

Abstract
Motivation: The application of information encoded in molecular networks for prognostic purposes is a crucial objective of systems biomedicine. This approach has not been widely investigated in the cardiovascular research area. Within this area, the prediction of clinical outcomes after suffering a heart attack would represent a significant step forward. We developed a new quantitative prediction-based method for this prognostic problem based on the discovery of clinically relevant transcriptional association networks. This method integrates regression trees and clinical class-specific networks, and can be applied to other clinical domains.
Results: Before analyzing our cardiovascular disease dataset, we tested the usefulness of our approach on a benchmark dataset with control and disease patients. We also compared it to several algorithms to infer transcriptional association networks and classification models. Comparative results provided evidence of the prediction power of our approach. Next, we discovered new models for predicting good and bad outcomes after myocardial infarction. Using blood-derived gene expression data, our models reported areas under the receiver operating characteristic curve above 0.70. Our model could also outperform different techniques based on co-expressed gene modules. We also predicted processes that may represent novel therapeutic targets for heart disease, such as the synthesis of leucine and isoleucine.
Availability: The SATuRNo software is freely available at http://www.lsi.us.es/isanepo/toolsSaturno/.
Contact: inepomuceno@us.es
Supplementary information: Supplementary data are available at Bioinformatics online.
1 BACKGROUND
A crucial objective of systems biomedicine is the application of information encoded in molecular networks for prognostic purposes. Standard approaches to biomarker discovery are based on the identification of differentially expressed genes or proteins. However, the multifactorial nature of common complex diseases limits a discovery process that relies on the assumption that genes act independently. Furthermore, it is known that powerful prognostic biomarkers may be encoded by genes that are not highly differentially expressed across control and disease patients (Azuaje et al., 2010) and vice versa, and differentially expressed genes are not always strong biomarkers (Devaux et al., 2010). A systems-level approach can provide insights into the interplay of genes and their association with clinical phenotypes. In comparison with cancer research, network-based prognostic approaches have not been widely investigated in the cardiovascular research area. Despite advances driven by functional genomics, there is a need to propose new prognostic methodologies that exploit the outcomes of systems biology in cardiovascular research. Within this area, the prediction of clinical outcomes after suffering a heart attack and personalized medicine would represent a significant contribution to translational research (Azuaje et al., 2009). In this article, we report a new supervised prediction method for this prognostic problem, which is based on the discovery of clinically relevant transcriptional association networks.
There are several models to infer gene–gene association networks from microarray data. These models range from relatively straightforward correlation-based methods to more sophisticated models, such as Bayesian network models. In standard correlation-based methods, the Pearson's coefficient has been used to extract gene–gene dependencies (D'Haeseleer et al., 1998). Different versions of this method exist, such as one by Obayashi and Kinoshita (2009) that uses correlation ranks instead of correlation values. In De la Fuente et al. (2004), the authors used Partial Pearson's correlation to extract associations between pairs of genes when this association can be explained by means of a third gene. Other methods are based on Gaussian graphical models. In these models, two genes are related if and only if their association can be explained by other genes in the dataset (Dobra et al., 2004). These methods are based on pairwise measurements and concepts of statistical independence. Additionally, there are other methods that analyze prior knowledge. For example, protein–protein interactions have been integrated with gene expression profiles to obtain networks as functional modules (Ulitsky and Shamir, 2007).
In this article, we analyzed gene expression biosignatures relevant to the prediction of clinical outcome after myocardial infarction (MI). The motivation of this study is to characterize two different groups of patients, who exhibit different clinical outcomes. In this context, the benefit of a network-based approach is 2-fold: (i) to improve systems-based understanding of the biological problems through quantitative descriptions of functional associations relevant to phenotypes and (ii) based on such knowledge, to contribute new classification models of disease. Our method also provides qualitative descriptions of mechanisms defining clinical classes that can be interpreted by clinical experts. In particular, we aimed to provide novel understandings of the molecular mechanisms that may drive ventricular dysfunction in post-myocardial infarction patients. Before reporting these results, we first present a comparative analysis performed on a published benchmark dataset.
Our method discovers transcriptional association networks with prognostic value for each clinical category. The networks are inferred from microarray data based on an unsupervised learning algorithm reported in Nepomuceno-Chamorro et al. (2010). This algorithm estimates the dependency between genes for a localized subspace of expression profiles instead of global similarities. The latter is the case of traditional correlation-based methods. In this article, we present a supervised prediction version of the approach proposed by Nepomuceno-Chamorro et al. (2010). Our method is also freely available as a Java-based application: Supervised prognostic Approach Through Regression Networks (SATuRNo).
Because we are interested in characterizing phenotype-specific networks, we built them taking into account the gene expression changes that occur in patients with the same clinical category. Hence, genes that are included in both networks are relevant to characterize both groups of patients, although different gene expression relations may be involved. Those genes that are unique to each class may represent significant components altered in one of the clinical conditions, e.g. poor prognosis. In addition, network differences may be associated with lose or gain of function, or the presence of compensatory functional mechanisms. Furthermore, the resulting networks are relevant for their prediction ability. Given a new patient, the method can predict the clinical category of this patient using network-based information from each class. This patient will be assigned to the class whose network is the likeliest to estimate the observed gene expression pattern. Hence, class-specific networks capture gene expression changes observed in a clinical category, and together are used to differentiate between patients.
The remainder of this article is organized as follows. In Section 2, a summary of the method is presented together with an explanation of how the supervised prediction is carried out. Section 3 reports results and discussions using different datasets, network inference approaches and classification models. The prediction power, as well as potential clinical relevance, of our approach was estimated by several classification performance measures and functional characterizations. A key outcome of this investigation was the discovery of new biomarkers for distinguishing between clinical response categories in MI patients: good outcome (normal left ventricular function) and bad outcome (left ventricular dysfunction). The last section provides conclusions and possible future research directions.
2 METHODS
In this study, we aimed to discover new gene expression biosignatures relevant to the prediction of ventricular dysfunction after MI. To deal with this prognostic problem, we developed SATuRNo as a new supervised prediction method based on the discovery of clinically relevant transcriptional association networks. This means that SATuRNo discovers transcriptional association networks with prognostic value for each clinical category. The networks were inferred from microarray data based on an unsupervised learning algorithm reported in Nepomuceno-Chamorro et al. (2010). SATuRNo is a supervised prediction version of that algorithm.
Our method consists of two main steps (Fig. 1). The first step involves inferring clinically relevant gene association networks from gene expression data. Each network is inferred from gene expression of patients with the same clinical category. To infer these networks, the unsupervised learning algorithm analyzes each gene by taking into account the remaining genes as inputs to a mathematical model that estimates the expression value of that gene. The latter is done by means of linear regression functions using M5′ model tree algorithm (Witten and Frank, 2005). This technique focuses on building linear models to separate areas of the search space, i.e. optimal partitions of gene expression samples. Each linear model represents localized similarities, i.e. specific groups of sample–sample relationships. Furthermore, this technique constructs linear models under all samples (global similarity) if the optimal partition is defined by the complete set of gene expression samples. Consequently, we can state that this method favors more localized similarities over global similarities. The second step involves predicting the clinical category of a new patient (test mode) through the inferred networks. Each network is based on localized linear models, which estimate expression values of all the genes. The prediction is based on the relative error between the true (i.e. observed expression value in the dataset) and predicted expression values of those genes involved in the inferred networks.
Fig. 1.

Schematic view of the proposed method. The first step involves building clinically relevant gene association networks from gene expression data of patients with the same clinical category. These networks are built based on the linear models generated by the model tree induction algorithm called M5P (Witten and Frank, 2005), an extension of regression tree algorithm. The second step involves predicting the clinical category of a new patient through the inferred networks. The prediction is based on the relative error between the true and predicted gene expression values of those genes involved in the inferred networks.
2.1 Building networks
The first step involves the inference of class-specific networks. To this aim, genes from the microarray are analyzed in an iterative process. In each iteration, a gene is taken as a target gene and the remaining genes as inputs to a mathematical model that estimates the expression value of the target gene. These input genes are used for splitting the search space into subspaces, i.e. into subset of gene expression samples to analyze localized similarities. In each subspace, a localized linear model between the target gene and different inputs genes is built by applying model trees. M5′ is a model tree algorithm, an extension of the regression tree algorithm, which has linear regression functions at the leaves.
Before describing our method in more detail, basic definitions are provided. Let M be a microarray dataset, i.e. the measurements space, which can be defined as: M = (C, G, L, Class) where C ={1, 2,… n} is a finite set of gene expression samples, G={1, 2,… m} is a finite set of genes, Class={c1, c2,… cp} is a finite set of clinical categories. Finally, L is a n×m gene expression matrix. The matrix L can be defined as: L=(vij) with vij representing the observed expression value of gene j under the sample i. Throughout the article, we will refer to gene expression samples as patients and to clinical categories as classes.
The class-specific network discovery algorithm is implemented as follows. First, the microarray data are divided into p parts, each of them represents the microarray set Mi formed by patients of the clinical class ci ∈ Class, with 1 ≤ i ≤ p. The aim of this partition is to build the underlying class-specific network that represents each group of patients with the same clinical class ci. Second, a network is built from each Mi. This is done by generating a forest of trees, i.e. a model tree is built for each gene gj ∈ G with 1 ≤ j ≤ m. Previous research conducted by (Zhang et al., 2003) has shown that this approach may be more biologically interpretable than a random forest (Breiman, 2001) or a single tree. In our method, the resulting forest of trees is called FT, and it can be defined as FT={MT1, MT2,… MTm} where MTj is the model tree built for the target gene gj. We used the M5′ algorithm to build model trees (Witten and Frank, 2005). M5′ is an extension of regression tree algorithms that constructs tree-based piecewise linear models, i.e. it constructs several linear models at the same time, each of them identified by a leaf in a tree. In this way, the method favors more localized similarities over global similarities.
Finally, the FT is pruned taken into account a threshold value θ. This pruning process consists in removing those MTj with relative error ε≥θ. This error can be defined as follows:
where a is the true target gene expression value, 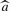 is the estimated target gene expression value for a patient i on the dataset. Each model tree belonging to FTθ estimates or predicts the target gene expression value by means of the linear models detected in its leaves. Gene association sets A={(gx, gy)} are defined as follow:
is the estimated target gene expression value for a patient i on the dataset. Each model tree belonging to FTθ estimates or predicts the target gene expression value by means of the linear models detected in its leaves. Gene association sets A={(gx, gy)} are defined as follow:
gx is the target gene of a MTx with ε≤θ, i.e. its model tree is not removed after the pruning phase.
gy is a gene that belongs to a linear model from MTx, i.e. one of the independent variable of a linear model defined as LM : gx = ∑k akgk.
2.2 Network-based patient classification
This second step involves predicting the clinical class of a new patient through the inferred clinical class-specific networks. The new patient can be classified into one of the p classes after p networks have been built.
Let GNi be a network from a ci class with (1≤i≤p), and let LMGNi be the set of linear models that constitute this network, i.e. those representing the set of associations A in network GNi. Given a new patient, the relative error between the predicted and the true gene expression values defined by each linear model from LMGNi is calculated. The mean value of such relative prediction errors are used to assign patients to classes.
The computing cost of building the forest of trees is m times the cost of building a M5′ tree, i.e. O(m2nlog(n)), where m is the number of genes and n the number of patients. Extracting the gene–gene associations is an iterative process which has a linear complexity O(m). Finally, the procedure of estimating the expression value of each target gene has a linear complexity. Consequently, the overall computing cost of our methodology is O(m2nlog(n)).
2.3 Datasets
Before evaluating our method on our cardiovascular research problem, we tested our approach on a published benchmark dataset.
2.3.1 The benchmark dataset.
As a benchmark dataset, we used the dataset reported in Dunckley et al. (2006) that consists of 13 control and 20 Alzheimer's disease (AD) brain tissue samples. This single cell gene expression dataset includes 35 722 gene probesets. We compared our method to several transcriptional association network algorithms and classification models. The experiments reported here focus on a pre-processed version of this dataset, which included a total of 1663 genes (Dunckley et al., 2006) as described in Ray et al. (2008).
2.3.2 The heart dataset.
This dataset was generated at the Laboratory of Cardiovascular Research and the Centre Hospitalier of Luxembourg. This research was approved by the local ethics committees and written informed consent was obtained from all patients. This dataset includes 32 patients with acute MI. Gene expression data were extracted from blood samples obtained on the day of MI. The clinical outcome of these patients, i.e. their prognostic class, was evaluated after 30 days post-MI by means of the ejection fraction (EF). The EF is an indicator of the blood pumping capacity of the heart and is measured by echocardiography. Our dataset included 16 patients with good prognosis (EF > 40%) and 16 with ventricular dysfunction, i.e. bad prognosis (EF ≤ 40%). Throughout the article, we will refer to this dataset as the heart dataset, and to the clinical outcomes as good and bad prognosis classes, respectively.
2.4 Software and experimental setting
SATuRNo was implemented as a Java stand-alone application and a prototype can be downloaded from http://www.lsi.us.es/isanepo/toolsSaturno/. The leave-one-out cross-validation (LOOCV) was applied to estimate the classification performance of all the models investigated. The LOOCV technique implies to run the software as many times as the number of patients included in the input microarray dataset. Each SATuRNo's run was executed in one of the nodes of a computing platform that used the Sun Grid Engine queuing system. Each node was a dual-core processor with 4 GB memory. Furthermore, we have run the Windows version tools of PPC and Matisse on a personal computer with dual-core and 4 GB memory.
3 RESULTS AND DISCUSSION
3.1 Benchmark analysis
We first assessed the predictive performance of our approach on the benchmark dataset. In this and subsequent disease-driven analyses, we compared our approach to other published techniques on the basis of two tasks: network inference and classification using the inferred networks. Consequently, to compare the predictive capability of our approach against other methods, we implemented prognostic models that differ in the way class-specific transcriptional association networks are inferred, and in the supervised classification methodology applied on the network information.
For network inference, we applied a Pearson correlation (PC)-based method (?), Partial PC-based method (De la Fuente et al., 2004) and the Matisse tool (Ulitsky and Shamir, 2007). Matisse detects functional modules using gene expression and protein interaction network data. After building class-specific networks, the genes involved in these networks were used as inputs to several classifiers: nearest neighbours (IB1), decision trees (C4.5 algorithm) and Naive Bayes classifiers.
Results reported here were obtained with a threshold value θ = 15%, i.e. the model trees with relative error greater than θ were removed. The threshold value theta was set within the LOOCV procedure. This parameter was optimized: (i) to achieve good classification performance and (ii) to obtain compact networks with a tractable size that can facilitate expert interpretation and future experimental validation. The parameters of the benchmark network inference methods discussed here were set to generate networks of sizes (34 nodes) comparable to those obtained with our approach. In Table 1, several standard classification performance measures are shown, such as the representative classification accuracy (Acc.), the true positive rate (TPR), false positive rate (FPR), specificity, sensitivity and the area under the receiver operating characteristic curve (AUC).
Table 1.
Results of the benchmark dataset: comparative analysis
| Method classifier | Our approach (SATuRNo) | PC-based method |
Partial PC-based method |
Matisse method |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IB1 | C4.5 | NB | IB1 | C4.5 | NB | IB1 | C4.5 | NB | ||
| Number of genes | 34 | 29 |
79 |
20 |
||||||
| Representative Acc. | 90.9% | 87.87% | 78.78% | 87.87% | 93.93% | 63.63% | 84.84% | 90.9% | 87.87% | 87.87% |
| Weighted Avg. TPR | 0.90 | 0.87 | 0.78 | 0.87 | 0.93 | 0.63 | 0.84 | 0.90 | 0.87 | 0.87 |
| Weighted Avg. FPR | 0.08 | 0.16 | 0.24 | 0.13 | 0.06 | 0.39 | 0.15 | 0.14 | 0.13 | 0.13 |
| Specificity | 0.84 | 0.76 | 0.69 | 0.84 | 0.92 | 0.53 | 0.84 | 0.76 | 0.84 | 0.84 |
| Sensitivity | 0.95 | 0.95 | 0.85 | 0.90 | 0.95 | 0.70 | 0.85 | 1 | 0.90 | 0.90 |
| AUC | 0.89 | 0.86 | 0.70 | 0.91 | 0.937 | 0.64 | 0.92 | 0.88 | 0.94 | 0.93 |
We compared our approach to other published techniques on the basis of two tasks: network inference and classification using the inferred networks. For network inference, we applied a PC-based method (?), Partial PC-based method (De la Fuente et al., 2004) and the Matisse tool (Ulitsky and Shamir, 2007). After building class-specific networks, the genes involved in these networks were used as inputs to several classifiers: nearest neighbors (IB1), decision trees (C4.5 algorithm) and Naive Bayes classifiers. Several measures as representative accuracy, TPR, FPR, specificity, sensitivity and AUC values are shown. The representative accuracy is the proportion of correctly classified patients. The TPR and FPR are the weighted average true and positive rate. The specificity is the proportion of control patients, which were recognized as control category. The sensitivity is the proportion of disease patients, which were recognized as disease category. Finally, the AUC values represent the area under the receiver operating characteristic curve. Avg., average.
In general, we showed that our approach can outperform the other methodologies. For example, our approach showed one of the highest Acc. values (90.9%). Only the Partial PC-based method obtained a better performance when combined with the IB1 algorithm. However, Partial PC-based method obtained the same sensitivity value. In other words, all poor prognosis patients tend to be recognized as having a poor clinical response. The fact that only Partial PC-based method obtained a better performance when combined with the IB1 algorithm suggests that the networks estimated by our approach can provide the basis for relatively accurate classification models.
We also compared key topological parameters of the networks produced by these methods. The number of nodes, edges, connected components, diameter and density are shown in Table 1 of Supplementary Material. Note that the density is low in all cases, i.e. the ratio of the number of edges and the number of possible edges is similar. The intersections between the resulting networks are shown in Tables 2–4 of Supplementary Material. Note than only the resulting networks from SATuRNo and CoExpress (http://bioinformatics.lu/CoExpress) has gene–gene associations in common. The remaining possible intersections have genes but not associations in common.
Table 2.
Genes in common between the networks from heart dataset
| Gene name | Full name | Location | Type |
|---|---|---|---|
| BANF1 | Barrier-to-autointegration factor | N | O |
| RPS4Y1 | 40S ribosomal protein S4 | ||
| Y isoform 1 | C | O | |
| OBFC2B | SOSS complex subunit B1 | U | O |
| APOF | Apolipoprotein F | ES | T |
| CCNO | Cyclin-O | N | TR |
| LOC125595 | gene model ab initio | U | U |
| HIST1H2AE | Histone H2A type 1-B/E | N | O |
| AK091188-2203 | – | U | U |
The overlap between networks from good and bad prognosis is small and it can be observed at the node level only, i.e. there are not edges in common. ES, extracellular space; C, cytoplasm; N, nucleus; U, unknown; O, other; T, transporter; TR, transcription regulator.
Table 4.
Heart dataset: comparison with others classifiers
| Method classifier | SATuRNo | PC-based method |
||
|---|---|---|---|---|
| IB1 | C4.5 | NB | ||
| Representative Acc | 72.41% | 65.62% | 50% | 53.12% |
| Specificity | 0.78 | 0.68 | 0.56 | 0.50 |
| Sensitivity | 0.67 | 0.62 | 0.43 | 0.56 |
| AUC | 0.72 | 0.65 | 0.39 | 0.59 |
Comparison between the performance of SATuRNo against different classification models whose inputs represented the genes detected by PC-based method (CoExpress).
3.2 Systems-based prognosis after MI
The method identified, from an original input set of 15 307 genes, networks with 17 edges (gene–gene associations) and 19 nodes (genes) in patients with good prognosis, and a network with 59 edges and 48 genes in patients with bad prognosis (Fig. 2). The bad and good prognosis networks were built from 12 and 5 linear models, respectively. Representative Acc. values of 72% were obtained (LOOCV). These class-specific networks have eight genes in common (Table 2). In the case of the bad prognosis network, its 8 genes were involved in 23 transcriptional associations, whereas in the case of good prognosis its 8 genes included 13 different associations with other genes. It is worth mention that these networks have a tractable size that facilitates expert interpretation and future validations. Furthermore, the good prognosis network inferred by our method is smaller than that from the bad prognosis category. This indicates that the latter group requires more information to be adequately characterized than the good prognosis group. Either, this may suggest that the bad prognosis group is more heterogeneous (genetically or clinically) than the good prognosis one, or that the larger number of associations in the bad prognosis group reflects a possible compensatory mechanism for the disruption of molecular pathways.
Fig. 2.

Clinically relevant gene association networks obtained from the heart dataset. Both networks were built from a microarray with 15 307 genes and the forest of trees was pruned using the threshold value θ = 15. Furthermore, the representative accuracy of these prognostic transcriptional networks was 72% (LOOCV).
In this analysis, a forest of model trees (MT) was built in which MT with a relative error ε greater than 15% were removed. As explained above, this pruning has been made in order to select model trees with low prediction errors. This threshold value was the one that consistently generated relatively small networks with reasonable classification Acc. We also obtained other prognosis models with higher Acc. values, but with relatively very large networks (more than 500 nodes).
Using the CoExpress tool (?), we applied the PC-based method and we compared results with the networks provided by our approach. Several topological parameters (i.e. diameter, number of genes and number of associations) are shown in Table 3. The networks obtained by PC-based method, with 0.95 as a threshold correlation value, have a huge number of nodes and edges in comparison with the networks obtained by our approach. Note that larger networks are obtained for less stringent threshold values. The capacity of our method to infer compact networks makes it particularly suitable to expert interpretation and future experimental validations.
Table 3.
Topological network parameters from heart dataset
| Method | Bad prognosis |
Good prognosis |
||||
|---|---|---|---|---|---|---|
| Nodes | Edges | Diameter | Nodes | Edges | Diameter | |
| PC-based method | 4297 | 16 407 | 21 | 3322 | 6228 | 29 |
| SATuRNo | 48 | 59 | 8 | 19 | 17 | 4 |
The networks obtained by PC-based method, with 0.95 as a threshold correlation value, have a huge number of nodes and edges in comparison with the networks obtained by our approach.
We compared the predictive capability of our approach against standard classification models based on gene expression data. We trained several classifiers using the datasets under study. We trained ‘lazy learning’ classifiers, classifiers based on decision trees and probabilistic classifiers based on Bayesian statistics. Finally, we trained support vector machine classifiers, which have been shown to be powerful and robust models in cancer and other research areas [see (Chu and Wang, 2005) or (Lee and Lee, 2003)]. Although SATuRNo did not outperform all the classifiers, it showed higher classification accuracy than most of the models, including support vector machine classifiers (see Supplementary Material with details in Table 5). This result suggests that networks estimated by our approach can provide the basis for relatively accurate classification models. These results encouraged us to investigate our method as a new strategy to discover potential biomarkers of clinical outcome after myocardial infarction. This is specially motivated by the fact that our method, unlike traditional approaches, can provide mechanistic insights of processes and associations underlying the clinical conditions through specific network-based visualizations.
We also compared the results reported by our approach against different classification models, whose inputs represented the genes detected by CoExpress. Our approach showed better classification performance as estimated by different indicators (Table 4), including the maximum AUC. The partial PC-based and Matisse methods were unsuitable to generate inputs to classification models because of the large number of genes in the input microarray (more than 15 000 genes). We used the software tools (Windows version) provided by De la Fuente et al. (2004) and Ulitsky and Shamir (2007). These experiments were run on a dual core personal computer with 4 GB memory. We emphasize that it was not our goal to provide a new optimal implementation of the PPC and Matisse method to run in a parallelized way.
3.2.1 Gene Ontology analysis
The resulting networks were analyzed in the context of Gene Ontology (GO) with the BINGO system (Maere et al., 2005). Detection of statistically overrepresented GO terms was done with the hypergeometric test, multiple-testing adjustments with the Benjamini and Hochberg false discovery rate and a significance level α=0.05.
This analysis did not identify GO biological process terms as significantly overrepresented in the bad prognosis network discovered by our approach. In the good prognosis network, this analysis identified the following GO biological process as significantly overrepresented: isoleucine catabolic process (GO id: 6550) and leucine biosynthetic process (GO id: 9098). These processes are particularly relevant in the context of remodeling after myocardial infarction. Indeed, the small leucine-rich protein biglycan, a component of the extracellular matrix of the heart, plays a pivotal role in cardiac remodeling, notably through the formation of a collagen matrix that aids in scar formation and in the preservation of left ventricular function (Westermann et al., 2008). Another small leucine-rich proteoglycan, decorin, prevents cardiac fibrosis, thereby positively affecting left ventricular remodeling (Li et al., 2009). Therefore, our networks, which were inferred from gene expression profiles derived from bad and good prognosis patients, identified potential prognostic biomarkers. Bad and good prognosis classes referred to patient groups with and without ventricular dysfunction after suffering a heart attack, respectively.
3.2.2 Literature and pathway knowledge mining
To further determine the potential biomedical relevance of our class-specific networks, we performed large-scale mining of the literature using the Agilent Literature Search (http://www.agilent.com/labs/research/litsearch.html) and DAVID Tools. The former is a Cytoscape plug-in (Shannon et al., 2003) that identifies published gene–disease associations encoded in PubMed abstracts. We implemented a search constrained by the keywords ‘heart failure’, ‘cardiovascular disease’ and ‘myocardial infarction’. The genes EDF1, BCAM and DLK1 were found to be associated with these search terms. The David tool (Huang et al., 2007a, b; Sherman et al., 2007), given a list of genes, allowed us to search for gene–disease associations in the Genetic Association Database (Becker et al., 2004) and the OMIM database (Hamosh et al., 2005). In this case only an association between BCAM and cardiovascular disease was found. The genes APOF (which appears in the two networks) and DCTN1 (good prognosis network) have previously been associated with neurological disorders including Alzheimer's disease (Kabbara et al., 2004; Vilarino-Guell et al., 2009). Although these associations will require further experimental and computational analyses, these findings are consistent with research that have found functional links between cardiovascular disease and Alzheimer's disease (Rosendorff et al., 2007; Stewart, 1998).
Finally, we applied the Ingenuity Pathway Analysis (IPA) (Ingenuity® Systems, Redwood City, CA, www.ingenuity.com ) to explore additional associations between our prognostic networks and molecular pathways. Examples of significant associations detected are as follows: Cell Cycle, Hematological Systems Development and Function, Hematopoiesis (P = 10−22 Fisher's exact test) and Cell Death, Hematological Disease, Immunological Disease (P = 10−19). From good prognosis, IPA reported Cardiac Edema, Cardiovascular Disease (P=0.001) among others. For more information see Supplementary Table S6 and S7. Furthermore, IPA highlighted several genes as known disease biomarkers: BCAM (breast cancer), RPS4Y1 and HINT1 (Ewing's sarcoma) and SRNP (bladder cancer).
3.3 General remarks
The outcome of algorithms based on building trees is influenced by a stopping criterion. The stopping criterion of the M5′ algorithm is as follows: the space is splitted unless the subset of samples contains very few cases or their values vary slightly. Also note that models based on decision trees have been shown to be useful to infer biologically meaningful gene association networks using microarray data (Soinov et al., 2003). This method used a supervised learning approach to address this question by building decision tree-related classifiers, which predict the state of a gene from the expression data of other genes. Soinov's method differs from ours in the sense that the former applies a transformation procedure to set the state of a gene as ‘expressed more than average’ and ‘expressed less than average’ before applying the supervised learning method, i.e. each gene is discretized before predicting its state. Moreover, Soinov et al. (2003) reported results for a small group of yeast gene expression dataset.
Finally, although it offers advantages for supporting clinical decision making, we acknowledge that our method is not suitable for the task of estimating detailed or dynamic representations of gene regulatory networks. Other methods specifically designed for this purpose are recommended [Meinshausen et al. (2006) and Wille and Buhlmann (2006)].
4 CONCLUSIONS
We presented a new supervised prediction method for prognostic applications. In particular, our method allowed us to discover potential novel biomarkers (Fig. 2) and systems-level mechanistic insights in cardiovascular research. Our method is based on the discovery of clinically relevant transcriptional association networks. It generates new hypotheses about clinically relevant interactions among genes using gene expression data and regression trees. We also demonstrated that, unlike traditional techniques, our method can discover small biologically meaningful networks, which facilitate human expert interpretation and targeted validations. Furthermore, our method allows the automated classification of patients into clinical categories. We also detected biological processes, such as the synthesis of leucine and isoleucine, which may be used to characterize and possibly treat the development of ventricular dysfunction after MI.
Our method can be applied to other biomedical applications and its software implementation, SATuRNo, is freely available. We also tested our approach on a published benchmark dataset with control and disease patients, and compared it to several algorithms to infer transcriptional association networks and classification models. These comparative results provided additional evidence of the prediction power of our approach.
In principle, the developed methodology can be used to infer clinically relevant networks using different types of molecular data, such as microarray gene expression and proteomics data. New versions of SATuRNo will be available in the future. The predictions reported here will require further in vitro investigations and their independent validation in larger patient cohorts.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Jose Riquelme for valuable feedback, and Monica Ray for providing the benchmark dataset (Ray et al., 2008). This work made extensive use of the distributed computing platform at Centro Científico de Andalucía (CICA). We thank Claudio Arjona at CICA for technical support.
Funding: In Luxembourg this research was in part supported by Fonds National de la Recherche (Luxembourg); Société pour la Recherche sur les Maladies Cardiovasculaires; Ministére de la Culture; de l'Enseignement Supérieur et de la Recherche. In Spain, it was in part supported by the Spanish Ministry of Science and Innovation under Grant TIN2007–68084–C02–00, and by the Plan Propio of the University of Seville.
Conflict of Interest: none declared.
REFERENCES
- Azuaje F, et al. Computational biology for cardiovascular biomarker discovery. Brief. Bioinformatics. 2009;10:367–377. doi: 10.1093/bib/bbp008. [DOI] [PubMed] [Google Scholar]
- Azuaje F, et al. Coordinated modular functionality and prognostic potential of a heart failure biomarker-driven interaction network. BMC Syst. Biol. 2010;4:60. doi: 10.1186/1752-0509-4-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker KG, et al. The genetic association database. Nat. Genet. 2004;36:431–432. doi: 10.1038/ng0504-431. [DOI] [PubMed] [Google Scholar]
- Breiman L. Random forests. Mach. Learn. 2001;45:5–32. [Google Scholar]
- Chu F, Wang L. Applications of support vector machines to cancer classification with microarray data. Int. J. Neural Syst. 2005;15:475–484. doi: 10.1142/S0129065705000396. [DOI] [PubMed] [Google Scholar]
- De la Fuente A, et al. Discovery of meaningful associations in genomic data using partial correlation coefficients. Bioinformatics. 2004;20:3565–3574. doi: 10.1093/bioinformatics/bth445. [DOI] [PubMed] [Google Scholar]
- Devaux Y, et al. Integrated protein network and microarray analysis to identify potential biomarkers after myocardial infarction. Funct. Integrat. Genomics. 2010;10:329–337. doi: 10.1007/s10142-010-0169-0. [DOI] [PubMed] [Google Scholar]
- D'Haeseleer P, et al. Mining the gene expression matrix: inferring gene relationships from large scale gene expression data. Proceedings of the Second International Workshop on Information Processing in Cell and Tissues. 1998:203–212. [Google Scholar]
- Dobra A, et al. Sparse graphical models for exploring gene expression data. J. Multivar. Anal. 2004;90:196–212. [Google Scholar]
- Dunckley T, et al. Gene expression correlates of neurofibrillary tangles in alzheimer's disease. Neurobiol. Aging. 2006;27:1359–1371. doi: 10.1016/j.neurobiolaging.2005.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamosh A, et al. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:D514–D517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, et al. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007a:1–7. doi: 10.1093/nar/gkm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, et al. The david gene functional classification tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007b;8:R183. doi: 10.1186/gb-2007-8-9-r183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabbara A, et al. Exclusion of CYP46 and APOM as candidate genes for Alzheimer's disease in a French population. Neurosci. Lett. 2004;363:139–143. doi: 10.1016/j.neulet.2004.03.066. [DOI] [PubMed] [Google Scholar]
- Lee Y, Lee C-K. Classification of multiple cancer types by multicategory support vector machines using gene expression data. Bioinformatics. 2003;19:1132–1139. doi: 10.1093/bioinformatics/btg102. [DOI] [PubMed] [Google Scholar]
- Li L, et al. Postinfarction gene therapy with adenoviral vector expressing decorin mitigates cardiac remodeling and dysfunction. J. Physiol. Heart Circ. Physiol. 2009;297:H1504–1513. doi: 10.1152/ajpheart.00194.2009. [DOI] [PubMed] [Google Scholar]
- Maere S, et al. BiNGO: a Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- Meinshausen N, et al. High dimensional graphs and variable selection with the lasso. Ann. Stat. 2006;34:1436–1462. [Google Scholar]
- Nepomuceno-Chamorro I, et al. Inferring gene regression networks with model trees. BMC Bioinformatics. 2010;11:517. doi: 10.1186/1471-2105-11-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obayashi T, Kinoshita K. Rank of correlation coefficient as a comparable measure for biological significance of gene coexpression. DNA Res. 2009;16:249–260. doi: 10.1093/dnares/dsp016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray M, et al. Variations in the transcriptome of alzheimer's disease reveal molecular networks involved in cardiovascular diseases. Genome Biol. 2008;9:R148. doi: 10.1186/gb-2008-9-10-r148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosendorff C, et al. Cardiovascular risk factors for Alzheimer's disease. Am. J. Geriatr. Cardiol. 2007;16:143–149. doi: 10.1111/j.1076-7460.2007.06696.x. [DOI] [PubMed] [Google Scholar]
- Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman B, et al. David knowledgebase: a gene-centered database integrating heterogeneous gene annotation resources to facilitate high-throughput gene functional analysis. BMC Bioinformatics. 2007;8:426. doi: 10.1186/1471-2105-8-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soinov L, et al. Towards reconstruction of gene networks from expression data by supervised learning. Genome Biol. 2003;4:R6. doi: 10.1186/gb-2003-4-1-r6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart R. Cardiovascular factors in Alzheimer's disease. J. Neurol., Neurosurg. Psych. 1998;65:143–147. doi: 10.1136/jnnp.65.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, Shamir R. Identification of functional modules using network topology and high-throughput data. BMC Syst. Biol. 2007;1:8. doi: 10.1186/1752-0509-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilarino-Guell C, et al. Characterization of DCTN1 genetic variability in neurodegeneration. Neurology. 2009;72:2024–2028. doi: 10.1212/WNL.0b013e3181a92c4c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann D, et al. Role of left ventricular stiffness in heart failure with normal ejection fraction. Circulation. 2008;117:2051. doi: 10.1161/CIRCULATIONAHA.107.716886. [DOI] [PubMed] [Google Scholar]
- Wille A, Buhlmann P. Low-order conditional independence graphs for inferring genetic networks. Stat. Appl. Genet. Mol. Biol. 2006;5:1170. doi: 10.2202/1544-6115.1170. [DOI] [PubMed] [Google Scholar]
- Witten I, Frank E. Data Mining: Practical Machine Learning Tools and Techniques with Java Implementations. San Francisco, USA: Morgan Kaufmann; 2005. [Google Scholar]
- Zhang H, et al. Cell and tumor classification using gene expression data: construction of forests. Proc. Natl Acad. Sci. USA. 2003;100:4168–4172. doi: 10.1073/pnas.0230559100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.