

Synopsis
Drug-induced movement disorders have dramatically declined with the widespread use of second generation antipsychotics but remain important in clinical practice and for understanding antipsychotic pharmacology. The diagnosis and management of dystonia, parkinsonism, akathisia, catatonia, neuroleptic malignant syndrome and tardive dyskinesia are reviewed in relation to the decreased liability of the second generation antipsychotics contrasted with evidence from the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Schizophrenia Trial. Data from the CATIE trial imply that advantages of second generation antipsychotics in significantly reducing extrapyramidal side effects compared with haloperidol may be diminished when compared with modest doses of lower-potency first generation drugs, that the dichotomy between first and second generation drugs may be oversimplified, and that antipsychotics could be conceptualized as a single drug class with a spectrum of risk for movement disorders depending upon receptor binding affinities and individual patient susceptibility.
Keywords: antipsychotic drugs, schizophrenia, tardive dyskinesia, catatonia, neuroleptic malignant syndrome, akathisia, parkinsonism, dystonia
Introduction
From its inception, the pharmacology of antipsychotic drugs was inextricably linked to the movement inhibiting or “neuroleptic” properties of these agents. 1 Chlorpromazine and other early antipsychotics were originally thought to be useful primarily for calming psychomotor excitement rather than specific antipsychotic effects. 2 Parkinsonism and other extrapyramidal side effects (EPS) were observed frequently but considered to be indicators that therapeutic doses of antipsychotics had been achieved.
However, it soon became clear that EPS can be mistaken for or worsen psychotic symptoms, are sometimes irreversible or lethal, necessitate additional burdensome side effects from antiparkinsonian agents, can be disfiguring and stigmatizing, and have been shown to influence compliance, relapse and rehospitalization. 2-4 As a result, EPS dominated concerns about tolerability of antipsychotic drugs and fueled efforts for new drug research and development.
In 1988, Kane et al. reported that clozapine had broader efficacy in schizophrenia with negligible EPS, stimulating the search for other second generation antipsychotics (SGAs) with less toxicity.5 Industry-sponsored clinical trials suggested that newer SGAs were superior to first generation antipsychotics (FGAs) in reducing psychotic symptoms and causing fewer EPS. 6-13 Cumulative and convincing evidence confirming reduced liability for EPS with SGAs contributed to their market dominance and fostered the concept of “atypicality” in their mechanism of action. 14-21
Subsequent studies raised questions about the advantages of SGAs seen in earlier trials. Although haloperidol was a very reasonable choice as a comparator in industry-sponsored trials because of its widespread use in clinical practice, new evidence suggested that the advantages of SGAs in reducing EPS were diminished when lower doses or lower-potency FGAs are used, or if prophylactic antiparkinsonian drugs are administered.19, 22-27
In view of these conflicting findings, the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Schizophrenia Trial offered an opportunity to address the liability for movement disorders between first and second generation antipsychotics.28-30 The strengths of the CATIE study include its large sample size, diverse representation of clinical settings, independence from industry sponsorship, and a well-controlled, double-blind, head-to-head treatment comparison. The rationale, design, methods and statistical analysis of the CATIE trial have been described previously.28 Briefly, CATIE was designed to address the overall effectiveness between four SGAs (olanzapine, risperidone, quetiapine and ziprasidone) and a mid-potency FGA (perphenazine) including the influence of specific EPS on tolerability and effectiveness.
Although EPS have been extensively reviewed, here we provide an update on diagnosis and management with insights derived from the CATIE trial. In addition, we cover the often neglected movement disorders of drug-induced catatonia and neuroleptic malignant syndrome (NMS).
Dystonia
Dystonia is an acute, alarming involuntary movement disorder that can be painful and distressing, and erodes patient trust and compliance. 31 It is characterized by briefly sustained or intermittent spasms or contractions of antagonistic muscle groups resulting in twisting and repetitive movements or postures. Drug-induced dystonia can affect any muscle group, but most commonly involves the head, neck, jaw, eyes and mouth resulting in spasmodic torticollis, retro- or anterocollis, trismus and dental trauma, forced jaw-opening or dislocation, grimacing, blepharospasm, tongue biting, protrusion or twisting, and distortion of the lips.32-34 It is not action or sensory stimulus dependent. More subtle signs, including muscle cramps or tightness of the jaw and tongue with difficulty speaking or chewing, may precede dystonia or occur alone. At the other extreme, dystonia may present as an oculogyric crisis or with other forced eye movements, or with dysarthria, dysphagia and potentially lethal respiratory stridor if pharyngeal or laryngeal musculature is affected. Less frequently observed, dystonia may affect axial, truncal or limb movements occasionally leading to camptocormia, pleurothotonus (Pisa syndrome) or opisthotonus.
The differential diagnosis of dystonia is extensive including primary genetic disorders and secondary forms including neurodegenerative disorders, structural abnormalities of the brain, and metabolic and toxic etiologies.34-37 Drug-induced dystonia is distinguished by recent antipsychotic treatment, negative family history, focal and non-progressive course, and absence of associated neurologic signs. The co-occurrence of dystonia or other EPS with behavioral disturbances narrows the differential to disorders associated with both features, e.g., Huntington’s and Wilson’s disease, neuroacanthocytosis, etc. Although most often associated with the initiation of antipsychotics, dystonia may also be seen if dosages are increased, a second antipsychotic is added, for the few days each time after long-acting injectable antipsychotics are administered, 34 if another drug, e.g., paroxetine, is added that inhibits antipsychotic metabolism, or following the discontinuation of antiparkinsonian agents. Dystonia also occurs in a tardive form, which may respond to anticholinergic treatment, but first appears or worsens rather than resolves when antipsychotic drugs are discontinued. The relationship between the acute and tardive forms is unknown. Apart from antipsychotic drugs, dopamine antagonists used as anti-emetics or sedatives (prochlorperazine, metoclopramide), anticonvulsants, antimalarials, stimulants and serotonergic agents have been implicated. Dystonias are encountered with levodopa during the course of treatment of Parkinson’s disease. Toxins (manganese, carbon monoxide, carbon disulfide) have also been implicated. Finally, dystonia was erroneously ascribed to psychogenic factors for much of the twentieth century, and patients with drug-induced dystonia still may be dismissed as hysterical if clinicians are unaware that it is often intermittent and fluctuates with stress and relaxation. However, some patients may feign symptoms to avoid taking antipsychotic drugs or to obtain anticholinergic drugs for euphoric effects.33
Dystonia is the earliest EPS to be seen after antipsychotic drug administration. Usually observed within a few hours of a single dose especially following parenteral administration, dystonias may appear after a delay of several hours to a few days. 32 In 95% of cases, dystonia appears within the first five days of treatment after a drug is started or increased in dosage. 32, 33 Dystonic reactions last a few seconds or several hours and are often recurrent even after a single drug dose. After drug discontinuation, dystonia resolves within 24-48 hours. 34
Dystonia is less frequent than parkinsonism or akathisia, occurring in 2% to 5% of patients generally. 32-34 However, in young men receiving high potency antipsychotics parenterally, the frequency approaches 90% in some series. 32
Patient risk factors for dystonia include age, male sex, race, previous dystonic reactions, family history of dystonia, cocaine use, mood disorders, hypocalcemia, hypoparathyroidism, hyperthyroidism and dehydration.33, 34, 38 Children and young adults are highly vulnerable, whereas drug-induced dystonia is rare over 45 years of age. 33
Drug dosage and potency have been associated with risk of dystonia.33, 34, 39 Moderate to high doses of antipsychotics are associated with dystonia, whereas low or very high doses are less often involved. 38 Antipsychotics with weak dopamine antagonism and prominent anticholinergic effects diminish the risk of dystonia, while the newer SGAs appear to have reduced liability as well. 14, 15 In a sample of published industry-sponsored trials, haloperidol had up to four times greater risk of causing dystonia than SGAs, 6, 8, 40 with clozapine unlikely to cause it at all.
There were only six cases of acute dystonia in the CATIE trial (6/1460 or 0.4%) that were not present at baseline, four of which resulted in treatment discontinuation. Of these six cases, none were receiving olanzapine, one was receiving perphenazine, one was receiving quetiapine, one was receiving risperidone, and three were receiving ziprasidone. 30 The CATIE data suggest that in older patients with chronic schizophrenia, use of a mid-potency FGA at modest doses presents no greater risk for dystonia than SGAs.
Moreover, Satterthwaite et al. performed a meta-analysis of studies comparing intramuscular haloperidol versus three injectable SGAs, and found that while the SGAs were significantly less likely to induce dystonia, this advantage was abolished when an anticholinergic agent was administered with haloperidol. 31 However, the use of prophylactic anticholinergic medications to prevent dystonia had been controversial prior to the introduction of the SGAs due to atropinic side effects. But in young or other high risk patients receiving parenteral, high potency antipsychotics, or in paranoid or other patients ambivalent about treatment, the benefits of preventing a dystonic reaction far outweigh potential risks. 41
Dystonia is responsive within 10 to 20 minutes to anticholinergic or antihistaminic agents administered intramuscularly and repeated if necessary. Intravenous administration is necessary only in an emergency, e.g., for laryngeal stridor. Benzodiazepines have been used in some cases. If response is not achieved, a search for underlying disorders, e.g., hypocalcemia, should be conducted or tardive dystonia considered.42 After dystonia is suppressed, oral anticholinergics are continued for 24 to 48 hours if the antipsychotic is discontinued, or for at least several days if antipsychotic treatment is continued with gradual tapering to prevent recurrence. However, at-risk patients who tolerate anticholinergic drugs may need continued prophylaxis.
The pathophysiology of drug-induced dystonia is unknown. 32, 35, 43 It remains unclear whether excessive dopaminergic activity from a compensatory increase in turnover following drug-induced receptor blockade causes dystonia as antipsychotic drug levels diminish, or whether dystonia results from dopamine antagonism per se. 32 Recent clarification of the genetics of primary dystonia may shed light on mechanisms of drug-induced forms in light of some reports suggesting familial predisposition. 34, 37
Parkinsonism
Drug-induced parkinsonism is a subacute syndrome that mimics Parkinson’s disease. Though less alarming than dystonia, it is more common, more difficult to treat and can be the cause of significant disability during maintenance treatment especially in the elderly. Bradykinesia is accompanied by masked facies, reduced arm swing, slowed initiation of activities, soft speech and flexed posture. 32 Bilateral and symmetrical rigidity of neck, trunk and extremities appears with cog-wheeling. Resting or action tremors are also observed symmetrically and can be generalized or take the form of a focal peri-oral tremor (rabbit syndrome). Patients may also experience sialorrhea, and postural or gait disturbances.
It is important to differentiate drug-induced parkinsonism from negative or withdrawal symptoms of schizophrenia and psychomotor retardation associated with depression. It can be difficult to distinguish drug-induced from idiopathic Parkinson’s disease. Parkinson’s disease is usually asymmetric in symptom presentation, has a slow progressive course, may have associated dysautonomia, should precede treatment and not resolve after antipsychotic drugs are discontinued, and may show nigrostriatal degeneration on dopamine transporter scans and sympathetic dysregulation on 123IMIBG cardiac scintigraphy. 44 Interestingly, olfactory deficits have recently been reported in drug-induced parkinsonism.45 The differential diagnosis also includes essential tremor and other causes of parkinsonism, including vascular parkinsonism which also tends to be asymmetric, and parkinsonism caused by other drugs including anti-emetics, tetrabenazine, and reserpine.
Although dopamine receptor blockade occurs within a few hours after administration of antipsychotic drugs, the onset of parkinsonism may be delayed from days to weeks, with 50% to 75% of cases occurring within one month and 90% within three months. 32 Parkinsonism may also occur after doses are increased, a second antipsychotic is added, anticholinergic drugs are discontinued, or another drug is added that reduces central dopamine activity or increases plasma levels of the antipsychotic. In most cases, symptoms are reversible in days or weeks, but occasionally, especially in the elderly, or if long-acting injectable antipsychotics are used, symptoms may last for months. In about 15% of cases, parkinsonism may persist, raising the possibility of underlying Parkinson’s disease. 44
The incidence of drug-induced parkinsonism is variable depending upon risk of the population studied, sensitivity of diagnosis and potency of the drugs used, but has been estimated to occur in at least 10 to 15% of patients treated in routine practice with FGAs. 32 It is the second most common cause of parkinsonism after Parkinson’s disease. The risk of drug-induced parkinsonism has been associated with increasing age, female gender, dementia, HIV infection, and pre-existing extrapyramidal disease or family history of Parkinson’s disease.32, 44, 46
Although parkinsonism has correlated with increased dosages, potency and reduced anticholinergic properties of antipsychotics,47 dose response relationships have not always been clear in view of differences in individual susceptibility. In industry-sponsored trials, haloperidol has been associated with two to four times the risk of parkinsonism compared with SGAs (22% to 38% versus 4% to 14%). 6, 8, 14, 15, 40, 48, 49 But judging by severe motor worsening experienced by patients with Parkinson’s disease after receiving antipsychotics, even SGAs may cause significant parkinsonism in susceptible individuals except for clozapine and quetiapine. 14, 15 Patients with Lewy body dementia may experience a potentially lethal “neuroleptic sensitivity syndrome” when exposed to FGAs or SGAs.
In contrast to previous studies using haloperidol as the representative FGA, there were no significant differences in the CATIE trial between perphenazine and SGAs in the proportion of patients exhibiting parkinsonism defined as a mean score of one or more on the Simpson-Angus Extrapyramidal Signs Scale (SAS) (Table 1). 28, 50 However, significantly more patients discontinued perphenazine (8%) than SGAs (2% to 4%) as a result of all EPS combined, and perphenazine (10%) had a significantly higher rate of concomitant anticholinergic drug use relative to SGAs (3% to 9%). Quetiapine itself was associated with a higher rate of anticholinergic side effects (31% versus 20 to 25%), and conversely, had the lowest rate of concomitant anticholinergic drugs.
Table 1.
Initial analysis of CATIE outcome measures related to EPS 28
| Outcome | Olanzapine | Quetiapine | Risperidone | Perphenazine | Ziprasidone | P Value |
|---|---|---|---|---|---|---|
| Discontinuations due to extrapyramidal symptoms*; | ||||||
| 8/336 (2) | 10/337 (3) | 11/341 (3) | 22/261 (8) | 7/185 (4) | 0.002 | |
| Extrapyramidal side effects*‡; | ||||||
| Parkinsonism | 16/240 (7) | 10/247 (4) | 20/238 (8) | 15/243 (6) | 6/129 (5) | 0.50 |
| Akathisia | 12/234 (5) | 12/248 (5) | 12/240 (6) | 16/241 (7) | 13/132 (10) | 0.19 |
| TD | 32/236 (14) | 30/236 (13) | 38/238 (16) | 41/237 (17) | 18/126 (14) | 0.23 |
| Anticholinergic side effects*€; | ||||||
| 79/336 (24) | 105/337 (31) | 84/341 (25) | 57/261 (22) | 37/185 (20) | <0.01 | |
| Anticholinergic medications added*; | ||||||
| 25/336 (7) | 11/337 (3) | 32/341 (9) | 26/261 (10) | 14/185 (8) | 0.01 | |
Number/total number of patients (%);
Parkinsonism percentages= the number of patients with an SAS mean score ≥1, with a mean score <1at baseline and at least one post-baseline assessment; Akathisia percentages= the number of patients with a BAS global score ≥3, with a global score <3 at baseline and at least one post-baseline assessment; TD percentages= the number of patients with an AIMS global score ≥2, with a global score <2 at baseline and at least one post-baseline assessment. Patients with TD at baseline were excluded from all EPS assessments.
Urinary hesitancy, dry mouth, constipation
From Caroff SN, Miller, D.D., Rosenheck, R.A. Extrapyramidal side effects. In: Stroup TS, Lieberman, J.A., ed. The Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Schizophrenia Trial: How does it inform practice, policy, and research? Cambridge: Cambridge University Press; 2010:156-172, with permission pending.30
In a more sensitive analysis of the CATIE data, examination of the proportion of patients showing no evidence of parkinsonism at baseline who met at least one of three criteria for parkinsonism (scored at least one moderate symptom or two mild symptoms on the SAS scale, treatment discontinuation for parkinsonism, treatment with antiparkinsonian medication) revealed again no significant differences between treatment groups (Table 2). 29, 30 Rates of parkinsonism were 37 to 44% among the four SGAs and 37% for the FGA perphenazine.
Table 2.
Second analysis of observed EPS events for patients without the events at baseline29
| Extrapyramidal event | Olanzapine | Quetiapine | Risperidone | Perphenazine | Ziprasidone |
|---|---|---|---|---|---|
| Any parkinsonian event | 70/201 (35)* | 55/187 (29) | 71/191 (37) | 48/160 (30) | 31/98 (32) |
| Any akathisia event | 52/238 (22) | 42/250 (17) | 61/244 (25) | 51/207 (25) | 26/130 (20) |
| Tardive dyskinesia (S-K) | 2/182 (1) | 8/179 (5) | 4/179 (2) | 6/183 (3) | 3/89 (3) |
| Tardive dyskinesia (mS-K) | 20/216 (9) | 19/222 (9) | 21/220 (10) | 26/221 (12) | 10/120 (8) |
Number/total number of patients without the extrapyramidal symptom at baseline (%)
Any parkinsonian event includes meeting SAS score criteria, discontinuing treatment or adding a medication for parkinsonism.
Any akathisia event includes meeting BAS score criteria, discontinuing treatment or adding a medication for akathisia.
S-K= Schooler Kane criteria; mS-K= Modified Schooler Kane criteria requiring only one post-baseline assessment. 90
From Caroff SN, Miller, D.D., Rosenheck, R.A. Extrapyramidal side effects. In: Stroup TS, Lieberman, J.A., ed. The Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Schizophrenia Trial: How does it inform practice, policy, and research? Cambridge: Cambridge University Press; 2010:156-172, with permission pending.30
Prophylaxis of parkinsonism with anticholinergic drugs is less compelling than for dystonia and introduces significant risk of atropinic toxicity. Given the delayed onset, close monitoring for parkinsonian symptoms with prompt consideration of lowering dosages or switching to lower risk antipsychotics takes precedence, albeit with attendant risk of relapse. If a given antipsychotic is effective and cannot be changed, and if parkinsonism persists, treatment may include anticholinergic drugs or amantadine. However, there is limited controlled evidence for the use of these agents. 42 Specific dopaminergic therapy is ineffective, due to ongoing drug-induced blockade of dopamine receptors, and raises the risk of worsening psychotic symptoms. Once patients have been maintained on adjunctive antiparkinsonian therapy for three to six months, cautious tapering may be attempted.32 However, several studies have shown 62% to 96% of patients may experience worsening parkinsonism following antiparkinsonian drug discontinuation. 51
The mechanisms underlying drug-induced parkinsonism parallel Parkinson’s disease. Antipsychotics induce a functional dopamine deficiency in the corpus striatum by blocking dopamine receptors. Their liability for inducing parkinsonism therefore is the product of dopamine receptor binding affinity balanced by affinity for blocking muscarinic cholinergic receptors. 47 Several causative genes and susceptibility factors for Parkinson’s disease have been identified recently implicating various proteinopathies, but these have not been studied in drug-induced variants. 52
Akathisia
Akathisia is another common EPS. 32, 46, 53-55 Akathisia is distinct in being defined by subjective as well as objective features, more often affecting the lower extremities, remaining a problem even with SGAs, and being less responsive to treatment. Subjectively, patients complain of inner tension, restlessness, anxiety, urge to move and inability to sit still, and drawing sensations in the legs. Observable motor features are complex, semi-purposeful and repetitive, including foot shuffling or tapping, shifting of weight, rocking, pacing incessantly and even running. Although the severity of these sensations varies with stress and arousal, they can become intolerable and have been associated with violence and suicide. 53, 55
Acute drug-induced akathisia must be distinguished from tardive akathisia, neurodegenerative conditions, and drug intoxication and withdrawal states. Restlessness can be observed with other medications including serotonin uptake inhibitors, calcium channel blockers, anti-emetics, anti-vertigo agents or sedatives. Akathisia resembles restless legs syndrome (Ekbom’s syndrome) which occurs during relaxation, rest or sleep mostly in the evening or night. Misdiagnosis of restless legs syndrome can lead to inappropriate prescription of dopamine agonists which could worsen psychosis leading to increased antipsychotic use further compounding akathisia. 54 Finally, distinguishing akathisia from agitation and anxiety is challenging but crucial in determining whether to continue antipsychotic treatment.
Akathisia may begin within several days after treatment but usually increases with duration of treatment, occurring in up to 50% of cases within one month and 90% of cases within three months. 32 Akathisia should resolve after drug discontinuation, but could temporarily worsen or persist in withdrawal or tardive forms.
Estimates of the incidence of akathisia vary between 21% to 75% across studies of FGAs, with an estimated prevalence on average of at least 20% to 35% of patients in routine practice, depending upon the susceptibility of the sample population, sensitivity of diagnosis, and the potency of drug treatment. 53
Patient risk factors for akathisia have not been well established, but increasing age, female sex, negative symptoms, cognitive dysfunction, iron deficiency, prior akathisia, concomitant parkinsonism, and mood disorders may entail greater risk. 54, 55 In most but not all clinical trials, SGAs have resulted in a significantly lower incidence of akathisia compared with FGAs. 53 In a representative sample of trials, akathisia developed at an incidence rate of about two to seven times higher with haloperidol (15% to 40%) compared with SGAs (0% to 12%). 6, 8, 14, 15, 40, 48, 49
In the CATIE study, there were no significant differences among treatment groups in the incidence of akathisia defined by scores of three more on the global assessment of the Barnes Akathisia Rating Scale (BAS) (Table 1). 28, 56 On subsequent analysis, examination of the proportion of patients showing no evidence of akathisia at baseline who met at least one of three criteria for akathisia (scored at least mild symptoms on the BAS global item, akathisia given as the reason for starting any medication, or discontinued antipsychotic medication due to akathisia) revealed no significant differences between treatment groups (Table 2). 29, 30 Rates of akathisia ranged from 26% to 34% for the SGAs and 35% for perphenazine.
There are no data on prophylaxis and given its subacute onset, close observation for early signs is the best preventive measure. Once developed, akathisia should prompt reassessment of antipsychotic therapy, with reduction in dosage, discontinuation, or switching to a less potent dopamine antagonist, all of which incur the risk of psychotic exacerbation or relapse. Evidence of treatments derives mostly from small, short-term clinical trials without active comparative groups. 55, 57 Beta-adrenergic blockers that are lipophilic and target beta2-receptors have been effective in some studies though limited by hypotension, bradycardia and medical contraindications. Anticholinergics have been used, but are felt to be most effective in the presence of concomitant parkinsonism. Benzodiazepines have been useful due to their anxiolytic and sedative properties. Amantadine may be effective in some cases. Recently, 5-HT2A receptor antagonists have attracted interest, with mirtazepine showing equal efficacy in treating akathisia compared to propranolol with better tolerability.55
The pathophysiology of akathisia remains unknown but dopamine antagonism underlying antipsychotic-induced akathisia, and treatment of restless legs syndrome with dopamine agonists, underscore the importance of dopamine-dependent mechanisms. Responses to anticholinergics, and beta-adrenergic and serotonergic blockers, suggest a role for other neurotransmitters as well. Apart from exploratory reports of dopamine receptor polymorphisms in drug-induced akathisia, there have been preliminary studies of genetic predisposition to restless legs syndrome which is highly familial and may shed light on underlying mechanisms. 58, 59
Catatonia
Catatonia is the least recognized movement disorder associated with antipsychotic drugs. The neglect is not surprising, given the lack of consensus on the definitions of behaviors considered catatonic and the boundaries of the syndrome itself.60 Jaspers defined catatonia as constituting “psychomotor” behaviors that fall between those with clear motor origins on the one hand, e.g., tremor, and behaviors stemming from known psychological states, e.g., hyperactivity in mania.61 Catatonia is conceptualized as a syndrome comprised of disorders of movement, speech and volition.
Catatonic symptoms that have been associated with antipsychotics include akinesia, stupor, and mutism (akinetic mutism), and less often catalepsy and waxy flexibility.62, 63 The more complex and qualitative catatonic behaviors typical of schizophrenia (stereotypies, echophenomena, verbigeration, automatic obedience) are rarely seen in drug-induced cases. In an unknown percentage of patients, antipsychotics could transform pre-existing catatonia into a more malignant form, consistent with NMS. 64, 65
The differential diagnosis of catatonia includes a broad range of neurodegenerative, developmental, metabolic, toxic, infectious and structural conditions affecting brain function.66 Catatonia overlaps and may be obscured by drug-induced parkinsonism. Catatonia also occurs in schizophrenia and mood disorders leading to the conundrum of the “catatonic dilemma”,67 in which it may be difficult to distinguish catatonia due to the psychiatric disorder requiring antipsychotic drugs from the treatment itself. This is best resolved by discontinuing antipsychotics leading to resolution of the iatrogenic cases.
Acute catatonia develops within hours to days after drug exposure and is expected to resolve in a similar period of time after drug discontinuation. In some cases, e.g., after resolution of NMS, mixed catatonic/parkinsonian features may persist for weeks or even months if not treated with electroconvulsive therapy (ECT).68 The onset of catatonia has been reported to occur with antipsychotics after discontinuation of benzodiazepines or antiparkinsonian agents. The incidence of drug-induced catatonia is unknown. Stubner et al reported only five cases of “catatonic neuroleptic syndrome” among 86,439 patients receiving antipsychotics in a retrospective drug surveillance program.69
Patient-related risk factors for drug-induced catatonia include past episodes and pre-existing catatonic symptoms observed in schizophrenia or mood disorders. Catatonia is mostly observed in association with high potency drugs.63 The relative liability between FGAs and SGAs has not been studied. However, SGAs are not without risk considering published case reports documenting both occurrence or worsening of catatonia with the newer drugs.14 However, SGAs have also been proposed as treatments for catatonia.70 In animal models, SGAs show an advantage over FGAs in reducing the risk of catalepsy,14 raising the possibility that these newer agents would be less likely to cause catatonia, and may be particularly useful in patients with pre-existing risk of catatonia. In fact, use of SGAs in trials that included patients with catatonic schizophrenia did not result in adverse effects in this subgroup and was beneficial in two of the studies. 14
There are no data on whether prophylaxis with benzodiazepines or other agents would prevent catatonia in vulnerable patients. A more conservative approach would be to avoid using antipsychotics and specifically treat pre-existing catatonia with benzodiazepines or ECT in patients presenting with the syndrome. For patients at risk based on past history, clinicians should consider using other agents for the underlying psychiatric disorder, e.g., lithium for mania. However, patients who require antipsychotic treatment merit careful observation and monitoring for incipient symptoms. Treatment of drug-induced catatonia has not been rigorously studied, but must include prompt discontinuation of the offending agent as patients with immobility and non-responsiveness are at high risk for medical complications, or may progress to NMS. Specific treatment includes benzodiazepines, but recent evidence has reinforced the utility of amantadine or memantine.63, 71 Use of direct dopaminergic agonists have not been studied. Patients who fail to respond to these measures may benefit from ECT, which offers the advantage of resolving both catatonia and the underlying psychosis or mood disorder.
The pathophysiology of drug-induced catatonia is unknown, but may involve drug effects on parallel dopamine pathways in basal ganglia-thalamocortical circuits subserving motor, arousal, volitional and imitative behaviors.72 The efficacy in anecdotal reports of benzodiazepines, amantadine and memantine suggest potential interactions with GABA-ergic and glutamate pathways. Several investigators have studied the genetic underpinnings of different types of catatonia which may correspond to drug sensitivity,73, 74 while Kanes has reported intriguing findings of genetic influences on sensitivity to drug-induced catatonia in strains of mice, mapped to chromosomes 4, 9 and 15.75
Neuroleptic malignant syndrome
NMS represents an extremely rare but potentially lethal form of EPS combining features of advanced parkinsonism and catatonia. 76-80 NMS was initially misdiagnosed as an idiopathic form of catatonia stemming from psychosis, often leading to tragic consequences of continued or even more aggressive antipsychotic treatment, with recognition as a drug-induced side effect delayed until the late 1950’s, and then not accepted worldwide until the 1980’s. 76 Classic signs are elevated temperatures from moderate to life-threatening hyperthermia, generalized rigidity with tremors, altered consciousness with catatonia, and autonomic instability. Rigidity is described as “lead-pipe”, tremors are often generalized, and other motor findings have included dyskinesias, myoclonus, dysarthria and dysphagia. In extreme form, NMS presents as a hypermetabolic crisis with muscle enzyme elevations (creatine phosphokinase, median elevations 800 IU/L), 81 myoglobinuria, leukocytosis, metabolic acidosis, hypoxia, elevated serum catecholamines, and low serum iron levels.79
The differential diagnosis of NMS includes other disorders with elevated temperatures and encephalopathy, such as malignant catatonia due to psychosis, 65 central nervous system infections, 82 benign extrapyramidal side effects, agitated delirium, heatstroke, 83 serotonin syndrome, and withdrawal from dopamine agonists, sedatives, or alcohol. In the peri-operative setting, NMS may be confused with malignant hyperthermia of anesthesia.84 NMS may result from treatment with dopamine antagonists used in medical settings. Although no laboratory test is diagnostic for NMS, thorough assessment and neuroimaging studies are often necessary to exclude other serious medical conditions.
NMS may develop explosively within hours but usually evolves over days; about two-thirds of cases occur during the first 1–2 weeks after drug initiation. 79 Once dopamine-blocking drugs are withheld, two-thirds of NMS cases resolve within 1–2 weeks, with an average duration of 7–10 days. 77 Patients may experience prolonged symptoms if injectable long-acting drugs are implicated. Occasional patients develop a residual catatonic/parkinsonian state after acute metabolic symptoms subside that can last for weeks to months unless ECT is administered. 68 NMS is potentially fatal due to renal failure, sudden cardiorespiratory arrest, disseminated intravascular coagulation, pulmonary emboli, or aspiration pneumonia.
The incidence of NMS is about 0.02% among patients treated with antipsychotic drugs.79 Potential risk factors have been investigated in small case control studies, and include dehydration, exhaustion, agitation, catatonia, previous episodes, and high doses of high-potency drugs given parenterally at a rapid rate.77 The effect of concurrent administration of multiple antipsychotics, lithium and selective serotonin or serotonin–norepinephrine reuptake inhibitors in enhancing the risk of NMS has been suggested but is unproven. 85 Most of these factors are common in psychiatric practice and therefore of limited value in predicting the rare occurrence of NMS, and do not outweigh the value of antipsychotic drugs when indicated.
NMS has been associated with all currently marketed antipsychotic drugs, but correlates with the use of high potency agents. Haloperidol has accounted for about half of all reported cases. The SGAs have also been implicated in isolated case reports, but a few large scale surveys suggest reduced risk compared with FGAs.79 There are reports of atypical or milder forms of NMS associated with SGAs, but in fact, NMS has always varied in severity even with FGAs.
Treatment consists of early diagnosis, discontinuing dopamine antagonists, and supportive medical care. Benzodiazepines, dopamine agonists, dantrolene, and ECT have been advocated, but randomized, controlled trials comparing these agents with supportive care have not been done and may not be feasible because NMS is rare, often self-limited after drug discontinuation, and heterogeneous in presentation, course, and outcome. 78 We have proposed that these agents may be considered empirically in individual cases, based on symptoms, severity, and duration of the episode. 78
Several lines of evidence strongly implicate drug-induced dopamine receptor blockade as the primary triggering mechanism in the pathogenesis of NMS. 72 These include the dopaminergic antagonist properties of all triggering agents, correlation of risk with increased drug potency, reports of beneficial effects of dopamine agonists, reduced cerebrospinal fluid levels of homovanillic acid in affected patients, correlations of NMS symptoms with dopamine pathways in basal ganglia-thalamocortical neurocircuits, and most convincingly, the fact that NMS is clinically indistinguishable from the “parkinsonian-hyperthermia syndrome” resulting from discontinuation or loss of efficacy of dopaminergic agents in patients with Parkinson’s disease. However, other neurotransmitter systems, autonomic and peripheral neuromuscular hypotheses have also been proposed. 86 Genetic studies have been reported in a small number of cases, examining associations between NMS and genetic polymorphisms for ryanodine and dopamine receptors, and the cytochrome oxidase system but without consistent results.
Tardive dyskinesia
In contrast to acute EPS, TD is insidious in onset, arises only after prolonged treatment and is often masked by ongoing treatment. In addition, TD is irreversible in most cases but usually mild, whereas acute EPS are transient but unmistakable and incapacitating. Even so, TD can become socially disfiguring and severe enough to compromise eating, speaking, breathing or ambulation.
TD presents as a polymorphous involuntary movement disorder.32, 87, 88 In its most common form, TD is characterized by involuntary, non-rhythmic, repetitive, purposeless hyperkinetic movements. Most often, TD affects orofacial and lingual musculature (“buccolinguomasticatory syndrome”) with chewing or bruxism of the jaw, protrusion, curling or twisting of the tongue, lip smacking, puckering, sucking and pursing, retraction, grimacing or bridling of the mouth, bulging of the cheeks, or eye blinking and blepharospasm. Choreoathetoid movements of the fingers, hands, upper or lower extremities are common. Axial symptoms affecting the neck, shoulders, spine or pelvis may be observed. Dyskinesias can affect breathing, swallowing or speech.
TD may present with other than choreoathetoid symptoms that can be difficult to distinguish from acute EPS. These may co-exist with classic TD symptoms, but represent separate subtypes with increased risk of progression, persistence and severe disability. For example, tardive dystonia, estimated to occur in up to 1% to 4% of treated patients,42 may be more generalized and disabling than TD, and may respond to anticholinergic agents. Akathisia, as well as other movement disorders, also occur as tardive variants. 89 Dyskinesias increase with emotional arousal, activation or distraction, and diminish with relaxation, sleep, or volitional effort. As a result, symptoms of TD fluctuate over time, such that repeated measurements are necessary for reliable assessment of severity and persistence.
Although different diagnostic schemes for TD have been proposed, criteria proposed by Schooler and Kane have been widely accepted. 90 These authors require at least three months of exposure to antipsychotics, ratings of at least moderate severity in one or more body areas, or ratings of at least mild severity in two or more body areas, and the absence of other conditions that might cause dyskinesias. Ratings are obtained using the Abnormal Involuntary Movement Scale (AIMS). 91
A careful neurologic evaluation is indicated for all patients with new-onset dyskinesias. Clues to neurological causes include family history, sudden onset or progressive course, associated medical or neurological abnormalities, and asymmetry. The differential diagnosis of TD includes acute EPS, transient withdrawal dyskinesias, spontaneous dyskinesias associated with schizophrenia and aging, Wilson’s disease, Huntington’s disease, Sydenham’s chorea, chorea gravidarum, Fahr’s syndrome, systemic lupus, senile chorea, Meige syndrome, edentulous chorea, infarction or lesions of the basal ganglia, post-anoxic and encephalitic states, Tourette’s syndrome, torsion dystonia, spasmodic torticollis, hyperthyroidism, hypoparathyroidism, and conversion disorder. Several drugs and toxins are associated with dyskinesias including caffeine, phenytoin, estrogens, levodopa, antidepressants, antihistamines, stimulants, and manganese poisoning.
The onset of TD occurs insidiously over three months or more of treatment and may begin with tic-like movements or increased eye blink frequency. TD is often suppressed or masked by ongoing antipsychotic treatment, becoming apparent only when treatment is reduced, switched or discontinued.
The natural course of TD is unclear. Early studies showed that withdrawal of antipsychotics may lead to an initial worsening of TD in 33% to 53% of patients (unmasking or withdrawal dyskinesia), but with long-term follow-up, 36% to 55% of patients eventually improve, which led to recommendations for drug reduction or withdrawal. 92 However, complete and permanent reversibility beyond the withdrawal period is rare; Glazer et al found that only 2% of patients showed complete reversal of TD after drug discontinuation. 93, 94 In a meta-analysis of treatments for TD, Soares and McGrath 95 reported that 37.3% of patients assigned to placebo across studies showed some improvement in TD, but concluded that insufficient evidence existed to support drug cessation or reduction as effective treatments for TD, especially when contrasted with robust evidence for the risk of psychotic relapse. 96
Data on the change in prevalence of TD during continued treatment have been inconclusive with some studies showing an increase, others a decrease, or no change at all.97 However, prevalence rates obscure the dynamics of TD in individual patients. Roughly 50% of patients have persistent TD symptoms, 10% to 30% have a reduction and 10% to 30% show increased symptoms during treatment. 98 Long-term studies estimated that from 2% to 23% of patients show loss of observable TD symptoms during treatment with FGAs.97 Similarly, studies of SGAs have shown reduction of TD ratings, with some showing greater reductions, less, or no difference, compared with first generation agents. 97 Improved outcome of TD correlates in some studies with younger age, lower doses, reduced duration of drug treatment and dyskinesia, and increased length of follow-up.
In the CATIE trial, there was a significant decline in ratings of TD severity among 200 patients with TD at baseline, but there were no significant differences between SGAs in the decline in AIMS scores. 97 Fifty-five percent of these patients met criteria for TD at two consecutive visits post-baseline, 76% met criteria at some or all post-baseline visits, 24% did not meet criteria at any subsequent visit, 32% showed ≥50% decrease and 7% showed ≥50% increase in AIMS scores. Thus, similar to past evidence on the course of TD during treatment with FGAs or SGAs, most patients showed either persistence or fluctuation in observable symptoms.
The frequency of TD occurring has been extensively studied but with varied results affected by spontaneous dyskinesias, sensitivity and definitions of diagnosis, susceptibility of the patient sample, fluctuation in TD symptoms, the dynamics of emergence and remission, and the influence of drug treatment on suppression and unmasking of TD. Estimates of the cross-sectional prevalence of TD with FGA antipsychotics range from 3% to 62% with a mean of 24%. 87, 99 Several studies have shown a cumulative incidence of TD of about 5% annually.87 In studies of first-episode patients with limited prior drug exposure, the incidence of TD was 6% to 12% in the first year even when low doses of antipsychotics were used.100, 101 The annual incidence of TD in patients over the age of 45 years was 25% to 30% after one year of treatment.102
Previous studies of TD risk have suggested an association with increasing age, female gender, psychiatric diagnosis, longer duration of antipsychotic treatment, higher cumulative drug doses, concomitant drug treatments, higher ratings of negative symptoms and thought disorder, greater cognitive impairments, presence of acute EPS, and diabetes. The patients with TD at baseline in the CATIE trial were found to be significantly older, and had been treated with an antipsychotic significantly longer, were more likely to be currently treated with an FGA and to be currently treated with an anticholinergic agent, had greater neurocognitive impairment, higher levels of psychopathology, and higher ratings of parkinsonism and akathisia compared with non-TD patients.103 Gender, race, and ethnicity were not differentially distributed between patients with TD versus those without TD. Patients with diabetes or hypertension did not have higher rates of TD. However, alcohol and drug abuse or dependence were significantly associated with TD.
Differences in liability for TD between FGAs and SGAs have been studied extensively. In contrast to the incidence of TD with FGAs, numerous industry sponsored trials of SGAs found a significant six to 12 fold reduction in risk for TD. 12, 27, 104-106 Correll et al, in a meta-analysis, found a 4.6% reduction in the risk of TD for SGAs compared with studies using haloperidol, but later found only a 1.6% difference in attributable risk when studies using mid-potency FGAs were included. 12, 27 It remains unlikely that clozapine causes TD. 107
In contrast to industry studies using haloperidol, there were no significant differences among groups in the CATIE trial receiving perphenazine or four SGAs in the incidence of TD defined by scores of two or more on the AIMS global severity score (Table 1).28 In a second analysis, patients were considered to have persistent TD if they met full Schooler-Kane (S-K) criteria. 29, 90 Analyses were also conducted using modified S-K criteria such that meeting the AIMS criteria on only one assessment was required, i.e., “probable” TD. Few patients who had no evidence of TD at baseline met full S-K TD criteria during treatment (1.1% to 4.5% receiving SGAs and 3.3% receiving perphenazine (Table 2). The proportion of patients who met modified S-K TD criteria ranged from 8.3% to 9.6% with SGAs and 11.8% for perphenazine. There were no statistically significant differences between treatment groups on any TD indicator.
Since there is no uniformly effective treatment for TD, it is important to minimize the risk of TD by early detection (Figure 1). Some preventive principles are to confirm the indication for antipsychotics, use conservative doses opting for lower potency agents, inform patients and caregivers of risk, assess on a regular basis for incipient signs, consider differential diagnosis and reconsider drug treatment if symptoms emerge.
Figure 1.
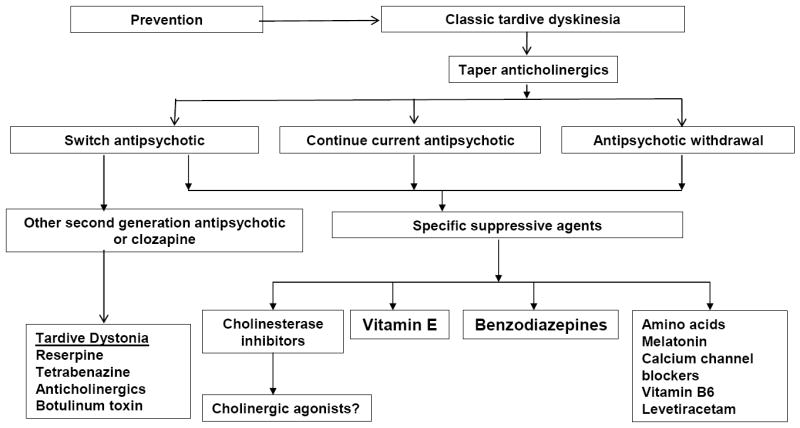
Proposed treatment algorithm for tardive dyskinesia.
The first step in treatment is the decision on antipsychotic treatment. Although drug withdrawal had been recommended in the past as increasing the odds of resolution of TD, about 33% to 53% of patients will experience worsening of dyskinesias initially, 36% to 55% may show improvement over time,92 but few will show complete resolution of symptoms,94 and the risk of psychotic relapse is 53% within nine months.96
A second option in a patient with good control of psychotic symptoms is to decide not to change the antipsychotic, try to gradually reduce the dose, inform patients and caregivers of risks, document the decision and monitor carefully. In most cases, TD is not progressive even with continued antipsychotic treatment, although symptoms may worsen in a few cases (7% in the CATIE trial).97 Another alternative is to switch antipsychotics; more potent antipsychotics, like haloperidol, can be used to suppress disabling symptoms of TD in about 67% of patients. 98 SGAs have also been associated with reduction of TD symptoms; 14, 15 results of the CATIE trial indicated that while most patients treated with SGAs show a persistent (34%) or fluctuating course (42%), 24% did not meet criteria on any visit at follow-up, and 32% showed greater than 50% reduction in AIMS scores.97 Clozapine has been recommended for suppressing tardive dystonia. While there have been speculations that SGAs may increase the possibility of remission on an active or passive basis, existing data are inconclusive whether recovery rather than suppression occurs during treatment with SGAs or FGAs.
After discontinuing or optimizing antipsychotic therapy, there are a large number of agents that have been tested in the treatment of TD (Figure 1).98, 108 These include dopamine depleting agents, dopamine agonists, noradrenergic agonists and antagonists, GABAergic drugs (benzodiazepines, valproate, baclofen, levetiracetam), lithium, calcium channel blockers, serotonergic drugs, vitamins (vitamin E), branched-chain amino acids, neuropeptides, cholinergic precursors and cholinesterase inhibitors, ECT, and botulinum toxin or surgical intervention (for tardive dystonia). Anticholinergic agents can worsen TD (expect for tardive dystonia) with improvement noted in 60% of cases after discontinuation.98
Several hypotheses have been proposed to explain the pathophysiology of TD including dopamine receptor hypersensitivity, GABA insufficiency, and structural damage resulting from increased catecholamine metabolism and oxidative free radical production. 109 Another hypothesis proposes that TD results from damage to striatal cholinergic interneurons due to the loss of dopamine mediated inhibition. 110 If correct, this implies that cholinesterase inhibitors or cholinergic agonists may be effective in suppressing TD by directly enhancing post-synaptic cholinergic activity, thereby compensating for the loss of pre-synaptic cholinergic neurons. Several preliminary trials have explored the use of cholinesterase inhibitors with mixed results. 111-113 However, this hypothesis is supported by animal and human evidence correlating cholinergic mechanisms with the delay in onset, irreversibility, age-related risk, worsening with anticholinergic drugs, and reduced risk with the newer SGA drugs, suggesting further investigation of cholinergic mechanisms may be worthwhile.
As a genetic basis for TD has been assumed, an increasing number of candidate genes have been selected for study. Using the CATIE trial sample, a total of 128 candidate genes selected from the literature were studied in 710 subjects (207 or 29.2% with TD). 114 No single marker or haplotype association reached statistical significance after adjustment for multiple comparisons, therefore providing no support for either novel or prior associations from the literature. In a genome wide association study of the CATIE sample, Aberg et al genotyped 738 schizophrenia patients for 492K SNPs in association with scale scores for EPS, and found two SNPs and one SNP reaching genomewide significance for parkinsonism and TD, respectively.115 Though these findings require replication, they demonstrate the potential of candidate gene and genomewide association studies to discover genetic pathways that mediate TD.
Conclusions
Whether the newer SGA drugs have reduced liability for EPS because of weaker dopamine receptor binding affinity or a more complex “atypical” mechanism affecting multiple neurotransmitters, their dominance in clinical practice has effectively and significantly reduced the frequency of these side effects. However, the association between antipsychotic drugs and EPS remains important for several reasons; (1) questions about the cost effectiveness of the SGAs, i.e., whether their reduced liability for EPS outweighs their higher costs, are legitimate; (2) tens of thousands of patients previously exposed to more potent agents have TD, which remains untreatable and irreversible; (3) there is some risk of acute EPS and TD with SGAs; (4) haloperidol and other FGAs are still used in psychiatry, in medical settings for delirium and agitation, and in developing nations; (5) better understanding of the pathophysiology of these disorders may provide insights into the mechanism of action of antipsychotics and the bases of psychotic disorders.
In summary of specific EPS, dystonia is less frequent than before but may be encountered with high potency drugs. Parkinsonism is quite debilitating but is more likely to be seen when antipsychotics are used in elderly patients. Akathisia remains a problem even with newer SGA drugs. TD is most often mild but irreversible with limited options for treatment. Catatonia is rare and associated with high potency drugs. NMS is extremely rare, but early diagnosis is crucial for a positive outcome. EPS are less likely to occur with SGAs compared with haloperidol, but clozapine and quetiapine, perhaps because of fast dissociation of dopamine receptor binding, are least likely to induce EPS.
Finally, the CATIE schizophrenia trial has provided additional perspective on the relative risks and clinical value of the SGAs and FGAs. The study showed that perphenazine, a mid-potency FGA used at modest doses, was not significantly different from four SGAs in the risk of developing EPS. It is fair to conclude that the significant advantages of SGAs over haloperidol in risk of EPS shown in previous trials are diminished when modest doses of a low or mid-potency FGA is used as the comparator. This conclusion is now supported by several published studies.19, 22, 23, 26, 27 These findings imply that haloperidol is not synonymous with “FGA”, and by extension, the dichotomy between FGAs and SGAs and the concept of “atypicality” based on EPS liability may be oversimplified. Instead, antipsychotic drugs could be conceptualized as a single drug class with a spectrum of risk for EPS depending upon receptor binding affinities, especially for dopamine and acetylcholine receptors, and individual patient susceptibility.
Acknowledgments
This material is based upon work supported in part by the Department of Veterans Affairs, Veterans Health Administration, Office of Research Development, with resources and the use of facilities at the Philadelphia Veterans Affairs Medical Center. The content of this work does not represent the views of the Department of Veterans Affairs or the United States Government. Dr. Caroff served as a consultant for Eli Lilly & Company and received research grants from Ortho-McNeil Neurologics and Bristol-Myers Squibb. The other authors have nothing to disclose. Data cited from previous publications of the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) project were based on work supported by the National Institute of Mental Health (NO1 MH90001).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Preskorn SH. The evolution of antipsychotic drug therapy: reserpine, chlorpromazine, and haloperidol. J Psychiatr Pract. 2007 Jul;13(4):253–257. doi: 10.1097/01.pra.0000281486.34817.8b. [DOI] [PubMed] [Google Scholar]
- 2.Rifkin A. Extrapyramidal side effects: a historical perspective. J Clin Psychiatry. 1987 Sep;48(Suppl):3–6. [PubMed] [Google Scholar]
- 3.Van Putten T. Why do schizophrenic patients refuse to take their drugs? Arch Gen Psychiatry. 1975;31:67–72. doi: 10.1001/archpsyc.1974.01760130049008. [DOI] [PubMed] [Google Scholar]
- 4.Caroff S. The Neuroleptic malignant syndrome. J Clin Psychiatry. 1980;41:79–83. [PubMed] [Google Scholar]
- 5.Kane J, Honigfeld G, Singer J, Meltzer H. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988 Sep;45(9):789–796. doi: 10.1001/archpsyc.1988.01800330013001. [DOI] [PubMed] [Google Scholar]
- 6.Arvanitis LA, Miller BG. Multiple fixed doses of “Seroquel” (quetiapine) in patients with acute exacerbation of schizophrenia: a comparison with haloperidol and placebo. The Seroquel Trial 13 Study Group. Biol Psychiatry. 1997 Aug 15;42(4):233–246. doi: 10.1016/s0006-3223(97)00190-x. [DOI] [PubMed] [Google Scholar]
- 7.Potkin SG, Saha AR, Kujawa MJ, et al. Aripiprazole, an antipsychotic with a novel mechanism of action, and risperidone vs placebo in patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry. 2003 Jul;60(7):681–690. doi: 10.1001/archpsyc.60.7.681. [DOI] [PubMed] [Google Scholar]
- 8.Tollefson GD, Beasley CM, Jr, Tran PV, et al. Olanzapine versus haloperidol in the treatment of schizophrenia and schizoaffective and schizophreniform disorders: results of an international collaborative trial. Am J Psychiatry. 1997 Apr;154(4):457–465. doi: 10.1176/ajp.154.4.457. [DOI] [PubMed] [Google Scholar]
- 9.Marder SR, Meibach RC. Risperidone in the treatment of schizophrenia. Am J Psychiatry. 1994 Jun;151(6):825–835. doi: 10.1176/ajp.151.6.825. [DOI] [PubMed] [Google Scholar]
- 10.Daniel DG, Zimbroff DL, Potkin SG, Reeves KR, Harrigan EP, Lakshminarayanan M. Ziprasidone 80 mg/day and 160 mg/day in the acute exacerbation of schizophrenia and schizoaffective disorder: a 6-week placebo-controlled trial. Ziprasidone Study Group. Neuropsychopharmacology. 1999 May;20(5):491–505. doi: 10.1016/S0893-133X(98)00090-6. [DOI] [PubMed] [Google Scholar]
- 11.Dossenbach M, Arango-Davila C, Silva Ibarra H, et al. Response and relapse in patients with schizophrenia treated with olanzapine, risperidone, quetiapine, or haloperidol: 12-month follow-up of the Intercontinental Schizophrenia Outpatient Health Outcomes (IC-SOHO) study. J Clin Psychiatry. 2005 Aug;66(8):1021–1030. doi: 10.4088/jcp.v66n0810. [DOI] [PubMed] [Google Scholar]
- 12.Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry. 2004 Mar;161(3):414–425. doi: 10.1176/appi.ajp.161.3.414. [DOI] [PubMed] [Google Scholar]
- 13.Tenback DE, van Harten PN, Slooff CJ, Belger MA, van Os J. Effects of antipsychotic treatment on tardive dyskinesia: a 6-month evaluation of patients from the European Schizophrenia Outpatient Health Outcomes (SOHO) Study. J Clin Psychiatry. 2005 Sep;66(9):1130–1133. [PubMed] [Google Scholar]
- 14.Caroff SN, Mann SC, Campbell EC, Sullivan KA. Movement disorders associated with atypical antipsychotic drugs. J Clin Psychiatry. 2002;63(Suppl 4):12–19. [PubMed] [Google Scholar]
- 15.Tarsy D, Baldessarini RJ, Tarazi FI. Effects of newer antipsychotics on extrapyramidal function. CNS Drugs. 2002;16(1):23–45. doi: 10.2165/00023210-200216010-00003. [DOI] [PubMed] [Google Scholar]
- 16.Kane JM, Woerner M, Lieberman J. Tardive dyskinesia: prevalence, incidence, and risk factors. J Clin Psychopharmacol. 1988 Aug;8(4 Suppl):52S–56S. [PubMed] [Google Scholar]
- 17.Glazer WM. Expected incidence of tardive dyskinesia associated with atypical antipsychotics. J Clin Psychiatry. 2000;61(Suppl 4):21–26. [PubMed] [Google Scholar]
- 18.Glazer WM. Extrapyramidal side effects, tardive dyskinesia, and the concept of atypicality. J Clin Psychiatry. 2000;61(Suppl 3):16–21. [PubMed] [Google Scholar]
- 19.Leucht S, Pitschel-Walz G, Abraham D, Kissling W. Efficacy and extrapyramidal side-effects of the new antipsychotics olanzapine, quetiapine, risperidone, and sertindole compared to conventional antipsychotics and placebo. A meta-analysis of randomized controlled trials. Schizophr Res. 1999 Jan 4;35(1):51–68. doi: 10.1016/s0920-9964(98)00105-4. [DOI] [PubMed] [Google Scholar]
- 20.Meltzer HY. The mechanism of action of novel antipsychotic drugs. Schizophr Bull. 1991;17(2):263–287. doi: 10.1093/schbul/17.2.263. [DOI] [PubMed] [Google Scholar]
- 21.Davis JM, Chen N, Glick ID. A meta-analysis of the efficacy of second-generation antipsychotics. Arch Gen Psychiatry. 2003 Jun;60(6):553–564. doi: 10.1001/archpsyc.60.6.553. [DOI] [PubMed] [Google Scholar]
- 22.Geddes J, Freemantle N, Harrison P, Bebbington P. Atypical antipsychotics in the treatment of schizophrenia: systematic overview and meta-regression analysis. BMJ. 2000 Dec 2;321(7273):1371–1376. doi: 10.1136/bmj.321.7273.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leucht S, Wahlbeck K, Hamann J, Kissling W. New generation antipsychotics versus low-potency conventional antipsychotics: a systematic review and meta-analysis. Lancet. 2003 May 10;361(9369):1581–1589. doi: 10.1016/S0140-6736(03)13306-5. [DOI] [PubMed] [Google Scholar]
- 24.Rosenheck R, Perlick D, Bingham S, et al. Effectiveness and cost of olanzapine and haloperidol in the treatment of schizophrenia: a randomized controlled trial. JAMA. 2003 Nov 26;290(20):2693–2702. doi: 10.1001/jama.290.20.2693. [DOI] [PubMed] [Google Scholar]
- 25.Hugenholtz GW, Heerdink ER, Stolker JJ, Meijer WE, Egberts AC, Nolen WA. Haloperidol dose when used as active comparator in randomized controlled trials with atypical antipsychotics in schizophrenia: comparison with officially recommended doses. J Clin Psychiatry. 2006 Jun;67(6):897–903. doi: 10.4088/jcp.v67n0606. [DOI] [PubMed] [Google Scholar]
- 26.Jones PB, Barnes TR, Davies L, et al. Randomized controlled trial of the effect on Quality of Life of second- vs first-generation antipsychotic drugs in schizophrenia: Cost Utility of the Latest Antipsychotic Drugs in Schizophrenia Study (CUtLASS 1) Arch Gen Psychiatry. 2006 Oct;63(10):1079–1087. doi: 10.1001/archpsyc.63.10.1079. [DOI] [PubMed] [Google Scholar]
- 27.Correll CU, Schenk EM. Tardive dyskinesia and new antipsychotics. Curr Opin Psychiatry. 2008 Mar;21(2):151–156. doi: 10.1097/YCO.0b013e3282f53132. [DOI] [PubMed] [Google Scholar]
- 28.Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005 Sep 22;353(12):1209–1223. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- 29.Miller DD, Caroff SN, Davis SM, et al. Extrapyramidal side-effects of antipsychotics in a randomised trial. Br J Psychiatry. 2008 Oct;193(4):279–288. doi: 10.1192/bjp.bp.108.050088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caroff SN, Miller DD, Rosenheck RA. Extrapyramidal side effects. In: Stroup TS, Lieberman JA, editors. The Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Schizophrenia Trial:How does it inform practice, policy, and research? Cambridge: Cambridge University Press; 2010. pp. 156–172. [Google Scholar]
- 31.Satterthwaite TD, Wolf DH, Rosenheck RA, Gur RE, Caroff SN. A meta-analysis of the risk of acute extrapyramidal symptoms with intramuscular antipsychotics for the treatment of agitation. J Clin Psychiatry. 2008 Dec;69(12):1869–1879. doi: 10.4088/jcp.v69n1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tarsy D. Neuroleptic-induced extrapyramidal reactions: classification, description, and diagnosis. Clin Neuropharmacol. 1983;6(Suppl 1):S9–26. [PubMed] [Google Scholar]
- 33.van Harten PN, Hoek HW, Kahn RS. Acute dystonia induced by drug treatment. BMJ. 1999 Sep 4;319(7210):623–626. doi: 10.1136/bmj.319.7210.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rupniak NM, Jenner P, Marsden CD. Acute dystonia induced by neuroleptic drugs. Psychopharmacology (Berl) 1986;88(4):403–419. doi: 10.1007/BF00178501. [DOI] [PubMed] [Google Scholar]
- 35.Tarsy D, Simon DK. Dystonia. N Engl J Med. 2006 Aug 24;355(8):818–829. doi: 10.1056/NEJMra055549. [DOI] [PubMed] [Google Scholar]
- 36.Marsden CD, Quinn NP. The dystonias. BMJ. 1990 Jan 20;300(6718):139–144. doi: 10.1136/bmj.300.6718.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nemeth AH. The genetics of primary dystonias and related disorders. Brain. 2002 Apr;125(Pt 4):695–721. doi: 10.1093/brain/awf090. [DOI] [PubMed] [Google Scholar]
- 38.Keepers GA, Casey DE. Prediction of neuroleptic-induced dystonia. J Clin Psychopharmacol. 1987 Oct;7(5):342–345. [PubMed] [Google Scholar]
- 39.Keepers GA, Clappison VJ, Casey DE. Initial anticholinergic prophylaxis for neuroleptic-induced extrapyramidal syndromes. Arch Gen Psychiatry. 1983 Oct;40(10):1113–1117. doi: 10.1001/archpsyc.1983.01790090075012. [DOI] [PubMed] [Google Scholar]
- 40.Simpson GM, Lindenmayer JP. Extrapyramidal symptoms in patients treated with risperidone. J Clin Psychopharmacol. 1997 Jun;17(3):194–201. doi: 10.1097/00004714-199706000-00010. [DOI] [PubMed] [Google Scholar]
- 41.Arana GW, Goff DC, Baldessarini RJ, Keepers GA. Efficacy of anticholinergic prophylaxis for neuroleptic-induced acute dystonia. Am J Psychiatry. 1988 Aug;145(8):993–996. doi: 10.1176/ajp.145.8.993. [DOI] [PubMed] [Google Scholar]
- 42.Dayalu P, Chou KL. Antipsychotic-induced extrapyramidal symptoms and their management. Expert Opin Pharmacother. 2008 Jun;9(9):1451–1462. doi: 10.1517/14656566.9.9.1451. [DOI] [PubMed] [Google Scholar]
- 43.Berardelli A, Rothwell JC, Hallett M, Thompson PD, Manfredi M, Marsden CD. The pathophysiology of primary dystonia. Brain. 1998 Jul;121(Pt 7):1195–1212. doi: 10.1093/brain/121.7.1195. [DOI] [PubMed] [Google Scholar]
- 44.Thanvi B, Treadwell S. Drug induced parkinsonism: a common cause of parkinsonism in older people. Postgrad Med J. 2009 Jun;85(1004):322–326. doi: 10.1136/pgmj.2008.073312. [DOI] [PubMed] [Google Scholar]
- 45.Bovi T, Antonini A, Ottaviani S, et al. The status of olfactory function and the striatal dopaminergic system in drug-induced parkinsonism. J Neurol. Jul 16; doi: 10.1007/s00415-010-5631-3. [DOI] [PubMed] [Google Scholar]
- 46.Gelenberg AJ. General principles of treatment of extrapyramidal syndromes. Clin Neuropharmacol. 1983;6(Suppl 1):S52–56. doi: 10.1097/00002826-198300061-00007. [DOI] [PubMed] [Google Scholar]
- 47.Snyder S, Greenberg D, Yamamura HI. Antischizophrenic drugs and brain cholinergic receptors. Affinity for muscarinic sites predicts extrapyramidal effects. Arch Gen Psychiatry. 1974 Jul;31(1):58–61. doi: 10.1001/archpsyc.1974.01760130040006. [DOI] [PubMed] [Google Scholar]
- 48.Hirsch SR, Kissling W, Bauml J, Power A, O’Connor R. A 28-week comparison of ziprasidone and haloperidol in outpatients with stable schizophrenia. J Clin Psychiatry. 2002 Jun;63(6):516–523. doi: 10.4088/jcp.v63n0609. [DOI] [PubMed] [Google Scholar]
- 49.Barnes TR, McPhillips MA. Critical analysis and comparison of the side-effect and safety profiles of the new antipsychotics. Br J Psychiatry Suppl. 1999(38):34–43. [PubMed] [Google Scholar]
- 50.Simpson GM, Angus JWS. A rating scale for extrapyramidal side effects. Acta Psychiatr Scand. 1970;212:11–19. doi: 10.1111/j.1600-0447.1970.tb02066.x. [DOI] [PubMed] [Google Scholar]
- 51.Gelenberg AJ. Treating extrapyramidal reactions: some current issues. J Clin Psychiatry. 1987 Sep;48(Suppl):24–27. [PubMed] [Google Scholar]
- 52.Bras JM, Singleton A. Genetic susceptibility in Parkinson’s disease. Biochim Biophys Acta. 2009 Jul;1792(7):597–603. doi: 10.1016/j.bbadis.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kane JM, Fleischhacker WW, Hansen L, Perlis R, Pikalov A, 3rd, Assuncao-Talbott S. Akathisia: an updated review focusing on second-generation antipsychotics. J Clin Psychiatry. 2009 May;70(5):627–643. doi: 10.4088/JCP.08r04210. [DOI] [PubMed] [Google Scholar]
- 54.Bratti IM, Kane JM, Marder SR. Chronic restlessness with antipsychotics. Am J Psychiatry. 2007 Nov;164(11):1648–1654. doi: 10.1176/appi.ajp.2007.07071150. [DOI] [PubMed] [Google Scholar]
- 55.Poyurovsky M. Acute antipsychotic-induced akathisia revisited. Br J Psychiatry. Feb;196(2):89–91. doi: 10.1192/bjp.bp.109.070540. [DOI] [PubMed] [Google Scholar]
- 56.Barnes TR. A rating scale for drug-induced akathisia. Br J Psychiatry. 1989 May;154:672–676. doi: 10.1192/bjp.154.5.672. [DOI] [PubMed] [Google Scholar]
- 57.Miller CH, Fleischhacker WW. Managing antipsychotic-induced acute and chronic akathisia. Drug Saf. 2000 Jan;22(1):73–81. doi: 10.2165/00002018-200022010-00006. [DOI] [PubMed] [Google Scholar]
- 58.Eichhammer P, Albus M, Borrmann-Hassenbach M, et al. Association of dopamine D3-receptor gene variants with neuroleptic induced akathisia in schizophrenic patients: a generalization of Steen’s study on DRD3 and tardive dyskinesia. Am J Med Genet. 2000 Apr 3;96(2):187–191. doi: 10.1002/(sici)1096-8628(20000403)96:2<187::aid-ajmg13>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 59.Pichler I, Hicks AA, Pramstaller PP. Restless legs syndrome: an update on genetics and future perspectives. Clin Genet. 2008 Apr;73(4):297–305. doi: 10.1111/j.1399-0004.2007.00937.x. [DOI] [PubMed] [Google Scholar]
- 60.Ungvari GS, Caroff SN, Gerevich J. The catatonia conundrum: evidence of psychomotor phenomena as a symptom dimension in psychotic disorders. Schizophr Bull. Mar;36(2):231–238. doi: 10.1093/schbul/sbp105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jaspers K. General Psychopathology. Manchester, England: Manchester University Press; 1963. [Google Scholar]
- 62.Lopez-Canino A, Francis A. Drug-induced catatonia. In: Caroff SN, Mann SC, Francis A, Fricchione GL, editors. Catatonia: From Psychopathology to Neurobiology. Washington, D.C: American Psychiatric Press, Inc.; 2004. pp. 129–139. [Google Scholar]
- 63.Gelenberg AJ, Mandel MR. Catatonic reactions to high-potency neuroleptic drugs. Arch Gen Psychiatry. 1977 Aug;34(8):947–950. doi: 10.1001/archpsyc.1977.01770200085010. [DOI] [PubMed] [Google Scholar]
- 64.White DA, Robins AH. An analysis of 17 catatonic patients diagnosed with neuroleptic malignant syndrome. CNS Spectr. 2000 Jul;5(7):58–65. doi: 10.1017/s1092852900013419. [DOI] [PubMed] [Google Scholar]
- 65.Mann SC, Caroff SN, Bleier HR, Welz WK, Kling MA, Hayashida M. Lethal catatonia. Am J Psychiatry. 1986 Nov;143(11):1374–1381. doi: 10.1176/ajp.143.11.1374. [DOI] [PubMed] [Google Scholar]
- 66.Gelenberg AJ. The catatonic syndrome. Lancet. 1976 Jun 19;1(7973):1339–1341. doi: 10.1016/s0140-6736(76)92669-6. [DOI] [PubMed] [Google Scholar]
- 67.Brenner I, Rheuban WJ. The catatonic dilemma. Am J Psychiatry. 1978 Oct;135(10):1242–1243. doi: 10.1176/ajp.135.10.1242. [DOI] [PubMed] [Google Scholar]
- 68.Caroff SN, Mann SC, Keck PE., Jr Francis A. Residual catatonic state following neuroleptic malignant syndrome. J Clin Psychopharmacol. 2000 Apr;20(2):257–259. doi: 10.1097/00004714-200004000-00021. [DOI] [PubMed] [Google Scholar]
- 69.Stubner S, Rustenbeck E, Grohmann R, et al. Severe and uncommon involuntary movement disorders due to psychotropic drugs. Pharmacopsychiatry. 2004 Mar;37(Suppl 1):S54–64. doi: 10.1055/s-2004-815511. [DOI] [PubMed] [Google Scholar]
- 70.Van Den Eede F, Van Hecke J, Van Dalfsen A, Van den Bossche B, Cosyns P, Sabbe BG. The use of atypical antipsychotics in the treatment of catatonia. Eur Psychiatry. 2005 Aug;20(5-6):422–429. doi: 10.1016/j.eurpsy.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 71.Carroll BT, Goforth HW, Thomas C, et al. Review of adjunctive glutamate antagonist therapy in the treatment of catatonic syndromes. J Neuropsychiatry Clin Neurosci. 2007 Fall;19(4):406–412. doi: 10.1176/jnp.2007.19.4.406. [DOI] [PubMed] [Google Scholar]
- 72.Mann SC, Caroff SN, Fricchione G, Campbell EC. Central dopamine hypoactivity and the pathogenesis of neuroleptic malignant syndrome. Psychiatric Annals. 2000;30:363–374. [Google Scholar]
- 73.Stober G. Genetics. In: Caroff SN, Mann SC, Francis A, Fricchione GL, editors. Catatonia: From Psychopathology to Neurobiology. Washington, D.C: American Psychiatric Press, Inc; 2004. pp. 173–187. [Google Scholar]
- 74.Kaiser R, Konneker M, Henneken M, et al. Dopamine D4 receptor 48-bp repeat polymorphism: no association with response to antipsychotic treatment, but association with catatonic schizophrenia. Mol Psychiatry. 2000 Jul;5(4):418–424. doi: 10.1038/sj.mp.4000729. [DOI] [PubMed] [Google Scholar]
- 75.Kanes SJ. Animal models. In: Caroff SN, Mann SC, Francis A, Fricchione GL, editors. Catatonia:From Psychopathology to Neurobiology. Washington, D.C: American Psychiatric Press, Inc.; 2004. pp. 189–200. [Google Scholar]
- 76.Caroff S. The Neuroleptic malignant syndrome. J Clin Psychiatry. 1980;41:79–83. [PubMed] [Google Scholar]
- 77.Caroff SN, Mann SC. Neuroleptic malignant syndrome. Med Clin North Am. 1993 Jan;77(1):185–202. doi: 10.1016/s0025-7125(16)30278-4. [DOI] [PubMed] [Google Scholar]
- 78.Strawn JR, Keck PE, Jr, Caroff SN. Neuroleptic malignant syndrome. Am J Psychiatry. 2007 Jun;164(6):870–876. doi: 10.1176/ajp.2007.164.6.870. [DOI] [PubMed] [Google Scholar]
- 79.Caroff SN. Neuroleptic malignant syndrome. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A, editors. Neuroleptic Malignant Syndrome and Related Conditions. Second. Washington, D.C: American Psychiatric Press, Inc.; 2003. pp. 1–44. [Google Scholar]
- 80.Caroff SN, Mann SC, Campbell EC, Berkowitz RM. Severe drug reactions. In: Ferrando SJ, Levenson JL, Owen JA, editors. Clinical Manual of Psychopharmacology in the Medically III. Washington, D.C: American Psychiatric Press, Inc.; 2010. pp. 39–77. [Google Scholar]
- 81.Meltzer HY, Cola PA, Parsa M. Marked elevations of serum creatine kinase activity associated with antipsychotic drug treatment. Neuropsychopharmacology. 1996 Oct;15(4):395–405. doi: 10.1016/0893-133X(95)00276-J. [DOI] [PubMed] [Google Scholar]
- 82.Caroff SN, Mann SC, McCarthy M, Naser J, Rynn M, Morrison M. Acute infectious encephalitis complicated by neuroleptic malignant syndrome. J Clin Psychopharmacol. 1998 Aug;18(4):349–351. doi: 10.1097/00004714-199808000-00022. [DOI] [PubMed] [Google Scholar]
- 83.Mann SC, Boger WP. Psychotropic drugs, summer heat and humidity, and hyperpyrexia: a danger restated. Am J Psychiatry. 1978 Sep;135(9):1097–1100. doi: 10.1176/ajp.135.9.1097. [DOI] [PubMed] [Google Scholar]
- 84.Caroff SN, Rosenberg H, Mann SC, Campbell EC, Gliatto MF, Sullivan KA. Neuroleptic malignant syndrome in the perioperative setting. Am J Anesthesiology. 2001;28:387–393. [Google Scholar]
- 85.Stevens DL. Association between selective serotonin-reuptake inhibitors, second-generation antipsychotics, and neuroleptic malignant syndrome. Ann Pharmacother. 2008 Sep;42(9):1290–1297. doi: 10.1345/aph.1L066. [DOI] [PubMed] [Google Scholar]
- 86.Gurrera RJ. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry. 1999 Feb;156(2):169–180. doi: 10.1176/ajp.156.2.169. [DOI] [PubMed] [Google Scholar]
- 87.Kane JM. Tardive dyskinesia: epidemiological and clinical presentation. In: Bloom FE, Kupfer DJ, editors. Psychopharmacology: the Fourth Generation of Progress. New York: Raven Press; 1995. pp. 1485–1495. [Google Scholar]
- 88.Casey DE. Neuroleptic drug-induced extrapyramidal syndromes and tardive dyskinesia. Schizophr Res. 1991 Mar-Apr;4(2):109–120. doi: 10.1016/0920-9964(91)90029-q. [DOI] [PubMed] [Google Scholar]
- 89.Burke RE, Kang UJ, Jankovic J, Miller LG, Fahn S. Tardive akathisia: an analysis of clinical features and response to open therapeutic trials. Mov Disord. 1989;4(2):157–175. doi: 10.1002/mds.870040208. [DOI] [PubMed] [Google Scholar]
- 90.Schooler NR, Kane JM. Research diagnoses for tardive dyskinesia. Arch Gen Psychiatry. 1982 Apr;39(4):486–487. doi: 10.1001/archpsyc.1982.04290040080014. [DOI] [PubMed] [Google Scholar]
- 91.Guy W. ECDEU Assessment Manual for Psychopharmacology Revised. Alcohol, Drug Abuse and Mental Health Administration, National Institute of Mental Health; 1976. Abnormal involuntary movement scale (AIMS) pp. 534–537. [Google Scholar]
- 92.Casey DE, Gerlach J. Tardive dyskinesia: what is the long-term outcome? In: Casey DE, Gardos G, editors. Tardive Dyskinesia and Neuroleptics: From Dogma to Reason. Washington, D.C.: American Psychiatric Press, Inc.; 1986. pp. 76–97. [Google Scholar]
- 93.Glazer WM, Moore DC, Schooler NR, Brenner LM, Morgenstern H. Tardive dyskinesia. A discontinuation study. Arch Gen Psychiatry. 1984 Jun;41(6):623–627. doi: 10.1001/archpsyc.1984.01790170097011. [DOI] [PubMed] [Google Scholar]
- 94.Glazer WM, Morgenstern H, Schooler N, Berkman CS, Moore DC. Predictors of improvement in tardive dyskinesia following discontinuation of neuroleptic medication. Br J Psychiatry. 1990 Oct;157:585–592. doi: 10.1192/bjp.157.4.585. [DOI] [PubMed] [Google Scholar]
- 95.Soares KV, McGrath JJ. The treatment of tardive dyskinesia--a systematic review and meta-analysis. Schizophr Res. 1999 Aug 23;39(1):1–16. doi: 10.1016/s0920-9964(99)00021-3. discussion 17-18. [DOI] [PubMed] [Google Scholar]
- 96.Gilbert PL, Harris MJ, McAdams LA, Jeste DV. Neuroleptic withdrawal in schizophrenic patients. A review of the literature. Arch Gen Psychiatry. 1995 Mar;52(3):173–188. doi: 10.1001/archpsyc.1995.03950150005001. [DOI] [PubMed] [Google Scholar]
- 97.Caroff SN, Davis VG, Miller DD, et al. Treatment outcomes of patients with tardive dyskinesia and chronic schizophrenia. J Clin Psychiatry. 2010 doi: 10.4088/JCP.09m05793yel. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Egan MF, Apud J, Wyatt RJ. Treatment of tardive dyskinesia. Schizophr Bull. 1997;23(4):583–609. doi: 10.1093/schbul/23.4.583. [DOI] [PubMed] [Google Scholar]
- 99.Woerner MG, Kane JM, Lieberman JA, et al. The prevalence of tardive dyskinesia. J Clin Psychopharmacol. 1991 Feb;11(1):34–42. [PubMed] [Google Scholar]
- 100.Chakos MH, Alvir JM, Woerner MG, et al. Incidence and correlates of tardive dyskinesia in first episode of schizophrenia. Arch Gen Psychiatry. 1996 Apr;53(4):313–319. doi: 10.1001/archpsyc.1996.01830040049009. [DOI] [PubMed] [Google Scholar]
- 101.Oosthuizen PP, Emsley RA, Maritz JS, Turner JA, Keyter N. Incidence of tardive dyskinesia in first-episode psychosis patients treated with low-dose haloperidol. J Clin Psychiatry. 2003 Sep;64(9):1075–1080. doi: 10.4088/jcp.v64n0913. [DOI] [PubMed] [Google Scholar]
- 102.Jeste DV. Tardive dyskinesia in older patients. J Clin Psychiatry. 2000;61(Suppl 4):27–32. [PubMed] [Google Scholar]
- 103.Miller DD, McEvoy JP, Davis SM, et al. Clinical correlates of tardive dyskinesia in schizophrenia: baseline data from the CATIE schizophrenia trial. Schizophr Res. 2005 Dec 1;80(1):33–43. doi: 10.1016/j.schres.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 104.Beasley CM, Dellva MA, Tamura RN, et al. Randomised double-blind comparison of the incidence of tardive dyskinesia in patients with schizophrenia during long-term treatment with olanzapine or haloperidol. Br J Psychiatry. 1999 Jan;174:23–30. doi: 10.1192/bjp.174.1.23. [DOI] [PubMed] [Google Scholar]
- 105.Jeste DV, Lacro JP, Bailey A, Rockwell E, Harris MJ, Caligiuri MP. Lower incidence of tardive dyskinesia with risperidone compared with haloperidol in older patients. J Am Geriatr Soc. 1999 Jun;47(6):716–719. doi: 10.1111/j.1532-5415.1999.tb01595.x. [DOI] [PubMed] [Google Scholar]
- 106.Kane JM. Tardive dyskinesia circa 2006. Am J Psychiatry. 2006 Aug;163(8):1316–1318. doi: 10.1176/ajp.2006.163.8.1316. [DOI] [PubMed] [Google Scholar]
- 107.Kane JM, Woerner MG, Pollack S, Safferman AZ, Lieberman JA. Does clozapine cause tardive dyskinesia? J Clin Psychiatry. 1993 Sep;54(9):327–330. [PubMed] [Google Scholar]
- 108.Jeste DV, Lohr JB, Clark K, Wyatt RJ. Pharmacological treatments of tardive dyskinesia in the 1980s. J Clin Psychopharmacol. 1988 Aug;8(4 Suppl):38S–48S. [PubMed] [Google Scholar]
- 109.Casey DE. Tardive dyskinesia: pathophysiology and animal models. J Clin Psychiatry. 2000;61(Suppl 4):5–9. [PubMed] [Google Scholar]
- 110.Miller R, Chouinard G. Loss of striatal cholinergic neurons as a basis for tardive and L-dopa-induced dyskinesias, neuroleptic-induced supersensitivity psychosis and refractory schizophrenia. Biol Psychiatry. 1993 Nov 15;34(10):713–738. doi: 10.1016/0006-3223(93)90044-e. [DOI] [PubMed] [Google Scholar]
- 111.Caroff SN, Campbell EC, Havey J, Sullivan KA, Mann SC, Gallop R. Treatment of tardive dyskinesia with donepezil: a pilot study. J Clin Psychiatry. 2001 Oct;62(10):772–775. doi: 10.4088/jcp.v62n1004. [DOI] [PubMed] [Google Scholar]
- 112.Caroff SN, Walker P, Campbell C, et al. Treatment of tardive dyskinesia with galantamine: a randomized controlled crossover trial. J Clin Psychiatry. 2007 Mar;68(3):410–415. doi: 10.4088/jcp.v68n0309. [DOI] [PubMed] [Google Scholar]
- 113.Caroff SN, Martine R, Kleiner-Fisman G, et al. Treatment of levodopa-induced dyskinesias with donepezil. Parkinsonism Relat Disord. 2006 May;12(4):261–263. doi: 10.1016/j.parkreldis.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 114.Tsai HT, Caroff SN, Miller DD, et al. A candidate gene study of tardive dyskinesia in the CATIE schizophrenia trial. Am J Med Genet B Neuropsychiatr Genet. 2009;5(153B):336–340. doi: 10.1002/ajmg.b.30981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Aberg K, Adkins DE, Bukszar J, et al. Genomewide association study of movement-related adverse antipsychotic effects. Biol Psychiatry. Feb 1;67(3):279–282. doi: 10.1016/j.biopsych.2009.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]