

Abstract
Background
The suppressed production of nitric oxide (NO), associated with endothelial dysfunction, is thought to be a cause of ischemia and reperfusion injury of the liver. But findings of the salutary effects of NO enhancement on such injury have been conflicting. In this study, we tested our hypothesis that NO enhancement would attenuate ischemic liver injury. For this purpose, an NO precursor, L-arginine, and a novel NO donor, FK409, were applied to a 2-hour total hepatic vascular exdusion model in dogs.
Study Design
L-arginine was administered IV at a dose of 100 mg/kg twice (n = 5), while 300 mg/kg twice of FK409 was infused continuously into the portal vein (n = 5). The drugs were given to the animals for 30 and 60 minutes before and after ischemia, respectively. Nontreated animals were used as the control (n = 10). Two-week survival, systemic and hepatic hemodynamics indices, liver function tests, energy metabolism, and histopathology were analyzed.
Results
Both treatments comparably augmented hepatic tissue blood flow, decreased liver enzyme release, and increased high-energy phosphate restoration during the reperfusion period, all of which contributed to rescuing all of the treated animals from the 2-hour total hepatic ischemia. In contrast, ischemia caused 70% mortality in the control group. Histologically, structural abnormality and neutrophil infiltration were markedly attenuated by the treatments. Systemic hypotension was observed in the animals treated with FK409, however.
Conclusions
Our data demonstrate that NO enhancement alleviates the liver injury caused by ischemia and reperfusion. The supplementation of L-arginine, rather than FK409, is considered more applicable to clinical use because of the absence of systemic adverse effects.
Vasoconstriction or microcirculatory disturbance induced by superoxide-mediated endothelial cell dysfunction has been considered one of major causes of liver injury associated with ischemia and subsequent reperfusion. Preischemic or postischemic administration of antioxidants, prostanoid-modulating drugs, adenosine-regulating agents, and antiendothelin antibody, all of which act as vasodilators, has been shown to attenuate postischemic hepatic damage.1-3 Nitric oxide (NO), an endothelium-derived relaxing factor with various beneficial biologic actions, is another therapeutic candidate that may protect the liver against the noxious insult. Findings of the salutary effects of NO on ischemia and reperfusion injury of the liver have been conflicting, however. Ma and associates4 demonstrated detrimental results with NO augmentation, but Jaeschke and colleagues5 and Wang and coworkers6 observed no important improvements by the treatment. Although Shiraishi and colleagues7 and Koeppel and associates8 described beneficial properties of NO, their observations were confined to the effects on hepatic blood flow or liver enzyme release.
In the present study, we tested our hypothesis that NO enhancement would attenuate vascular dysfunction, lessen neutrophil-related tissue iniury, and reduce ischemia and reperfusion injury of the liver. For this purpose, we tested an NO precursor, L-arginine, and a novel NO donor, FK409,9 in dogs subjected to 2-hour total hepatic vascular exclusion. Therapeutic effects of the agents were determined by the animals’ 2-week survival, the severity of liver injury, and hepatic and systemic hemodynamic indices.
METHODS
The study was conducted with the approval of the Institutional Animal Care and Use Committee of Hokkaido University and the University of Pittsburgh, and was maanaged according to guidelines issued by the National Institutes of Health and the Public Health Service Policy on the humane use and care of laboratory animals.
Animals
After overnight fasting, adult female beagle dogs, weighing 8.0 to 12.0 kg, were anesthetized with an IV injection of thiopental sodium (Abbott Laboratories, North Chicago, IL), 25 mg/kg. They were then incubated, and anesthesia was maintained by positive mechanical ventilation with a gas mixture of isoflurane (1.0%), nitrous oxide (2 L/min), and oxygen (2 L/min). The right carotid artery and the right jugular vein were cannulated for monitoring arterial and central venous pressures. ECGs and esophageal temperature were monitored during the operation. An electrolyte solution (Plasmalyte; Baxter, Deerfield, IL) was infused continuously into the animals at a rate of 20 ml/kg/h throughout the procedure. Arterial blood gas and electrolytes levels were measured frequently and corrected if necessary by administration of sodium bicarbonate, calcium dichloride, and potassium chloride.
Operative Procedures
After midline laparotomy, the liver was completely skeletonized by dissecting all of the suspensory ligaments and the retrohepatic vena cava from the posterior abdominal wall.1 During the total hepatic vascular exclusion, we used venovenous bypass to decompress the splanchnic venous beds and the infrahepatic IVC. The system was equipped with the centrifugal pump (Biomedicus 520D, Minetonka, MN) and Tygon tubings (Norton Industrial Plastics, Akron, OH) connecting the left femoral and the splenic and left jugular veins. The bypass flow was maintained at more than 50 mL/kg/min. Heparin sodium (Upjohn, Kalamazoo, MI), 50 U/kg, was administered IV 5 minutes before ischemia. Total hepatic vascular exclusion was achieved by cross-clamping the portal vein, the hepatic artery, and the IVC both above and below the liver. The exclusion was continued for 2 hours, and then the liver was reperfused by removing the clamps. Immediately after reperfusion, the bypass system was disconnected and a splenectomy was performed. The animals were allowed to eat and drink the morning after the operation and were observed for 2 weeks. Cefamandole nafate, 1 g, was given intraoperatively, and administration was continued for 3 days postoperatively.
Experimental Groups
Twenty dogs were randomly divided into three groups: group CT (n = l0), untreated control; group LA (n = 5), treated with L-arginine; and group FK (n = 5), treated with FK409. L-arginine (Sigma Chemical Co., St. Louis, MO) was dissolved in physiologic saline at a concentration of 50 mg/mL. The novel NO donor, FK409 (Fujisawa Pharmaceutical Co., Osaka, Japan), was also dissolved in physiologic saline at a concentration of 150 μg/mL. In group LA, L-arginine at a dose of 100 mg/kg was administered IV for 30 minutes before ischemia and for 60 minutes after ischemia, starting 5 minutes before reperfusion. In group FK, FK409 at a dose of 300 μg/kg was given continuously into the portal vein through a branch of the mesenteric veins for 30 minutes before ischemia and for 60 minutes after ischemia, beginning 5 minutes before reperfusion.
Determinations
The cytoprotective effects of the drugs were assessed by the following measures: 2-week animal survival, hepatic tissue blood flow (HTBF), liver enzyme release, serum total bile acid (TBA) and hyaluronic acid (HA) levels, tissue high-energy phosphate levels, and histopathologic findings. The influence of the drugs on systemic hemodynamic indices was evaluated by the mean arterial blood pressure (MAP), which was calculated according to the following equation:
The completeness of total hepatic vascular exclusion was confirmed by the indocyanine green (ICG) dye retention rate during ischemia. Briefly, ICG (Cardio-Green; Becton Dickinson Microbiology Systems, Cockeysville, MD) at a dose of 0.5 mg/kg was given IV 30 minutes after the onset of vascular exclusion, and 3 mL of heparinized arterial blood was collected 20 minutes after dye injection. The ICG retention rate was determined with the use of a spectrophotometer (Lambda; Perkin Elmer, Norwalk, CT) at a wavelength of 805 nm.
Hepatic tissue blood flow
HTBF was measured with use of a laser-Doppler flowmeter (Advance Laser Flowmeter, ALF21; Advance Company Ltd., Tokyo, Japan) before ischemia; 5, 30, 60, 90, and 120 minutes after ischemia; and 5, 15, 30, and 60 minutes after reperfusion. The measurements were repeated three times at three different lobes at each time point. The mean value of the measurements at respective time points was calculated and expressed as a percentage of the preischemic initial value. To avoid a misreading of HTBF measurements related to respiratory motion, we temporarily stopped the ventilator during the measurements.
Liver enzymes
Blood samples for assessment of the release of liver enzymes were collected serially before ischemia; just before reperfusion and at 5, 15, 30, and 60 minutes after reperfusion; 3,6, 12, and 24 hours afier reperfusion; and on postoperative days 2,3,5,7, 10, and 14. A Technicon RA500 autoanalyzer (Bayer, Tarrytown, NY) was used to determine serum levels of aspartate aminotransferase (AST), alanine aminotransferase, and LDH.
Total bile acid and hyaluronic acid
The blood samples for TBA and HA measurements were collected before ischemia, just before reperfision, and 1, 3, 6, 12, and 24 hours after reperfusion. The serum TBA level was measured by the enzymatic fluorimetric method with the Sterongnost-alpha Flu Kit (Nyegaad, Oslo, Norway).10 The serum HA level was determined by the sandwich-binding protein assay of Chichibu and colleagues.11
High-energy phosphates
Wedge liver biopsies were performed before ischemia, at the end of ischemia, and 15 and 60 minutes after reperfusion. Half of the specimen was immediately stored in liquid nitrogen for tissue biochemistry analysis, and the rest was fmed with buffered formalin for histopathologic analysis. Tissue levels of the adenine nucleotides ATP ADP and AMP were determined with a Waters highperformance liquid chromatography system (Model 510 pumps, Model 484 absorbance module, and Model 717 WISP system; Waters Chromatography Division, Millipore Corp., Milford, MA) at 254 nm (Waters Model 484, a tunable absorbance detector). 12 Total adenine nucleotide (TAN) and energy charge (EC) were calculated with the following equations, respectively13:
and
Histopathology
Formalin-fixed specimens were embedded in paraffin and stained with hematoxylin and eosin. Histopathologic analysis was performed by a single pathologist without knowledge of the groups or the timing of the tissue sampling. Histopathologic damage was assessed semiquantitatively based on the degree of sinusoidal congestion, sinusoidal cell derangement, hepatocyte injury, and hepatocyte necrosis, with each index scored as none = 0; mild = 1; moderate = 2; and severe = 3. The number of infiltrated polymorphonuclear cells (PMNs) per 1,000 hepatocyte nuclei was counted by observing the tissues stained by Leder’s method.14
Statistical Analysis
The data for continuous measurements were expressed as mean ± SEM in each of the three study groups of dogs. The animals’ survival rates were compared pairwise by Fisher’s exact probability test. Intergroup analysis was performed using one-way ANOVA. When ANOVA showed a significant difference (p < 0.05), a post hoc test was used to determine the p values for each group. A two-sided p value < 0.05 was considered statistically significant. These procedures were performed with the statistical program Statview (version 4.5; Abacus Concepts, Inc., Berkeley, CA) on a Macintosh computer (Apple Computer, Inc., Cupertino, CA).
RESULTS
General Conditions
Clinically, no bleeding tendency was observed in either the control or the treated animals throughout the procedure. The ICG retention rate was 94.82% ± 1.26%, 95.05% ± 1.33%, and 96.24% ± 1.15% in groups CT, LA, and FK, respectively, indicating the completeness of the total hepatic vascular exclusion.
Systemic Hernodynamics
Mean arterial pressures in groups CT and LA remained stable before ischemia. In contrast, remarkable hypotension developed in the animals treated with FK409, reaching the nadir within 10 minutes afier the initiation of drug infusion (Fig. 1). After the completion of preischemic FK409 administration, the blood pressure recovered rapidly and remained at a level similar to those in other two groups during the ischemic period. After reperfision, serious hypotension developed in all animals. Although the blood pressure was gradually elevated in group LA thereafter, significant hypotension was seen in groups CT and FK. Postreperfusion hypotension of group FK animals continued until the cessation of drug administration.
Figure 1.
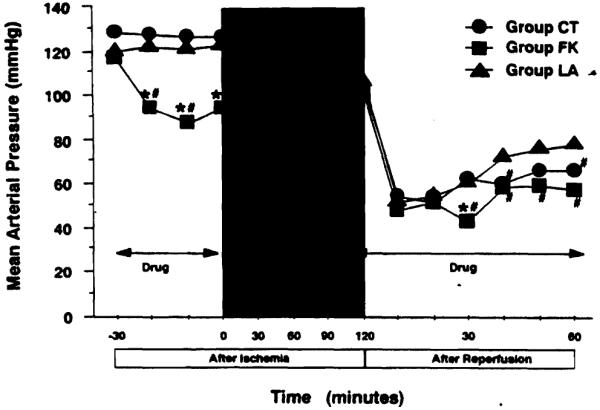
Changes in mean arterial pressure as an indicator of systemic hemodynamics. Mean arterial pressure was calculated according to the following equation: MAP = diastolic arterial pressure + (systolic arterial pressure − diastolic arterial pressure) × 1/3. Group CT, untreated control (n = 10); group LA, treated with L-arginine (n = 5); group FK, treated with FK409 (n = 5). *p < 0.05 versus group CT; #p < 0.05 versus group LA.
Hepatic Tissue Blood Flow
The changes of HTBF in each group are illustrated in Figure 2. The preischemic drug administration induced no remarkable changes in HTBF, representing approximately 100% of the initial value. Despite the complete ischemia, we noted false-positive tissue blood flow ranging from 8% to 11%, which was due to limitations of the Doppler flow measurement technique. Immediately after reperfusion, all Iivers in group CT developed outflow block, characterized by conspicuous swelling, mottled reperfusion, and hard consistency. The HTBF of group CT animals was approximately 25% of the initial value throughout the observation. In contrast, all livers treated with L-arginine or FK409 demonstrated smooth and fast revascularization, with HTBF in the range of 55% to 60%. The HTBF levels in groups LA and FK were two to three times higher than that in group CT throughout the reperfusion period.
Figure 2.
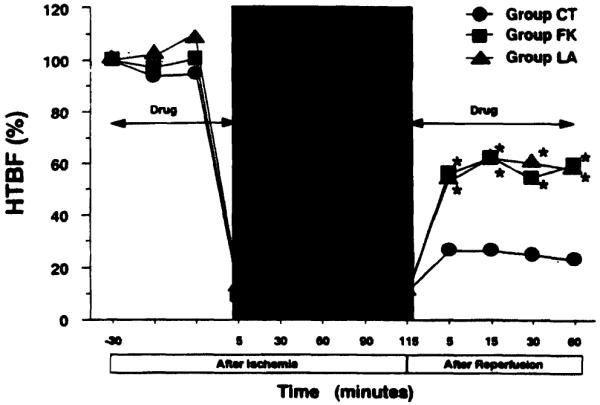
Changes in hepatic tissue blood flow (HTBF). Values are expressed as a percentage of the respective preischemic initial flow. Group CT, untreated control (n = 10); group LA, treated with L-arginine (n = 5); group FK, treated with FK409 (n = 5). *p < 0.05 versus group CT.
Animal Survival
The 2-hour warm ischemia of the liver was poorly tolerated by group CT animals, in which 7 of the 10 dogs died of liver failure; 6 died within 24 hours after reperfusion and 1 on the second postoperative day. In contrast, all of the animals treated with L-arginine or FK409 survived for 2 weeks. The 2-week survival rates of groups CT, LA, and FK were 30%, 100%, and 100%, respectively (group LA versus group CT, and group FK versus group CT, p < 0.05 for each comparison).
Liver Function Tests
Serum AST levels in control animals rose rapidly after reperfusion, reaching a peak value of 11,521 ± 1,014 U/L at 12 hours; peak AST levels in groups LA and FK were 3,593 ± 1,260 and 3,574 ± 1,154 U/L, respectively (Fig. 3). Compared with the control group, AST levels in the treated groups were significantly lower from 3 hours after reperfusion until postoperative day 3. No substantial difference was found in AST levels between the treated groups. Serum levels of alanine aminotransferase changed similarly to those of AST (Table 1). Serum LDH levels reached the peak value within 15 minutes after reperfusion and slowly declined thereafter. The elevation of LDH was markedly suppressed in groups LA and FK compared wid CT (Table 1).
Figure 3.
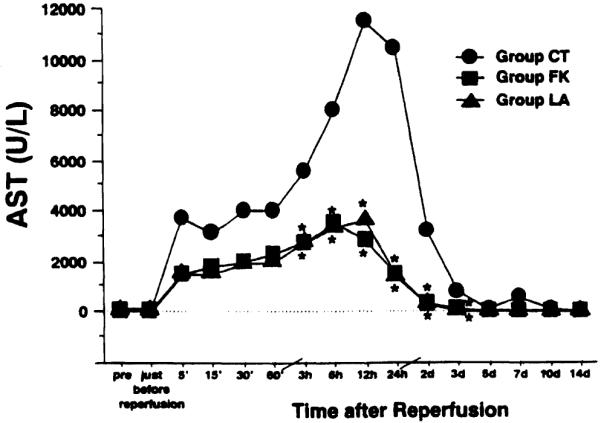
Changes in serum aspartate aminotransferase (ASV level after reperfusion. Group CT, untreated control (n = 10); group LA, treated with L-arginine (n = 5); group FK, treated with FK409 (n = 5). *p < 0.05 versus group CT.
Table 1.
Alanine Aminotransferase, Lactate Dehydrogenase, Total Bile Acid, and Hyaluronic Acid Levels After Reperfusion
| Measurement and group |
Before ischemia |
End of ischemia |
After Reperfusion |
||||
|---|---|---|---|---|---|---|---|
| l h | 3 h | 6 h | 12 h | 24 h | |||
| ALT | |||||||
| CT | 32 ± 4 | 33 ± 3 | 4,961 ± 798 | 6,576 ± 1,007 | 9,332 ± 1,128 | 13,563 ± 1,379 | 12,680 ± 1,451 |
| LA | 17 ± 3 | 21 ± 2 | 1,610 ± 373* | 2,277 ± 503* | 2,732 ± 601* | 3,214 ± 661* | 2,378 ± 517* |
| FK | 19 ± 6 | 26 ± 15 | 2,202 ± 758* | 2,814 ± 1139* | 3,766 ± 1,568* | 3,410 ± 1,521* | 2,789 ± 1,388* |
| LDH | |||||||
| CT | 81 ± 16 | 173 ± 27 | 3,385 ± 474 | 3,128 ± 479 | 2,906 ± 559 | 3,046 ± 634 | 1,441 ± 216 |
| LA | 72 ± 12 | 185 ± 52 | 1,391 ± 355* | 1,250 ± 304* | 968 ± 254* | 693 ± 186* | 261 ± 49* |
| FK | 121 ± 57 | 165 ± 99 | 1,991 ± 704 | 1,628 ± 623* | 1,142 ± 314* | 603 ± 192* | 343 ± 128* |
| TBA | |||||||
| CT | 0.3 ± 0.3 | 29.9 ± 6.3 | 48.5 ± 11.7 | 100.2 ± 11.0 | 133.8 ± 11.8 | 124.3 ± 13.1 | 175.4 ± 39.0 |
| LA | — | — | 70.6 ± 19.4 | 87.6 ± 25.1 | 58.0 ± 35.2* | 41.2 ± 22.4* | 20.2 ± 6.7* |
| FK | — | — | 49.6 ± 10.8 | 83.6 ± 23.2 | 68.8 ± 24.4* | 53.2 ± 13.4* | 53.4 ± 24.5* |
| HA | |||||||
| CT | 16.7 ± 6.5 | 348.9 ± 68.1 | 78.1 ± 27.5 | 61.4 ± 17.4 | 82.4 ± 30.9 | 112.6 ± 23.8 | 515.3 ± 167.5 |
| LA | 9.4 ± 4.3 | 371.2 ± 113.5 | 28.8 ± 6.5 | 42.3 ± 22.7 | 39.4 ± 13.6 | 36.2 ± 10.3* | 201.1 ± 50.0 |
| FK | 12.2 ± 7.5 | 261.0 ± 84.8 | 22.8 ± 13.1 | 74.0 ± 33.2 | 55.9 ± 26.8 | 47.1 ± 13.4* | 188.4 ± 60.2* |
ALT, alanine aminotransferase; CT, untreated control (n = 10); FK, group treated with FK409 (n = 5); HA, hyaluronic acid; LA, group tested with L-arginine (n = 5); LDH, lactate dehydrogenase; TBA, total bile acid.
p < 0.05 versus group CT. Data are expressed as mean ± SEM.
Total Bile Acid and Hpluronic Acid Levels
Serum HA levels exhibited two-phase changes; characterized by peaks at the end of ischemia and at 24 hours after reperfusion. Repeat elevation of HA levels during the reperfusion period was significantly suppressed in groups LA and FK compared with group CT (Table 1).
Serum TBA levels rose similarly in all groups until 3 hours after reperfusion (Table 1). The level in group CT animals rose progressively thereafter, but those of groups LA and FK animals gradually reverted to preoperative levels.
Energy Metabolism
Changes in the tissue levels of adenine nucleotides are summarized in Table 2. Two hours of total hepatic vascular exclusion induced significant decreases in tissue ATP and ADP levels, with a concomitant rise of AMP and decrease in TAN. AMP and TAN in groups LA and FK remained at significantly higher levels than those in the control group at the end of ischemia, however, suggesting the delayed decay of adenine nucleotides to purine catabolites during the ischemic period. Although high-energy phosphates were restored rather quickly in all groups after reperfusion, resynthesis of ATP, ADP and TAN was more rapid and significant in the animals treated with L-arginine and FK409 than in those in the control group. The energy charge showed no significant differences at any time point among the groups.
Table 2.
Tissue Concentrations of Adenine Nucleotides (nmol/g Tissue) During Ischemia and After Reperfusion
| Examination time | ATP | ADP | AMP | TAN | Energy charge |
|---|---|---|---|---|---|
| Before ischemia | |||||
| CT | 2,666.2 ± 310.6 | 894.0 ± 11.2 | 213.6 ± 29.0 | 3,773.7 ± 314.1 | 0.82 ± 0.02 |
| End of ischemia | |||||
| CT | 236.9 ± 45.2 | 335.0 ± 29.1 | 1,147.1 ± 107.2 | 1,718.9 ± 147.8 | 0.23 ± 0.03 |
| LA | 452.1 ± 187.3 | 665.0 ± 236.0 | 1,889.1 ± 185.9* | 2,925.7 ± 560.7* | 0.23 ± 0.04 |
| FK | 338.9 ± 104.7 | 491.1 ± 141.9 | 1,706.6 ± 53.0* | 2,536.8 ± 221.0 | 0.20 ± 0.05 |
| 15 min after reperfusion | |||||
| CT | 846.8 ± 170.2 | 409.9 ± 65.6 | 160.0 ± 17.8 | 1,416.8 ± 234.0 | 0.72 ± 0.04 |
| LA | 2,071.8 ± 338.6* | 738.3 ± 146.7 | 276.4 ± 55.2 | 3,086.5 ± 365.8* | 0.78 ± 0.04 |
| FK | 2,041.2 ± 593.3* | 866.0 ± 170.5* | 431.8 ± 121.5* | 3,339.2 ± 424.5* | 0.74 ± 0.08 |
| 60 min after reperfusion | |||||
| CT | 1,227.2 ± 138.3 | 329.4 ± 33.4 | 112.2 ± 14.7 | 1,668.8 ± 160.1 | 0.83 ± 0.02 |
| LA | 2,181.4 ± 504.1 | 768.1 ± 94.5* | 205.6 ± 47.7* | 3,155.1 ± 527.1* | 0.80 ± 0.02 |
| FK | 2,314.6 ± 401.5* | 774.6 ± 105.8* | 274.2 ± 24.5* | 3,363.4 ± 440.8* | 0.78 ± 0.02 |
p < 0.05 versus group CT. Data are expressed as mean ± SEM.
ADP, adenosine diphosphate; AMP, adenosine monophosphate; ATP, adenosine triphosphate; CT, untreated control (n = 10); FK, group treated with FK409 (n = 5); LA, group treated with L-arginine (n = 5), TAN, total adenine nucleotides.
Histopathology
Treatment with L-arginine or FK409 induced no evident histopathologic differences between the treated groups and the control group at the end of ischemia or 15 minutes after reperfusion. At 60 minutes after reperfusion, however, control livers were characterized by sinusoidal congestion, sinusoidal cell derangement, ischemic hepatocyte changes, and hepatocyte necrosis, all of which were significantly attenuated by the treatments (Fig. 4). In particular, the degrees of hepatocyte necrosis and ischemic hepatocyte changes were remarkably lessened in the treated groups (Fig. 5A).
Figure 4.

Histologic examination of liver tissue 60 minutes after reperfusion (hematoxylin and eosin, X25). (A) Untreated control liver, Although basic architecture was preserved, severe sinusoidal congestion, mild sinusoidal cell derangement, and mild hepacocyte injury and necrosis were found. (B) The liver treated with FK409. Although mild sinusoidal congestion and sinusoidal cell derangement were recognized, neither hepatocyte injury nor necrosis was found. (C) The liver treated with L-arginine. Mild sinusoidal cell derangement was found, but sinusoidal congestion and hepatocyte injury and necrosis wur not observed.
Figure 5.

(A) Hiologic damage score. Hiologic damage was assessed semiquantitatively based on the extent of sinusoidal congestion, sinusoidd derangement, hepatocyte injury, and hepatocyte necrosis. Each index was scored as none = 0; mild = 1; moderate = 2; and severe = 3. (B) Number of infiltrated polymorphonuclear cells (PMNs) in liver tissue. Data are expressed as mean ± SEM per 1,000 hepatocyte nuclie. Group CT, untreated control (n = 10); group LA, treated with L-arginine (n = 5); group FK, treated with FK409 (n = 5). *p < 0.05 versus group CT.
At the end of ischemia, no substantial changes were found in the number of infiltrated PMNs among the three groups. The PMN number in the control animals gradually and markedly increased after reperfusion, but the increases were significantly suppressed in groups LA and FK (Fig. 5B). No significant differences were seen in the number of infiltrated PMNs between the treated groups (Fig. 5B).
DISCUSSION
This study showed that hepatic warm ischemia for 2 hours caused severe liver injury after reperfusion, resulting in a 70% mortality rate for control animals. In contrast, enhancement of NO by L-arginine and a novel NO donor, FK409, exhibited a remarkable dleviating effect on such injury. Pre- and postischemic administration of these agents allowed 100% survival of the animals, augmented HTBF, enhanced bile acid metabolism, and lessened hepatocyte damage. In addition, the treatments prevented the decay of high-energy phosphates during ischemia and accelerated ATP restoration after reperhion. Hitopathologic alterations were markedly attenuated by the treatments, with less PMN infiltration into postischemic liver tissues.
NO is synthesized from NO synthases (NOS) through the L-arginine to L-ciuulline pathway.15 NOS are classified into two different types: calcium-dependent constitutive NOS and calcium-independent inducible NOS.16 Constitutive NOS are located in vascular endothelial cells and produce an appropriate amount of NO in response to circulatory environments,17 but inducible NOS synthesize NO excessively when stimulated by certain cytokines or endotoxin.18 NO has various biologic actions such as vasodilation (vascular smooth muscle relaxation) and inhibition of platelet aggregation; a mechanism for both involves the elevation of cyclic guanosine monophosphate following soluble guanylate cyclase activation by N0.19,20 NO also has inhibitory effects on the endothelium-neutrophil interaction, partially through down-regulation of the expression of adhesion molecules both on endothelium and neutrophiis21,22 and of the production of cytokines by inflammatory cells.23 In the liver, NO has been reported to exert hepatic sinusoidal dilation and to improve the microcirculation of this organ by altering the morphofunctional activity of fit-storing (Ito) cells.24
Our hypothesis in this study was based upon evidence that a decrease of NO production by injured endothelial cells through the attack of reactive oxygen species eliminate is one of the important mechanisms responsible for organ injury caused by ischemia and reperfusion.25 We experimented with elevation of the NO level by using the NO precursor L-arginine and a novel NO donor, FK409. To our knowledge, this is the first report to demonstrate not only that NO enhancement reduced ischemia and reperfusion injury of the liver, but also that treatment with L-arginine and FK409 rescued all dogs from the grave ischemic insult that caused 70% mortality in control animals.
Shiraishi and associates7 also demonstrated that portal injection of L-arginine at a dose of 10 mg/kg induced a temporal burst of inuahepatic NO after reperfusion and mitigated the injury of rat and porcine livers subjected to 1 hour of ischemia followed by reperfusion. On the other hand, by using an isolated rat liver perfusion model, Ma and colleagues4 found that the inhibition of NO production by Nω-nitro-L-arginine methyl ester hydrochloride (L-NAME) attenuated liver injury after ischemia. Alternatively, Jaeschke and coworkers5 observed no substantial effects of L-NAME treatment on rat liver ischemic injury. Although the conflicting results between our study and the others might be due to differences in animal species, models, and agents tested, the data from our preclinical study in dogs reveal the manifest protective effects of NO against ischemia and reperfusion injury in the liver.
Although the NO level was not determined directly in this study, we believe that L-arginine administration augmented NO production in sinusoidal endothelial cells. Mammalian vascular endothelial cells produce NO, L-ciulllline, and water fiom L-arginine and oxygen as substrates through the participation of NOS. Little information has been available as to the quantity of NO production by the cells aftcr exogenous L-arginine administration under normal conditions.26,27 It has been proved, however, that once the cells are injured, they take up L-arginine preferentially28 and produce NO depending on L-arginine availability.29 Kakumitsu and associates30 reported that exogenous L-arginine (30 g/body) selectively decreased the sinusoidal resistance and augmented the hepatopetal portal flow in cirrhotic patients who had deteriorated sinusoida cells compared with healthy subjects, who also demonstrated increased urinary excretion of an NO metabolite, nitrate. Huk and coauthors31 demonstrated that the administration of L-arginine maintained NO production during reperfusion in a rabbit skeletal muscle-ischemia model. In addition, the enantiomer of L-arginine (D-arginine), which has the same effect on humoral factors such as insulin and glucagon32 has been reported to have no effects on ischemia and reperfusion injury.33 These facts suggest that L-arginine administration in our study exerted its effects through enhanced NO production in the injured sinusoidal endothelial cells rather than through humoral effects. On the other hand, NO enhancement by FK409 administration is evident because the agent spontaneously releases NO in the blood at a linear ratio of 1:1 between its degradation and NO formation.
FK409 is a novel semisynthetic product of Streptomyces griseosporeus that acts as an NO donor, characterized by the rapid release of NO with a half-life of 6 minutes in canine blood.9,34,35 Both L-arginine and FK409 provided similar and comparable hepatoprotective effects in our model; however, these agents had different influences on systemic hemodynamic indices. L-arginine administration caused minimal systemic hemodynamic changes which was consistent with previous reports.26,36 In contrast, FK409 caused transient but significant hypotension limited to the drug infusion period, even,though this agent was given through the portal vein. Although the exact mechanism remains unclear, this phenomenon might be attributable to differences in the magnitude of NO production and in the organ specificity between the agents because the injured sinusoidal endothelial cells are thought to take up L-arginine and to produce NO eliminate.
Mechanisms by which NO provided hepatoprotective effects against ischemia and reperfusion injury were not fully determined in our study, but several possibilities can be considered. First, it could be due to amelioration of the microcirculatory disturbance by the vasodilatory effect of NO. In this srudy, the treatments with L-arginine or FK409 restored postischemic HTBF to two- to three fold that of the control group. NO has been proved to regulate hepatic sinusoidal circulation through morphofunctional influences on Ito cells,24 rather than through a direct effect on portal or arterial vascular smooth muscle cells. Feletou and coworkers37 showed that the canine portal mesenteric veins were insensitive to NO stimulation. Because NO has been shown to modulate other vasoactive substances such as endodthelin38 and prostanoids,39 NO-driven changes of these vasoactive substances might contribute, at least in part, to the improvement in the hepatic microcirculation after reperfusion.
Second, NO could exert hepatoprotective effects by inhibiting PMN adherence to the endothelium. PMNs, when adhered to endothelial cells and activated, have been demonstrated to aggravate liver damage by releasing myeloperoxidase, elastase, and proteolytic enzymes.40 NO itself inhibits PMN adhesion in vitro.41 Lefer and colleagues42 also demonstrated that an NO donor, SPM5 185, lessened PMN adhesion to the endothelium in a canine heart model, which is consistent with our observation that the treatments significantly reduced the number of PMNs in postischemic liver tissues. Because L-NAME, a NOS antagonist, inhibits NO production and up-regulates both P-selectin on the endothelium and integrins on PMNs, the reduced numbers of infiltrated PMNs in the treated groups in this study might have resulted from down-regulation of the expression of adhesion molecules by augmented NO.21,22
The third possible explanation for the alleviating effects of NO on liver ischemia and reperfusion injury relates to its superoxide scavenging property. Superoxide radicals are implicated as an initial and potential mediator of ischemia and reperhion injury.25 Although it is also known that NO can combine with superoxide radicals to form the more highly toxic free radical species, peroxynitrite,43 peroxynitrite is unlikely to be involved in our model because a very high level of peroxynitrite (ie, at least 500 μmol/L) is required to produce cytotoxic effects.44 In this study, the NO level was estimated to be 1 × l0−7 mol/L according to the pharmacokinetics FK409, which is 5,000 times lower than the toxic level of peroxynitrite. Most important, the presence of peroxynitrite has never been proved in an in vivo system.
Finally, an alternative explanation for the beneficial effects of the treatments might involve energy metabolism. The treatments maintained energy metabolism at the end of ischemia and promoted its restoration after reperfusion. NO modulates a key enzyme of the Krebs cycle (mitochondrial aconitase) and several mitochondrial electron transport casc ades.45,46 NO might act as an energy-sparing agent during ischemia. After reperfusion, the increase in HTBF by the treatments could contribute to the delivery of more nutrients and oxygen to the liver and improve the restoration of energy metabolism. Although the precise mechanism is not fully understood, favorable energy metabolism undoubtedly contributed to the better survival in the treated animals.
In conclusion, we demonstrated the marked hepatoprotective effects of L-arginine and FK409 against severe ischemia and reperfusion injury in canine livers. Both agents attenuated liver injury and rescued all animals from the critical insult. Although further studies are required to elucidate the exact mechanisms by which L-arginine and NO donors prevent ischemic liver injury, the supplementation of L-arginine, rather than FK409, appears to be a possible strategy in clinical settings because of its absence of adverse hypotensive effects.
Acknowledgment
The authors thank Dr Saitoh, Associate Professor, Department of Hygiene, Hokkaido University School of Medicine, for reviewing the statistical analyses.
Aided by research grants from the Veterans Administration and Project Grant no. DK-29961 from the National Institutes of Health, Bethesda, MD.
References
- 1.Todo S, Zhu Y, Zhang S, et al. Attenuation of ischemic liver injury by augmentation of endogenous adenosine. Transplantation. 1997;63:217–223. doi: 10.1097/00007890-199701270-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ishizaki N, Zhu Y, Zhang S, et al. Comparison of various lazaroid compounds for protection against ischemic liver injury. Transplantation. 1997;63:202–208. doi: 10.1097/00007890-199701270-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Urakami A, Todo S, Zhu Y, et al. Attenuation of ischemic liver injury by monoclonal anti-endorhclin antibody, AwETN40. J Am Coll Surg. 1997;185:358–364. [PMC free article] [PubMed] [Google Scholar]
- 4.Ma TT, Ischiropoulos H, Brass CA. Endotoxin-stimulated nitric oxide production increases injury and reduces rat liver chemiluminescence during reperfusion. Gastroenterology. 1995;108:463–469. doi: 10.1016/0016-5085(95)90075-6. [DOI] [PubMed] [Google Scholar]
- 5.Jaeschke H, Schini VB, Farhood A. Role of nitric oxide in the oxidant stress during ischemia/reperfusion injury of the liver. Life Sci. 1992;50:1797–1804. doi: 10.1016/0024-3205(92)90064-v. [DOI] [PubMed] [Google Scholar]
- 6.Wang Y, Mathews R, Guido DM, et al. Inhibition of nitric oxide synthesis aggravates reperfusion injury after hepatic ischemia and endotoxemia. Shock. 1995;4:282–288. doi: 10.1097/00024382-199510000-00009. [DOI] [PubMed] [Google Scholar]
- 7.Shiraishi M, Hiroyasu S, Nagahama M, et al. Role of exogenous L-arginine in hepatic ischemia-reperfusion injury. J Surg Res. 1997;69:429–434. doi: 10.1006/jsre.1997.5094. [DOI] [PubMed] [Google Scholar]
- 8.Koeppel TA, Thies JC, Schemmer P, et al. Inhibition of nitric oxide synthesis in ischemia and reperfusion of the rat liver is followed by impairment of hepatic microvascular blood flow. J Hepato1. 1997;27:163–169. doi: 10.1016/s0168-8278(97)80297-8. [DOI] [PubMed] [Google Scholar]
- 9.Hino M, Iwaki M, Okamoto M, et al. FK409, a novel vasodilator isolated from the acid-treated fermentation broth of streptomyces griseosporeus. J Antibiot (Tokyo) 1989;42:1578–1583. doi: 10.7164/antibiotics.42.1578. [DOI] [PubMed] [Google Scholar]
- 10.Mashige F, Imai K, Osuga T. A simple and sensitive assay of tool serum bile acids. Clin Chim Acta. 1976;70:79–86. doi: 10.1016/0009-8981(76)90007-3. [DOI] [PubMed] [Google Scholar]
- 11.Chichibu K, Matsuura T, Shichijo S, Yokoyama MM. Assay of serum hyaluronic acid in clinical application. Clin Chim Acta. 1989;181:317–324. doi: 10.1016/0009-8981(89)90237-4. [DOI] [PubMed] [Google Scholar]
- 12.Hamamoto I, Takaya S, Todo S, et al. Can adenine nucleotides predict primary nonfunction of the human liver homograft? Transplant. 1994;7:89–95. doi: 10.1007/bf00336468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Atkinson DE. The energy charge of the adenylate pool as a regulatory parameter: interaction with feedback modifiers. Biochemistry. 1968;7:4030–4034. doi: 10.1021/bi00851a033. [DOI] [PubMed] [Google Scholar]
- 14.Stevens A. Enzyme histochemistry: diagnostic applications. In: Bancroft JD, Stevens A, editors. Theory and Practice of Histological Techniques. Churchill Livingstone; New York: 1990. pp. 401–411. [Google Scholar]
- 15.Palmer RMJ, Ashton DS, Moncada S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature. 1988;333:664–666. doi: 10.1038/333664a0. [DOI] [PubMed] [Google Scholar]
- 16.Dinerman JL, Lowenstein CJ, Snyder SH. Molecular mechanisms of nitric oxide regulation. Potential relevance to cardiovascular disease. Circ Res. 1993;73:217–222. doi: 10.1161/01.res.73.2.217. [DOI] [PubMed] [Google Scholar]
- 17.Baek KJ, Thiel BA, Lucas S, Stuehr DJ. Macrophage nitric oxide synthase subunits. J Biol Chem. 1993;268:21120–21129. [PubMed] [Google Scholar]
- 18.Nussler AK, Billiar TR. Inflammation, immunoregulation, and inducible nitric oxide synthase. J Leukoc Biol. 1993;54:171–178. [PubMed] [Google Scholar]
- 19.Rapport RM, Murad F. Agonist-induced endothelium-dependent relaxation in rat thoracic aorta may be mediated through cGMP. Circ Res. 1983;52:352–357. doi: 10.1161/01.res.52.3.352. [DOI] [PubMed] [Google Scholar]
- 20.Radamoski MW, Palmer RMJ. Endogenous nitric oxide inhibits human platelet adhesion to vascular endothelium. Lancet. 1987;ii:1057–1058. doi: 10.1016/s0140-6736(87)91481-4. [DOI] [PubMed] [Google Scholar]
- 21.Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci USA. 1991;88:4651–4655. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davenpeck KL, Gauthier TW, Lefer AM. Inhibition of endothelil-derived nitric oxide promotes P-selectin expression and actions in the rat microcirculation. Gastroenterology. 1994;107:1050–1058. doi: 10.1016/0016-5085(94)90229-1. [DOI] [PubMed] [Google Scholar]
- 23.Harbrecht BG, Wang SC, Simmons RL, Billiar TR. Cyclic GMP and guanylate cyclase mediate lippolysaccharide-induced Kupffer cell tumor necrosis factor-a synthesis. J Leukoc Biol. 1995;57:297–302. doi: 10.1002/jlb.57.2.297. [DOI] [PubMed] [Google Scholar]
- 24.Kawada N, Tran-thi TA, Klein H, Decker K. The contraction of hepatic stellate (Ito) cells stimulated with vasoactive substances. Possible involvement of endothelin 1 and nitric oxide in the regulation of the sinusoidal tonus. Eur J Biochem. 1993;213:815–823. doi: 10.1111/j.1432-1033.1993.tb17824.x. [DOI] [PubMed] [Google Scholar]
- 25.Tsao PS, Lefer AM. Time course and mechanism of endothelial dysfunction in isolated ischemic- and hypoxic-perfused rat hearts. Am J Physiol. 1990;259:H1660–1666. doi: 10.1152/ajpheart.1990.259.6.H1660. [DOI] [PubMed] [Google Scholar]
- 26.Langle F, Steininger R, Waldmann E, et al. Improvement of cardiac output and liver blood flow and reduction of pulmonary vascular resistance by intravenous infusion of L-arginine during the early reperfusion period in pig liver transplantation. Transplantation. 1997;63:1225–1233. doi: 10.1097/00007890-199705150-00007. [DOI] [PubMed] [Google Scholar]
- 27.Rees D, Palmer M, Moncada S. Role of endothelium-derived nitric oxide in the regulation of blood pressure. Proc Natl Acad Sci USA. 1989;86:3375–3378. doi: 10.1073/pnas.86.9.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Girerd XJ, Hirsch AT, Cooke JP, et al. L-arginine augments endothelium-dcpendent vasodilation in cholesterol-fed rabbits. Circ Res. 1990;67:1301–1308. doi: 10.1161/01.res.67.6.1301. [DOI] [PubMed] [Google Scholar]
- 29.Fineman J, Chang R, Soifer S. L-arginine, a precursor of EDRF in vivo, produces pulmonary vasodilation in lambs. Am J Physiol. 1991;261:H1563–1569. doi: 10.1152/ajpheart.1991.261.5.H1563. [DOI] [PubMed] [Google Scholar]
- 30.Kakumitsu S, Shijo H, Yokovama M, et al. Effect of L-arginine on the systemic, mesenteric, and hepatic circulation in patients with cirrhosis. Hepatology. 1992;7:377–382. doi: 10.1002/hep.510270210. [DOI] [PubMed] [Google Scholar]
- 31.Huk I, Nanobashvili J, Neumayer C, et al. L-arginine treament alters the kinetics of nitric oxide and superoxide release and reduces ischemia/reperfusion injury in skeletal muscle. Circulation. 1997;96:667–675. doi: 10.1161/01.cir.96.2.667. [DOI] [PubMed] [Google Scholar]
- 32.Palmer JP, Walter RM, Ensick JW. Arginine-stimulated acute phase of insulin and glucagon secretion: I. In normal man. Diabetes. 1975;24:735–740. doi: 10.2337/diab.24.8.735. [DOI] [PubMed] [Google Scholar]
- 33.Weyrich AS, Ma XL, Lefer AM. The role of L-arginine in ameliorating reperfusion injury after myocardial ischemia in the cat. Circulation. 1992;86:279–288. doi: 10.1161/01.cir.86.1.279. [DOI] [PubMed] [Google Scholar]
- 34.Fukuyama S, Kita Y, Hirasawa Y, et al. A new nitric oxide (NO) releaser: spontaneous NO releax from FK409. Free Radic Res. 1995;23:443–452. doi: 10.3109/10715769509065265. [DOI] [PubMed] [Google Scholar]
- 35.Isono T, Sato N, Yamamoto T, et al. Tolerance to the vascular effect of a novel nitric oxide-donating vasodilator, FK409. Eur J Pharmacol. 1994;260:163–168. doi: 10.1016/0014-2999(94)90333-6. [DOI] [PubMed] [Google Scholar]
- 36.Aisaka K, Gross S, Griffth O, Levi R. NG-methyl-L-arginine, an inhibitor of endothelium-derived nitric oxide synthesis, is a potent pressor agent in the guinea pig: does nitric oxide regulate blood pressure in vivo? Biochem Biophys Res Commun. 1989;160:881–886. doi: 10.1016/0006-291x(89)92517-5. [DOI] [PubMed] [Google Scholar]
- 37.Feletou M, Hoeffner U, Vanhoutte P. Endothelium-derivcd relaxing factors do not affect the smooth muscle of portal-mesenteric veins. Blood Vessels. 1989;26:21–32. [PubMed] [Google Scholar]
- 38.Kourembanas S, McQuillan LP, Leung GK, Faller DV. Nitric oxide regulates the expression of vasoconstrictors and growth factors by vascular endothelium under both normoxia and hypoxia. J Clin Invest. 1993;92:99–104. doi: 10.1172/JCI116604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hotter G, Closa D, Pi F, et al. Nitric oxide and arachidonate metabolism in ischemia-reperfusion associated with pancreas transplantation. Transplantation. 1995;59:417–421. [PubMed] [Google Scholar]
- 40.Granger DN. Role of xanrhine oxide and granulocytes in ischemia-reperfusion injury. Am J Physiol. 1988;255:H1269–H1275. doi: 10.1152/ajpheart.1988.255.6.H1269. [DOI] [PubMed] [Google Scholar]
- 41.McCall T, Whittle BJR, Boughton-Smith NK, Moncada S. Inhibition of FMLP-induced aggregation of rabbit neutrophils by nitric oxide [abstract] Br J Pharmacol. 1988;95:517P. [Google Scholar]
- 42.Lefer DJ, Nakanishi K, Johnston WE, Vinten-Johansen J. Antineutrophil and myocardial protecting actions of a novel nitric oxide donor after acute myocardial ischemia and reperfusion in dogs. Circulation. 1993;88:2337–2350. doi: 10.1161/01.cir.88.5.2337. [DOI] [PubMed] [Google Scholar]
- 43.Beckman JS, Beckman TW, Chen J, et al. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Radi R, Beckman JS, Bush KM, Freeman BA. Peroxyynitrite-induced membrane lipid peroxidation: the cytotoxic potential of superoxide and nitric oxide. Arch Biochem Biophys. 1991;2:481–487. doi: 10.1016/0003-9861(91)90224-7. [DOI] [PubMed] [Google Scholar]
- 45.Stuehr DJ, Nathan CF. Nitric oxide: a macrophage product responsible for cytostsis and respiratory inhibition in tumor cells. J Exp Med. 1989;169:1543–1555. doi: 10.1084/jem.169.5.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stadler J, Billiar TR, Curran RD, et al. Effect of endogenous and exogenous nitric oxide on mitochondrial respiration of rat hepatocyces. Am J Physiol. 1991;260:C910–C916. doi: 10.1152/ajpcell.1991.260.5.C910. [DOI] [PubMed] [Google Scholar]