

Abstract
Summary: The advent of next-generation sequencing for functional genomics has given rise to quantities of sequence information that are often so large that they are difficult to handle. Moreover, sequence reads from a specific individual can contain sufficient information to potentially identify and genetically characterize that person, raising privacy concerns. In order to address these issues, we have developed the Mapped Read Format (MRF), a compact data summary format for both short and long read alignments that enables the anonymization of confidential sequence information, while allowing one to still carry out many functional genomics studies. We have developed a suite of tools (RSEQtools) that use this format for the analysis of RNA-Seq experiments. These tools consist of a set of modules that perform common tasks such as calculating gene expression values, generating signal tracks of mapped reads and segmenting that signal into actively transcribed regions. Moreover, the tools can readily be used to build customizable RNA-Seq workflows. In addition to the anonymization afforded by MRF, this format also facilitates the decoupling of the alignment of reads from downstream analyses.
Availability and implementation: RSEQtools is implemented in C and the source code is available at http://rseqtools.gersteinlab.org/.
Contact: lukas.habegger@yale.edu; mark.gerstein@yale.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
The advent of next-generation sequencing technologies has revolutionized the study of genomes and transcriptomes. In particular, the application of deep sequencing approaches to transcriptome profiling (RNA-Seq) is increasingly becoming the method of choice for studying the transcriptional landscape of cells (Hillier et al., 2009; Mortazavi et al., 2008; Wang et al., 2009). Typically, the first step in this analysis is the alignment of the sequence reads to a reference sequence set. Recently, a number of different alignment tools have been developed to map short reads in an efficient manner (Trapnell and Salzberg, 2009). While much progress has been made on this front, there is still a great need for a set of software tools that facilitate the downstream analysis of mapped RNA-Seq reads.
Further, two other issues remain to be addressed. First, the immense file size of next-generation sequencing data poses many challenges in terms of data processing, storage and sharing. Secondly, mechanisms to protect personal confidential genetic information need to be established. With the birth of personal genomics, sequencing data stems fundamentally from individuals, and this type of data cannot be distributed as easily because significant privacy concerns arise with sharing all the sequence variations of a particular individual (Greenbaum et al., 2008; Lowrance and Collins, 2007). One critical challenge for genomics, then, is to devise new data summaries that allow the sharing of large amounts of information from sequencing experiments without exposing the genotypic information of the underlying individual (Supplementary Material).
Although many data formats have been developed such as SAM (Li et al., 2009), there is no practical solution yet that addresses the privacy concerns when sharing large sequence alignment files. Addressing this challenge is precisely what we have endeavored to do in putting together the Mapped Read Format (MRF), a format that allows data summaries to be exchanged, enabling many aspects of the RNA-Seq calculation to be performed such as expression measurements, but that also detaches the actual sequence variation in a person into separate files. Further, it provides a very clear way of linking these two pieces of information so that the data summaries can be subsequently conjoined back to the original sequences for more in-depth analyses with potentially confidential data.
Here, we present an overview of a flexible suite of tools (RSEQtools) that are designed to facilitate easily customizable workflows and efficient pipeline building for the analysis of RNA-Seq experiments using this compact format (Fig. 1). Briefly, we first convert the aligned reads into MRF and thus decouple the alignment step from the downstream analyses. RSEQtools implements several modules using this standardized format for performing common RNA-Seq analyses, such as expression quantification, discovery of transcribed regions, coverage computations annotation manipulation, etc.
Fig. 1.

Schematic overview of RSEQtools. Mapped reads are first converted into MRF from common alignment tool output formats, including SAM. The resulting MRF files can be divided in two files: one with the alignment only and another with the corresponding sequence reads. The read identifiers provide a mapping between the two files. Then, several modules perform the downstream analyses independently from the mapping step, such as expression quantification, visualization of the mapped read and the calculation of annotation statistics, etc. Other tools have been developed based on this framework to perform more sophisticated analyses such as transcript assembly, isoform quantification (IQSeq, http://rnaseq.gersteinlab.org/IQSeq), fusion transcript identification (FusionSeq, http://rnaseq.gersteinlab.org/fusionseq), as well as aggregation and correlation of signal tracks (ACT, http://act.gersteinlab.org).
2 FEATURES AND METHODS
2.1 MRF and converters
MRF only stores a minimal set of information, i.e. information that cannot be derived from the MRF data itself. This has the advantage of keeping the format succinct, while still capturing the relevant information for most analyses. MRF consists of three components: comment lines (optional) denoted by a leading ‘#’ sign, a header line and the mapped reads. The header line specifies the data type of each column: AlignmentBlocks, Sequences, QualityScores and QueryID. The column type AlignmentBlocks is required and represents the mapped reads. Each alignment block contains the coordinates with respect to the reference genome to which the read aligns as well as the read coordinates. A read spanning multiple regions, e.g. multiple exons, is denoted by multiple alignment blocks that are separated by a comma. Paired-end reads can be represented by using a set of alignment blocks for each end, which are separated by the ‘|’ symbol. By using this format, it is straightforward to specify both gapped and paired-end alignments. The RSEQtools package includes various utilities to convert the output of several mapping tools into MRF. A converter for the commonly used SAM format is included as well. The first example below represents two paired-end reads where one end is spliced, whereas the second example shows two un-spliced single-end reads with their associated QueryIDs:
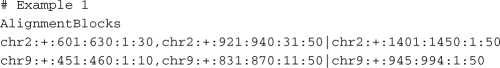

The optional types Sequences, QualityScores and QueryID provide additional information. In particular, the confidentiality issues can be addressed by generating two files: one including the alignments and a second one containing the sequences such as a FASTQ file. The former is useful for most analyses and can be publicly shared because it does not contain confidential information, whereas the latter can be subjected to a higher level of security and control. The two files can be conjoined, if necessary, by using the common QueryID as shown in Figure 1.
2.2 RNA-Seq analysis with RSEQtools
The RSEQtools suite contains a set of modules to perform a large variety of tasks including the quantification of expression values, manipulation of gene annotation sets, visualization of the mapped reads, generation of signal tracks, the identification of transcriptional active regions and several auxiliary utilities (Supplementary Table S1).
Genome annotation tools: to generate a splice junction library from any annotation set, we extract the genomic sequences of all the exons and synthetically create all splice junctions specified in the annotation set. This splice junction library can be used in combination with the reference sequences. A second tool is particularly useful when estimating expression levels. In order to capture the information of the various transcript isoforms, a ‘gene model’ is required. The module mergeTranscripts collapses the transcript isoforms into a single gene model by either taking the union or intersection of the exonic nucleotides.
Quantification of gene expression: one of the key features of RNA-Seq is the quantification of expression at different levels. Hence, a key module calculates the gene expression values for a given annotation set and a collection of mapped reads in MRF format. The annotation set specifies which ‘elements’ will be quantified. The program mrfQuantifier calculates RPKM (reads per kilobase per million mapped reads) values at the nucleotide level (Mortazavi et al., 2008). Briefly, for a given entry in the annotation set (typically an exon or gene model), the number of nucleotides from all the reads that overlap with this annotation entry are added up and then this count is normalized by sequence length of the annotation entry (per killobase) and by the total number of mapped nucleotides (per million). This calculation is not performed at the transcript level, which requires a more sophisticated analysis (Guttman et al., 2010; Trapnell et al., 2010).
Visualization of mapped reads: the RSEQtools package also contains various tools for visualizing the results in genome browsers, by means of wiggle (WIG) and bedGraph files, which are commonly used to represent a signal track of mapped reads. Also, a GFF file can be generated from MRF files to visualize splice junction reads (example in Fig. 1).
Identification of transcriptionally active regions (TARs): transcribed regions can be identified de novo by performing a maxGap/minRun segmentation (Kampa et al., 2004; Royce et al., 2005) from the signal files using the wigSegmenter program. Briefly, the signal is first thresholded to identify transcribed elements. Contiguous elements whose distance is less than ‘maxGap’ are joined together and then filtered if the final size is less than ‘minRun’. This type of analysis is particularly useful in discovering novel TARs such as small RNAs, etc.
MRF selection and auxiliary utilities: lastly, RSEQtools includes a set of utilities to easily manipulate MRF files and a collection of format conversion tools allowing for rapid pipeline development.
Implementation and run time: the modules of the RSEQtools suite were implemented in C and the code was optimized in order to efficiently handle large datasets. The importance of code scalability cannot be overemphasized in a time where datasets become increasingly large and easily exceed several gigabytes. For example, the conversion of an ELAND export file (uncompressed file size: ∼4 GB; total number of reads: ∼20 million; number of mapped reads: ∼12 million) to MRF takes ∼2 min and the resulting MRF file is significantly smaller (∼400 MB uncompressed, ∼130 MB compressed with gzip). Converting the same ELAND export file to SAM generates a file of ∼3.1 GB (uncompressed) and the corresponding BAM file has a size of ∼1.2 GB. The subsequent quantification of gene expression using mrfQuantifier requires 45 s to calculate estimates for about 20 000 genes.
In addition, the modularity of RSEQtools also enables the development of additional programs in any programming language and their seamless integration into this framework. Finally, most modules use STDIN and STDOUT to process the data, making them suitable to be integrated into an automated pipeline.
3 CONCLUSIONS
In summary, RSEQtools contains a number of useful and highly specific modules that can rapidly analyze RNA-Seq data. The MRF format has two major features: it allows the decoupling of downstream analysis from the mapping strategy and addresses the issue of confidentiality that is intrinsic in any sequencing experiments involving human subjects. By separating the actual sequencing reads from the alignments, MRF provides a mechanism to protect the private genotypic information of the underlying individual. Although this approach removes the most obvious genotypic features, other distinctive attributes do remain. First of all, the information in a MRF file is at least equivalent to that in traditional expression array, which can potentially identify the underlying individual. Secondly, some information about structural variants may be contained in the MRF file of an RNA-Seq experiment. However, it is not obvious how to extract genotypic information from a subset of structural variations just affecting genes. In addition, inferring structural variations from RNA-Seq data as opposed to DNA sequencing would be more complicated due to the presence of alternative splicing.
Another advantage of storing the alignments without the underlying sequences is that it saves space, especially as reads become longer. Moreover, a possible future extension is the development of a specific compression schema that could further reduce the size of the files. In addition, this data format could be easily applied to sequence alignments obtained from other high-throughput functional genomic assays such as ChIP-Seq or chromosome conformation capture (3C).
Supplementary Material
ACKNOWLEDGEMENT
We thank Raymond Auerbach for critical reading and editing as well as Wasay Hussain for testing of the software.
Funding: National Institutes of Health.
Conflict of Interest: none declared.
REFERENCES
- Greenbaum D, et al. Genomic anonymity: have we already lost it? Am. J. Bioeth. 2008;8:71–74. doi: 10.1080/15265160802478560. [DOI] [PubMed] [Google Scholar]
- Guttman M, et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 2010;28:503–510. doi: 10.1038/nbt.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier LW, et al. Massively parallel sequencing of the polyadenylated transcriptome of C.elegans. Genome Res. 2009;19:657–666. doi: 10.1101/gr.088112.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampa D, et al. Novel RNAs identified from an in-depth analysis of the transcriptome of human chromosomes 21 and 22. Genome Res. 2004;14:331–342. doi: 10.1101/gr.2094104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowrance WW, Collins FS. ETHICS: identifiability in genomic research. Science. 2007;317:600–602. doi: 10.1126/science.1147699. [DOI] [PubMed] [Google Scholar]
- Mortazavi A, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Method. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Royce TE, et al. Issues in the analysis of oligonucleotide tiling microarrays for transcript mapping. Trends Genet. 2005;21:466–475. doi: 10.1016/j.tig.2005.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Salzberg SL. How to map billions of short reads onto genomes. Nat. Biotechnol. 2009;27:455–457. doi: 10.1038/nbt0509-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, et al. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.