 has been developed. It is a high-specific activity 131I-MIBG and was shown to result in a lower frequency of adverse effects than regular 131I-MIBG 99. Treatment effects with drastically fewer unlabelled molecules may be a promising improvement for 131I-MIBG therapy. However, its efficacy remains to be evaluated.
Besides radiotherapy, targeting the NET system with ST analogs has been used in the treatment of metastatic PGL (90Y-DOTATOC). Success rate for radiolabelled octreotide analogues is not entirely clear yet, because only a few patients were treated by this means.
Treatment with unlabelled octreotide has also been done. However, it seems to be ineffective with respect to blood pressure, catecholamine- and Chromogranin A release 100. An explanation for the lack of effect may be the relatively low expression of the target of octreotide, the ST2a receptor. While the ST2a receptor was found in only 25 % of PGL samples, the ST3 receptor was found in more than 60 % of cells in 90 % of 52 PGL samples examined 101. Thus, targeting the ST3 receptor may prove a more beneficial approach in the treatment of metastatic PGL.
Treatment of metastatic PGL with the mTOR inhibitor everolimus was attempted with no significant benefit, and thus is not recommended 102, 103.
The most promising approach to date seems to be treatment with multiple tyrosine kinase inhibitors like sutinib. Reports on 5 patients with metastatic PGL treated with sutinib are currently available. All 5 patients showed at least a partial response 104–106. A phase II trial is currently being organized. Despite the promising results with sutinib, the tyrosine kinase inhibitor imatinib did not prove to be useful in two patients with metastatic PHEO 107.
A recent study showed that the interleukin 13 (IL-13) receptor α2 is expressed on the tumor cell’s surface in humans and in a mouse model. Treatment of PGL bearing mice with IL-13 coupled to a truncated Pseudomonas exotoxin lead to a significant decrease in tumor size 108. The compound has been successfully tested in several clinical trials 109. Targeting the IL-13 receptor may be a promising new strategy to treat metastatic and inoperable PGL.
Acknowledgments
Support: Funding was provided by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD.
Footnotes
DISCLOSURE STATEMENT: The authors have nothing to disclose.
Carbidopa has been shown to increase the tumor-to-background ratio of 18F-FDOPA uptake in PGL, particularly PHEO 68
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fränkel F. Ein Fall von doppelseitigem, völlig latent verlaufenen Nebennierentumor und gleichzeitiger Nephritis mit Veränderungen am Circulationsapparat und Retinitis. Virchows Archiv. 1886;103(2):244–263. [Google Scholar]
- 2.Pacak K, Eisenhofer G, Ahlman H, et al. Pheochromocytoma: recommendations for clinical practice from the First International Symposium. October 2005. Nat Clin Pract Endocrinol Metab. 2007 Feb;3(2):92–102. doi: 10.1038/ncpendmet0396. [DOI] [PubMed] [Google Scholar]
- 3.O’Riordain DS, William F, Young J, Grant CS, Carney JA, Heerden JAv. Clinical Spectrum and Outcome of Functional Extraadrenal Paraganglioma. World Journal of Surgery. 1996 Jul 21;20(7):916–922. doi: 10.1007/s002689900139. [DOI] [PubMed] [Google Scholar]
- 4.Opocher G, Schiavi F, Iacobone M, et al. Familial nonsyndromic pheochromocytoma. Ann N Y Acad Sci. 2006 Aug;1073:149–155. doi: 10.1196/annals.1353.015. [DOI] [PubMed] [Google Scholar]
- 5.Bryant J, Farmer J, Kessler LJ, Townsend RR, Nathanson KL. Pheochromocytoma: the expanding genetic differential diagnosis. J Natl Cancer Inst. 2003 Aug 20;95(16):1196–1204. doi: 10.1093/jnci/djg024. [DOI] [PubMed] [Google Scholar]
- 6.Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005 Aug 20-26;366(9486):665–675. doi: 10.1016/S0140-6736(05)67139-5. [DOI] [PubMed] [Google Scholar]
- 7.Hao H-X, Khalimonchuk O, Schraders M, et al. SDH5, a Gene Required for Flavination of Succinate Dehydrogenase, Is Mutated in Paraganglioma. Science. 2009 August 28;325(5944):1139–1142. doi: 10.1126/science.1175689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bayley JP, Kunst HP, Cascon A, et al. SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol. 2010 Jan 11; doi: 10.1016/S1470-2045(10)70007-3. [DOI] [PubMed] [Google Scholar]
- 9.Qin Y, Yao L, King EE, et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet. 2010 Mar;42(3):229–233. doi: 10.1038/ng.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaal J, Burnichon N, Korpershoek E, et al. Isocitrate Dehydrogenase Mutations Are Rare in Pheochromocytomas and Paragangliomas. J Clin Endocrinol Metab. 2009 Nov 13; doi: 10.1210/jc.2009-2170. [DOI] [PubMed] [Google Scholar]
- 11.Ladroue C, Carcenac R, Leporrier M, et al. PHD2 mutation and congenital erythrocytosis with paraganglioma. N Engl J Med. 2008 Dec 18;359(25):2685–2692. doi: 10.1056/NEJMoa0806277. [DOI] [PubMed] [Google Scholar]
- 12.Mannelli M, Castellano M, Schiavi F, et al. Clinically Guided Genetic Screening in a Large Cohort of Italian Patients with Pheochromocytomas and/or Functional or Nonfunctional Paragangliomas. J Clin Endocrinol Metab. 2009 May 1;94(5):1541–1547. doi: 10.1210/jc.2008-2419. [DOI] [PubMed] [Google Scholar]
- 13.Amar L, Bertherat J, Baudin E, et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol. 2005 Dec 1;23(34):8812–8818. doi: 10.1200/JCO.2005.03.1484. [DOI] [PubMed] [Google Scholar]
- 14.Gimenez-Roqueplo AP, Lehnert H, Mannelli M, et al. Phaeochromocytoma, new genes and screening strategies. Clin Endocrinol (Oxf) 2006 Dec;65(6):699–705. doi: 10.1111/j.1365-2265.2006.02714.x. [DOI] [PubMed] [Google Scholar]
- 15.Gimenez-Roqueplo AP, Favier J, Rustin P, et al. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003 Sep 1;63(17):5615–5621. [PubMed] [Google Scholar]
- 16.Timmers HJ, Kozupa A, Eisenhofer G, et al. Clinical presentations, biochemical phenotypes, and genotype-phenotype correlations in patients with SDHB-associated pheochromocytomas and paragangliomas. J Clin Endocrinol Metab. 2007 Jan 2; doi: 10.1210/jc.2006-2315. [DOI] [PubMed] [Google Scholar]
- 17.Boedeker CC, Neumann HP, Maier W, Bausch B, Schipper J, Ridder GJ. Malignant head and neck paragangliomas in SDHB mutation carriers. Otolaryngol Head Neck Surg. 2007 Jul;137(1):126–129. doi: 10.1016/j.otohns.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 18.Burnichon N, Rohmer V, Amar L, et al. The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J Clin Endocrinol Metab. 2009 Aug;94(8):2817–2827. doi: 10.1210/jc.2008-2504. [DOI] [PubMed] [Google Scholar]
- 19.Brouwers FM, Eisenhofer G, Tao JJ, et al. High frequency of SDHB germline mutations in patients with malignant catecholamine-producing paragangliomas: implications for genetic testing. J Clin Endocrinol Metab. 2006 Nov;91(11):4505–4509. doi: 10.1210/jc.2006-0423. [DOI] [PubMed] [Google Scholar]
- 20.Yu J-PK. Management of malignant pheochromocytoma. The Endocrinologist. 2002;12:291–299. [Google Scholar]
- 21.Lehnert H, Mundschenk J, Hahn K. Malignant pheochromocytoma. Front Horm Res. 2004;31:155–162. doi: 10.1159/000074663. [DOI] [PubMed] [Google Scholar]
- 22.Havekes B, Romijn JA, Eisenhofer G, Adams K, Pacak K. Update on pediatric pheochromocytoma. Pediatr Nephrol. 2009 May;24(5):943–950. doi: 10.1007/s00467-008-0888-9. [DOI] [PubMed] [Google Scholar]
- 23.Pham TH, Moir C, Thompson GB, et al. Pheochromocytoma and paraganglioma in children: a review of medical and surgical management at a tertiary care center. Pediatrics. 2006 Sep;118(3):1109–1117. doi: 10.1542/peds.2005-2299. [DOI] [PubMed] [Google Scholar]
- 24.Barotini M, Levin G, Sanso G. Characteristics of Pheochromocytoma in a 4- to 20-Year-Old Population. Annals of the New York Academy of Sciences. 2006;1073 doi: 10.1196/annals.1353.003. [DOI] [PubMed] [Google Scholar]; Pheochromocytoma First International Symposium; pp. 30–37. [Google Scholar]
- 25.Neumann HP, Bausch B, McWhinney SR, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002 May 9;346(19):1459–1466. doi: 10.1056/NEJMoa020152. [DOI] [PubMed] [Google Scholar]
- 26.Ciftci AO, Tanyel FC, Senocak ME, Buyukpamukcu N. Pheochromocytoma in children. J Pediatr Surg. 2001 Mar;36(3):447–452. doi: 10.1053/jpsu.2001.21612. [DOI] [PubMed] [Google Scholar]
- 27.Hammond PJ, Murphy D, Carachi R, Davidson DF, McIntosh D. Childhood phaeochromocytoma and paraganglioma: 100% incidence of genetic mutations and 100% survival. J Pediatr Surg. 2010 Feb;45(2):383–386. doi: 10.1016/j.jpedsurg.2009.10.082. [DOI] [PubMed] [Google Scholar]
- 28.Ganesh HK, Acharya SV, Goerge J, Bandgar TR, Menon PS, Shah NS. Pheochromocytoma in children and adolescents. Indian J Pediatr. 2009 Nov;76(11):1151–1153. doi: 10.1007/s12098-009-0287-7. [DOI] [PubMed] [Google Scholar]
- 29.Lee S, Nakamura E, Yang H, et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell. 2005 Aug;8(2):155–167. doi: 10.1016/j.ccr.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 30.Pollard PJ, Briere JJ, Alam NA, et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005 Aug 1;14(15):2231–2239. doi: 10.1093/hmg/ddi227. [DOI] [PubMed] [Google Scholar]
- 31.Selak MA, Armour SM, MacKenzie ED, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005 Jan;7(1):77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 32.Dahia PL, Ross KN, Wright ME, et al. A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet. 2005 Jul;1(1):72–80. doi: 10.1371/journal.pgen.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dahia PL, Hao K, Rogus J, et al. Novel pheochromocytoma susceptibility loci identified by integrative genomics. Cancer Res. 2005 Nov 1;65(21):9651–9658. doi: 10.1158/0008-5472.CAN-05-1427. [DOI] [PubMed] [Google Scholar]
- 34.Favier J, Briere JJ, Burnichon N, et al. The Warburg effect is genetically determined in inherited pheochromocytomas. PLoS One. 2009;4(9):e7094. doi: 10.1371/journal.pone.0007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mack FA, Rathmell WK, Arsham AM, Gnarra J, Keith B, Simon MC. Loss of pVHL is sufficient to cause HIF dysregulation in primary cells but does not promote tumor growth. Cancer Cell. 2003 Jan;3(1):75–88. doi: 10.1016/s1535-6108(02)00240-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kondo K, Kim WY, Lechpammer M, Kaelin WG., Jr Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol. 2003 Dec;1(3):E83. doi: 10.1371/journal.pbio.0000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoffman MA, Ohh M, Yang H, Klco JM, Ivan M, Kaelin WG., Jr von Hippel-Lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF. Hum Mol Genet. 2001 May 1;10(10):1019–1027. doi: 10.1093/hmg/10.10.1019. [DOI] [PubMed] [Google Scholar]
- 38.Dahia PL. Transcription association of VHL and SDH mutations link hypoxia and oxidoreductase signals in pheochromocytomas. Ann N Y Acad Sci. 2006 Aug;1073:208–220. doi: 10.1196/annals.1353.023. [DOI] [PubMed] [Google Scholar]
- 39.Srirangalingam U, Khoo B, Walker L, et al. Contrasting clinical manifestations of SDHB and VHL associated chromaffin tumours. J Endocr Relat Cancer. 2009 June 1;16(2):515–525. doi: 10.1677/ERC-08-0239. [DOI] [PubMed] [Google Scholar]
- 40.Amar L, Baudin E, Burnichon N, et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab. 2007 Oct;92(10):3822–3828. doi: 10.1210/jc.2007-0709. [DOI] [PubMed] [Google Scholar]
- 41.Pollard PJ, El-Bahrawy M, Poulsom R, et al. Expression of HIF-1{alpha}, HIF-2{alpha} (EPAS1), and Their Target Genes in Paraganglioma and Pheochromocytoma with VHL and SDH Mutations. J Clin Endocrinol Metab. 2006 November 1;91(11):4593–4598. doi: 10.1210/jc.2006-0920. [DOI] [PubMed] [Google Scholar]
- 42.Gimenez-Roqueplo AP, Favier J, Rustin P, et al. Functional consequences of a SDHB gene mutation in an apparently sporadic pheochromocytoma. J Clin Endocrinol Metab. 2002 Oct;87(10):4771–4774. doi: 10.1210/jc.2002-020525. [DOI] [PubMed] [Google Scholar]
- 43.Ishii T, Yasuda K, Akatsuka A, Hino O, Hartman PS, Ishii N. A mutation in the SDHC gene of complex II increases oxidative stress, resulting in apoptosis and tumorigenesis. Cancer Res. 2005 Jan 1;65(1):203–209. [PubMed] [Google Scholar]
- 44.Ishii N, Ishii T, Hartman PS. The role of the electron transport SDHC gene on lifespan and cancer. Mitochondrion. 2007 Feb-Apr;7(1–2):24–28. doi: 10.1016/j.mito.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 45.Smith EH, Janknecht R, Maher LJ., III Succinate inhibition of {alpha}-ketoglutarate-dependent enzymes in a yeast model of paraganglioma. Hum Mol Genet. 2007 December 15;16(24):3136–3148. doi: 10.1093/hmg/ddm275. [DOI] [PubMed] [Google Scholar]
- 46.Guzy RD, Sharma B, Bell E, Chandel NS, Schumacker PT. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol. 2008 Jan;28(2):718–731. doi: 10.1128/MCB.01338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cervera AM, Apostolova N, Crespo FL, Mata M, McCreath KJ. Cells silenced for SDHB expression display characteristic features of the tumor phenotype. Cancer Res. 2008 Jun 1;68(11):4058–4067. doi: 10.1158/0008-5472.CAN-07-5580. [DOI] [PubMed] [Google Scholar]
- 48.Bornemann M, Hill SC, Kidd GS., 2nd Lactic acidosis in pheochromocytoma. Ann Intern Med. 1986 Dec;105(6):880–882. doi: 10.7326/0003-4819-105-6-880. [DOI] [PubMed] [Google Scholar]
- 49.You MJ, Castrillon DH, Bastian BC, et al. Genetic analysis of Pten and Ink4a/Arf interactions in the suppression of tumorigenesis in mice. Proc Natl Acad Sci U S A. 2002 Feb 5;99(3):1455–1460. doi: 10.1073/pnas.022632099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kiss NB, Geli J, Lundberg F, et al. Methylation of the p16INK4A promoter is associated with malignant behavior in abdominal extra-adrenal paragangliomas but not pheochromocytomas. Endocr Relat Cancer. 2008 Jun;15(2):609–621. doi: 10.1677/ERC-07-0285. [DOI] [PubMed] [Google Scholar]
- 51.Geli J, Kiss N, Karimi M, et al. Global and regional CpG methylation in pheochromocytomas and abdominal paragangliomas: association to malignant behavior. Clin Cancer Res. 2008 May 1;14(9):2551–2559. doi: 10.1158/1078-0432.CCR-07-1867. [DOI] [PubMed] [Google Scholar]
- 52.Pacak K, Lenders JW, Eisenhofer G. Pheochromocytoma: diagnosis, localisation and treatment. Blackwell Publishing; 2007. [Google Scholar]
- 53.Eisenhofer G, Bornstein SR, Brouwers FM, et al. Malignant pheochromocytoma: current status and initiatives for future progress. Endocr Relat Cancer. 2004 Sep;11(3):423–436. doi: 10.1677/erc.1.00829. [DOI] [PubMed] [Google Scholar]
- 54.Carlsen E, Abdullah Z, Kazmi SM, Kousparos G. Pheochromocytomas, PASS, and immunohistochemistry. Horm Metab Res. 2009 Sep;41(9):715–719. doi: 10.1055/s-0029-1238274. [DOI] [PubMed] [Google Scholar]
- 55.Elder EE, Xu D, Hoog A, et al. KI-67 AND hTERT expression can aid in the distinction between malignant and benign pheochromocytoma and paraganglioma. Mod Pathol. 2003 Mar;16(3):246–255. doi: 10.1097/01.MP.0000056982.07160.E3. [DOI] [PubMed] [Google Scholar]
- 56.Mohammad Tavangar S, Shojaee A, Moradi Tabriz H, Heshmat R, Lashkari A, Azimi S. Immunohistochemical expression of Ki67, c-erbB-2, and c-kit antigens in benign and malignant pheochromocytoma. Pathol Res Pract. 2010 Feb 26; doi: 10.1016/j.prp.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 57.Thompson LD. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol. 2002 May;26(5):551–566. doi: 10.1097/00000478-200205000-00002. [DOI] [PubMed] [Google Scholar]
- 58.Wu D, Tischler AS, Lloyd RV, et al. Observer variation in the application of the Pheochromocytoma of the Adrenal Gland Scaled Score. Am J Surg Pathol. 2009 Apr;33(4):599–608. doi: 10.1097/PAS.0b013e318190d12e. [DOI] [PubMed] [Google Scholar]
- 59.van Nederveen FH, Gaal J, Favier J, et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncol. 2009 Aug;10(8):764–771. doi: 10.1016/S1470-2045(09)70164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sadow PM, Rumilla KM, Erickson LA, Lloyd RV. Stathmin expression in pheochromocytomas, paragangliomas, and in other endocrine tumors. Endocr Pathol. 2008 Summer;19(2):97–103. doi: 10.1007/s12022-008-9028-0. [DOI] [PubMed] [Google Scholar]
- 61.Hayry V, Salmenkivi K, Arola J, Heikkila P, Haglund C, Sariola H. High frequency of SNAIL-expressing cells confirms and predicts metastatic potential of phaeochromocytoma. Endocr Relat Cancer. 2009 Dec;16(4):1211–1218. doi: 10.1677/ERC-09-0049. [DOI] [PubMed] [Google Scholar]
- 62.Waldmann J, Slater EP, Langer P, et al. Expression of the transcription factor snail and its target gene twist are associated with malignancy in pheochromocytomas. Ann Surg Oncol. 2009 Jul;16(7):1997–2005. doi: 10.1245/s10434-009-0480-y. [DOI] [PubMed] [Google Scholar]
- 63.Pacak K, Eisenhofer G. An assessment of biochemical tests for the diagnosis of pheochromocytoma. Nat Clin Pract Endocrinol Metab. 2007 Nov;3(11):744–745. doi: 10.1038/ncpendmet0615. [DOI] [PubMed] [Google Scholar]
- 64.Lenders JW, Willemsen JJ, Eisenhofer G, et al. Is supine rest necessary before blood sampling for plasma metanephrines? Clin Chem. 2007 Feb;53(2):352–354. doi: 10.1373/clinchem.2006.076489. [DOI] [PubMed] [Google Scholar]
- 65.Timmers HJ, Pacak K, Huynh TT, et al. Biochemically silent abdominal paragangliomas in patients with mutations in the succinate dehydrogenase subunit B gene. J Clin Endocrinol Metab. 2008 Dec;93(12):4826–4832. doi: 10.1210/jc.2008-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Eisenhofer G, Goldstein DS, Sullivan P, et al. Biochemical and clinical manifestations of dopamine-producing paragangliomas: utility of plasma methoxytyramine. J Clin Endocrinol Metab. 2005 Apr;90(4):2068–2075. doi: 10.1210/jc.2004-2025. [DOI] [PubMed] [Google Scholar]
- 67.van Duinen N, Steenvoorden D, Kema IP, et al. Increased urinary excretion of 3-methoxytyramine in patients with head and neck paragangliomas. J Clin Endocrinol Metab. 2010 Jan;95(1):209–214. doi: 10.1210/jc.2009-1632. [DOI] [PubMed] [Google Scholar]
- 68.Timmers HJ, Hadi M, Carrasquillo JA, et al. The effects of carbidopa on uptake of 6-18F-Fluoro-L-DOPA in PET of pheochromocytoma and extraadrenal abdominal paraganglioma. J Nucl Med. 2007 Oct;48(10):1599–1606. doi: 10.2967/jnumed.107.042721. [DOI] [PubMed] [Google Scholar]
- 69.Havekes B, King K, Lai EW, Romijn JA, Corssmit EP, Pacak K. New imaging approaches to phaeochromocytomas and paragangliomas. Clin Endocrinol (Oxf) 2009 Jun 8; doi: 10.1111/j.1365-2265.2009.03648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Krenning EP, Kwekkeboom DJ, Pawels S, Kvols LK, Reubi JC. Somatostatin receptor scintigraphy. In: Freeman LM, editor. Nuclear Medicine Annual 1995. New York: Raven Press; 1995. pp. 242–244. [Google Scholar]
- 71.Cleary S, Brouwers FM, Eisenhofer G, et al. Expression of the noradrenaline transporter and phenylethanolamine N-methyltransferase in normal human adrenal gland and phaeochromocytoma. Cell Tissue Res. 2005 Dec;322(3):443–453. doi: 10.1007/s00441-005-0026-y. [DOI] [PubMed] [Google Scholar]
- 72.Eisenhofer G. The role of neuronal and extraneuronal plasma membrane transporters in the inactivation of peripheral catecholamines. Pharmacol Ther. 2001 Jul;91(1):35–62. doi: 10.1016/s0163-7258(01)00144-9. [DOI] [PubMed] [Google Scholar]
- 73.Epelbaum J, Bertherat J, Prevost G, et al. Molecular and pharmacological characterization of somatostatin receptor subtypes in adrenal, extraadrenal, and malignant pheochromocytomas. J Clin Endocrinol Metab. 1995 Jun;80(6):1837–1844. doi: 10.1210/jcem.80.6.7775631. [DOI] [PubMed] [Google Scholar]
- 74.Reubi JC, Waser B, Khosla S, et al. In vitro and in vivo detection of somatostatin receptors in pheochromocytomas and paragangliomas. J Clin Endocrinol Metab. 1992 May;74(5):1082–1089. doi: 10.1210/jcem.74.5.1349024. [DOI] [PubMed] [Google Scholar]
- 75.Timmers HJ, Kozupa A, Chen CC, et al. Superiority of fluorodeoxyglucose positron emission tomography to other functional imaging techniques in the evaluation of metastatic SDHB-associated pheochromocytoma and paraganglioma. J Clin Oncol. 2007 Jun 1;25(16):2262–2269. doi: 10.1200/JCO.2006.09.6297. [DOI] [PubMed] [Google Scholar]
- 76.Timmers HJ, Chen CC, Carrasquillo JA, et al. Comparison of 18F-fluoro-L-DOPA, 18F-fluoro-deoxyglucose, and 18F-fluorodopamine PET and 123I-MIBG scintigraphy in the localization of pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. 2009 Dec;94(12):4757–4767. doi: 10.1210/jc.2009-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zelinka T, Timmers HJ, Kozupa A, et al. Role of positron emission tomography and bone scintigraphy in the evaluation of bone involvement in metastatic pheochromocytoma and paraganglioma: specific implications for succinate dehydrogenase enzyme subunit B gene mutations. Endocr Relat Cancer. 2008 Mar;15(1):311–323. doi: 10.1677/ERC-07-0217. [DOI] [PubMed] [Google Scholar]
- 78.Ilias I, Yu J, Carrasquillo JA, et al. Superiority of 6-[18F]-fluorodopamine positron emission tomography versus [131I]-metaiodobenzylguanidine scintigraphy in the localization of metastatic pheochromocytoma. J Clin Endocrinol Metab. 2003 Sep;88(9):4083–4087. doi: 10.1210/jc.2003-030235. [DOI] [PubMed] [Google Scholar]
- 79.Fassnacht M, Kreissl MC, Weismann D, Allolio B. New targets and therapeutic approaches for endocrine malignancies. Pharmacol Ther. 2009 Jul;123(1):117–141. doi: 10.1016/j.pharmthera.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 80.Scholz T, Schulz C, Klose S, Lehnert H. Diagnostic management of benign and malignant pheochromocytoma. Exp Clin Endocrinol Diabetes. 2007 Mar;115(3):155–159. doi: 10.1055/s-2007-970410. [DOI] [PubMed] [Google Scholar]
- 81.Adjalle R, Plouin PF, Pacak K, Lehnert H. Treatment of malignant pheochromocytoma. Horm Metab Res. 2009 Sep;41(9):687–696. doi: 10.1055/s-0029-1231025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zografos GN, Vasiliadis G, Farfaras AN, Aggeli C, Digalakis M. Laparoscopic surgery for malignant adrenal tumors. JSLS. 2009 Apr-Jun;13(2):196–202. [PMC free article] [PubMed] [Google Scholar]
- 83.Pacak K. Preoperative management of the pheochromocytoma patient. J Clin Endocrinol Metab. 2007 Nov;92(11):4069–4079. doi: 10.1210/jc.2007-1720. [DOI] [PubMed] [Google Scholar]
- 84.Pacak K, Fojo T, Goldstein DS, et al. Radiofrequency ablation: a novel approach for treatment of metastatic pheochromocytoma. J Natl Cancer Inst. 2001 Apr 18;93(8):648–649. doi: 10.1093/jnci/93.8.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Venkatesan AM, Locklin J, Lai EW, et al. Radiofrequency ablation of metastatic pheochromocytoma. J Vasc Interv Radiol. 2009 Nov;20(11):1483–1490. doi: 10.1016/j.jvir.2009.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cozzi PJ, Englund R, Morris DL. Cryotherapy treatment of patients with hepatic metastases from neuroendocrine tumors. Cancer. 1995 Aug 1;76(3):501–509. doi: 10.1002/1097-0142(19950801)76:3<501::aid-cncr2820760322>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 87.Keiser Hr Fau - Goldstein DS, Goldstein Ds Fau - Wade JL, Wade Jl Fau - Douglas FL, Douglas Fl Fau - Averbuch SD, Averbuch SD. Treatment of malignant pheochromocytoma with combination chemotherapy. 19850705 DCOM- 19850705 (0194-911X (Print)).
- 88.Nomura K, Kimura H, Shimizu S, et al. Survival of patients with metastatic malignant pheochromocytoma and efficacy of combined cyclophosphamide, vincristine, and dacarbazine chemotherapy. J Clin Endocrinol Metab. 2009 Aug;94(8):2850–2856. doi: 10.1210/jc.2008-2697. [DOI] [PubMed] [Google Scholar]
- 89.He J, Makey D, Fojo T, et al. Successful chemotherapy of hepatic metastases in a case of succinate dehydrogenase subunit B-related paraganglioma. Endocrine. 2009 Oct;36(2):189–193. doi: 10.1007/s12020-009-9219-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huang H Fau - Abraham J, Abraham J Fau - Hung E, Hung E Fau - Averbuch S, et al. Treatment of malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: recommendation from a 22-year follow-up of 18 patients. 20081009 DCOM- 20081118 (0008-543X (Print)).
- 91.Kulke MH, Stuart K, Enzinger PC, et al. Phase II study of temozolomide and thalidomide in patients with metastatic neuroendocrine tumors. J Clin Oncol. 2006 Jan 20;24(3):401–406. doi: 10.1200/JCO.2005.03.6046. [DOI] [PubMed] [Google Scholar]
- 92.Kulke Mh Fau - Stuart K, Stuart K Fau - Enzinger PC, Enzinger Pc Fau - Ryan DP, et al. Phase II study of temozolomide and thalidomide in patients with metastatic neuroendocrine tumors. 20060119 DCOM- 20060209 (1527–7755 (Electronic)).
- 93.Keiser HR, Goldstein DS, Wade JL, Douglas FL, Averbuch SD. Treatment of malignant pheochromocytoma with combination chemotherapy. Hypertension. 1985 May-Jun;7(3 Pt 2):I18–24. doi: 10.1161/01.hyp.7.3_pt_2.i18. [DOI] [PubMed] [Google Scholar]
- 94.Loh KC, Fitzgerald PA, Matthay KK, Yeo PP, Price DC. The treatment of malignant pheochromocytoma with iodine-131 metaiodobenzylguanidine (131I-MIBG): a comprehensive review of 116 reported patients. J Endocrinol Invest. 1997 Dec;20(11):648–658. doi: 10.1007/BF03348026. [DOI] [PubMed] [Google Scholar]
- 95.Safford SD, Coleman RE, Gockerman JP, et al. Iodine -131 metaiodobenzylguanidine is an effective treatment for malignant pheochromocytoma and paraganglioma. Surgery. 2003 Dec;134(6):956–962. doi: 10.1016/s0039-6060(03)00426-4. discussion 962–953. [DOI] [PubMed] [Google Scholar]
- 96.Gonias S, Goldsby R, Matthay KK, et al. Phase II study of high-dose [131I]metaiodobenzylguanidine therapy for patients with metastatic pheochromocytoma and paraganglioma. J Clin Oncol. 2009 Sep 1;27(25):4162–4168. doi: 10.1200/JCO.2008.21.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gedik GK, Hoefnagel CA, Bais E, Olmos RA. 131I-MIBG therapy in metastatic phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging. 2008 Apr;35(4):725–733. doi: 10.1007/s00259-007-0652-6. [DOI] [PubMed] [Google Scholar]
- 98.Shilkrut M, Bar-Deroma R, Bar-Sela G, Berniger A, Kuten A. Low-dose iodine-131 metaiodobenzylguanidine therapy for patients with malignant pheochromocytoma and paraganglioma: single center experience. Am J Clin Oncol. 2010 Feb;33(1):79–82. doi: 10.1097/COC.0b013e31819e2c28. [DOI] [PubMed] [Google Scholar]
- 99.Coleman RE, Stubbs JB, Barrett JA, de la Guardia M, Lafrance N, Babich JW. Radiation dosimetry, pharmacokinetics, and safety of ultratrace Iobenguane I-131 in patients with malignant pheochromocytoma/paraganglioma or metastatic carcinoid. Cancer Biother Radiopharm. 2009 Aug;24(4):469–475. doi: 10.1089/cbr.2008.0584. [DOI] [PubMed] [Google Scholar]
- 100.Lamarre-Cliche M, Gimenez-Roqueplo AP, Billaud E, Baudin E, Luton JP, Plouin PF. Effects of slow-release octreotide on urinary metanephrine excretion and plasma chromogranin A and catecholamine levels in patients with malignant or recurrent phaeochromocytoma. Clin Endocrinol (Oxf) 2002 Nov;57(5):629–634. doi: 10.1046/j.1365-2265.2002.01658.x. [DOI] [PubMed] [Google Scholar]
- 101.Mundschenk J, Unger N, Schulz S, Hollt V, Steinke R, Lehnert H. Somatostatin receptor subtypes in human pheochromocytoma: subcellular expression pattern and functional relevance for octreotide scintigraphy. J Clin Endocrinol Metab. 2003 Nov;88(11):5150–5157. doi: 10.1210/jc.2003-030262. [DOI] [PubMed] [Google Scholar]
- 102.Druce MR, Kaltsas GA, Fraenkel M, Gross DJ, Grossman AB. Novel and evolving therapies in the treatment of malignant phaeochromocytoma: experience with the mTOR inhibitor everolimus (RAD001) Horm Metab Res. 2009 Sep;41(9):697–702. doi: 10.1055/s-0029-1220687. [DOI] [PubMed] [Google Scholar]
- 103.Chrisoulidou A, Kaltsas G, Ilias I, Grossman AB. The diagnosis and management of malignant phaeochromocytoma and paraganglioma. Endocr Relat Cancer. 2007 Sep;14(3):569–585. doi: 10.1677/ERC-07-0074. [DOI] [PubMed] [Google Scholar]
- 104.Joshua AM, Ezzat S, Asa SL, et al. Rationale and evidence for sunitinib in the treatment of malignant paraganglioma/pheochromocytoma. J Clin Endocrinol Metab. 2009 Jan;94(1):5–9. doi: 10.1210/jc.2008-1836. [DOI] [PubMed] [Google Scholar]
- 105.Jimenez C, Cabanillas ME, Santarpia L, et al. Use of the tyrosine kinase inhibitor sunitinib in a patient with von Hippel-Lindau disease: targeting angiogenic factors in pheochromocytoma and other von Hippel-Lindau disease-related tumors. J Clin Endocrinol Metab. 2009 Feb;94(2):386–391. doi: 10.1210/jc.2008-1972. [DOI] [PubMed] [Google Scholar]
- 106.Park K-S, Lee J-L, Ahn H, et al. Sunitinib, a Novel Therapy for Anthracycline- and Cisplatin-refractory Malignant Pheochromocytoma. Jpn J Clin Oncol. 2009 May 1;39(5):327–331. doi: 10.1093/jjco/hyp005. [DOI] [PubMed] [Google Scholar]
- 107.Gross DJ, Munter G, Bitan M, et al. The role of imatinib mesylate (Glivec) for treatment of patients with malignant endocrine tumors positive for c-kit or PDGF-R. Endocr Relat Cancer. 2006 Jun;13(2):535–540. doi: 10.1677/erc.1.01124. [DOI] [PubMed] [Google Scholar]
- 108.Lai EW, Joshi BH, Martiniova L, et al. Overexpression of interleukin-13 receptor-alpha2 in neuroendocrine malignant pheochromocytoma: a novel target for receptor directed anti-cancer therapy. J Clin Endocrinol Metab. 2009 Aug;94(8):2952–2957. doi: 10.1210/jc.2009-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Joshi BH, Hogaboam C, Dover P, Husain SR, Puri RK. Role of interleukin-13 in cancer, pulmonary fibrosis, and other T(H)2-type diseases. Vitam Horm. 2006;74:479–504. doi: 10.1016/S0083-6729(06)74019-5. [DOI] [PubMed] [Google Scholar]
- 110.King KS, Whatley MA, Alexopoulos DK, et al. The use of functional imaging in a patient with head and neck paragangliomas. J Clin Endocrinol Metab. 2010 Feb;95(2):481–482. doi: 10.1210/jc.2009-2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fitzgerald PA, Goldsby RE, Huberty JP, et al. Malignant pheochromocytomas and paragangliomas: a phase II study of therapy with high-dose 131I-metaiodobenzylguanidine (131I-MIBG) Ann N Y Acad Sci. 2006 Aug;1073:465–490. doi: 10.1196/annals.1353.050. [DOI] [PubMed] [Google Scholar]