

Abstract
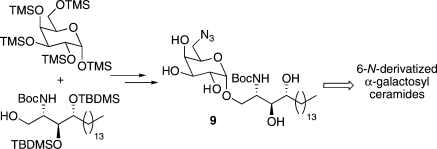
A concise route to the 6-azido-6-deoxy-α-galactosyl-phytosphingosine derivative 9 is reported. Orthogonal protection of the two amino groups allows elaboration of 9 into a range of 6-N-derivatized α-galactosyl ceramides by late-stage introduction of the acyl chain of the ceramide and the 6-N-group in the sugar headgroup. Biologically active glycolipids 6 and 8 have been synthesized to illustrate the applicability of the approach.
The synthetic glycolipid α-galactosyl ceramide (α-GalCer),(1) also known as KRN7000 (1) (Figure 1), has been shown to bind to the protein CD1d. Recognition of the resulting glycolipid−protein complex by T cell receptors (TCRs) located on the surface of invariant natural killer T (iNKT) cells leads to activation of the immune response through the release of a diverse range of cytokines, including both Th1 (IFNγ) and Th2 (IL-4) cytokines.2−4 The release of Th1 cytokines may contribute to antitumor and antimicrobial functions, while the release of Th2 cytokines is believed to play a role in alleviating autoimmune diseases5−7 such as multiple sclerosis(8) and arthritis.(9) When both Th1 and Th2 cytokines are released together, their effects oppose one another, which may induce mixed and unpredictable biological effects.(10) This is the case upon α-GalCer/CD1d activation of the immune response, which has complicated efforts to develop KRN7000 as a therapeutic agent. The search for analogues of this glycolipid, which induce a more biased Th1/Th2 response, is therefore a current focus of many immunological studies.
Figure 1.

Prototypical KRN7000 (1) and biologically active analogues 2, 3, 4, and 5.
A number of α-GalCer analogues have been reported that exhibit more skewed Th1/Th2 cytokine profiles compared with that elicited by α-GalCer 1 (Figure 1).(11) Truncation of the acyl(12) (2) and sphingosine8,13 (OCH (3)) chains and the incorporation of unsaturation in the acyl chain (α-GalCer C20:2 (4))(14) result in CD1d agonists that generate a more Th2-biased response. Examples of more Th1-biasing molecules are much rarer;15,16a the C-glycosyl analogue of KRN7000, α-C-GalCer (5), is one such molecule that has been shown to induce a useful Th1-biased response.(15)
There has been recent interest in galactosyl ceramides in which the hydroxyl group at the 6-position of the sugar headgroup has been modified (Figure 2).(16) Crystal structures of the CD1d-KRN7000 complex(17) and a TCR-KRN7000-CD1d complex(18) show that the 6-hydroxyl group is not involved directly in hydrogen bonding with either the CD1d protein or the TCR. Moreover, it has been shown that the TCR-glycolipid-CD1d interaction can tolerate derivatization at C6.16d,19 Savage has reported that GalCer 6 (PBS-57) offers improved solubility over KRN7000, effectively stains both mouse and human NKT cells, and stimulates cytokine release at low concentrations.(16b) Wang has synthesized the biotinylated α-GalCer analogue 7 to facilitate detection of this antigen in flow cytometry assays.(16c) In this study, the problematic low solubility of these α-GalCer analogues was remedied by incorporating a truncated acyl chain. Of particular interest, Van Calenbergh recently found that substituting the 6-hydroxyl group of 1 for a range of aryl ureas, such as 8, induced a more Th1-biased cytokine response than is observed with α-GalCer.(16a)
Figure 2.

Biologically active 6-derivatized α-galactosyl ceramides 6−8 and our target precursor 9.
Previously reported routes to 6-derivatized α-GalCer analogues are somewhat lengthy, involve extensive protecting group manipulation, and often suffer from poor overall yields. In addition, they do not allow the late-stage variation of the acyl chain; a change of acyl chain therefore requires a complete repeat of the synthetic sequence. Here we present a concise synthesis of the orthogonally protected diamino α-GalCer analogue 9 (Figure 2), which is primed for further elaboration to 6-N-derivatized galactosyl ceramides. The described route allows both the acyl chain of the ceramide unit and the 6-substituent of the sugar headgroup to be varied at a late stage of the synthesis, thus providing access to a potentially large and varied library of this class of compound.
The retrosynthesis of our target 9 is shown in Figure 3. With careful choice of protecting groups, it was envisaged that 9 (or the potentially more useful Boc-deprotected derivative 10) could be accessed in a single step from fully protected azide 11, which in turn could be accessed from alcohol 12, using a Mitsunobu reaction to install the azide. At this point we would require a selective monodeprotection of the primary 6-OH for which there was precedent from Fernández, who showed that 1,2,3,4-tetra-O-trimethylsilylgalactose could be obtained by treating 1,2,3,4,6-penta-O-trimethylsilyl-galactose 13 with acetic acid.(20) We therefore reasoned that alcohol 12 could be accessed from 14 in a similar manner. Glycoside 14 was further disconnected to glycosyl donor 15 and sphingosine acceptor 16.
Figure 3.

Retrosynthetic analysis of targets 9 and 10.
Trimethylsilyl groups were chosen as the protecting groups for the sugar unit owing to (1) their ease of attachment to the galactose starting material,20,23 (2) the ability to monodeprotect the primary silyl ether selectively,(20) (3) the ease with which the remaining silyl groups can be removed when required, and (4) the “arming” effect that silyl groups impart on the sugar donor in glycosylation reactions.(21)tert-Butyldimethylsilyl (TBDMS) groups were chosen as the protecting groups for the sphingosine acceptor. While conferring an increased level of stability during the synthesis, as well as providing an extra handle for 1H and 13C NMR spectroscopic analysis, we envisaged these bulkier silyl groups could also be removed along with the trimethylsilyl groups of the sugar head unit in a single step.
Per-TMS-protected galactose 13 was synthesized in quantitative yield by treating d-galactose with chlorotrimethylsilane and hexamethyldisilazane in the presence of pyridine.(22) Acceptor 16 was accessed in three steps from commercially available phytosphingosine 17 (Scheme 1): Boc protection afforded carbamate 18, which underwent 3-fold silylation of the triol functionality to provide silyl ether 19. Selective monodeprotection of the primary silyl ether in 19 with HF-pyridine(23) concluded the synthesis of glycosyl acceptor 16.
Scheme 1. Synthesis of Glycosyl Acceptor 16.

The key step in the synthesis of 9 employed Gervay−Hague’s elegant glycosylation methodology in which the glycosyl iodide donor 15 was generated in situ by treating per-TMS-protected galactose 13 with iodotrimethylsilane.(24) Glycosyl iodide 15 reacted with acceptor 16 in the presence of Bu4NI and Hünig’s base, to provide glycoside 20 exclusively as the α-anomer in 75% yield, thus providing a short and scalable route to galactosyl ceramide precursors (Scheme 2). With this method of glycosylation, the protecting groups are usually removed immediately via an acidic workup.(24) However, it was desirable in our case to leave the silyl protecting groups in the product intact and effect a selective monodeprotection of the primary trimethylsilyl group instead. To this end, following Fernández’s method for 6-trimethylsilyl ether deprotection,(20) treatment of 20 with acetic acid in acetone/methanol gave monodeprotected 21 in a satisfactory 78% yield, and provided the valuable handle for modifying the 6-position. Alcohol 21 was converted in 87% yield to azide 22 by a Mitsunobu reaction employing diphenylphosphoryl azide (DPPA), diisopropyl azodicarboxylate (DIAD), and triphenylphosphine.(25) Treating azide 22 with tetrabutylammonium fluoride (TBAF) successfully removed the five silyl protecting groups; however, the residual tetrabutylammonium byproduct could not be separated from the polar product by flash column chromatography. While the use of silica-supported TBAF ameliorated this problem, a better deprotection method involved stirring 22 with 10% trifluoroacetic acid (TFA) in dichloromethane overnight. Interestingly, while this method produced pentaol 9 exclusively, treatment of azide 22 with neat TFA for 10 min resulted in clean conversion to Boc-deprotected amine 10 (Scheme 2).
Scheme 2. Synthesis of α-Galactosides 9 and 10.

To illustrate the versatility of amine 10 we completed the synthesis of two biologically active 6-derivatized α-galactosyl ceramides 6 (PBS-57) and 8, which differ in both the acyl chain of the ceramide base and the nitrogen-containing functionality at the 6-position of the sugar headgroup (Scheme 3). Treating a biphasic mixture of amine 10 in THF/8 M aqueous NaOAc with nervonoyl chloride afforded 6-azido GalCer 23 in 81% yield. Subsequent Staudinger24d,26 reaction followed by treatment with acetyl chloride afforded PBS-57 (6).(16b) Alternatively, acylation of 10 with hexacosanoyl chloride, to provide amide 24, followed by Staudinger reduction24d,26 and then treatment with phenylisocyanate, afforded Th1-biasing α-GalCer derivative 8.(16a)
Scheme 3. Synthesis of α-Galactosyl Ceramides 6 and 8.

An even more convergent route to our target molecules would employ 6-azido galactose 25 in the key glycosylation step (Scheme 4). Azide 25 was synthesized by deprotecting the primary silyl ether in per-TMS-protected galactose 13 to afford alcohol 26, followed by Mitsunobu reaction with DPPA, DIAD, and PPh3. Following the same procedure as with per-TMS-protected galactose 13, 6-azido galactose 25 was treated with TMSI and the resulting glycosyl iodide 27 added to a solution of acceptor 16, Bu4NI, and Hünig’s base in dichloromethane. Unfortunately, this reaction was unsuccessful, only providing various TMS-deprotection products of 6-azido galactose 25. The failure of this sugar donor to undergo glycosylation can be understood by the electron-withdrawing nature of the azide group imparting a strongly deactivating effect.(27) Acceptors such as alcohol 16 already display attenuated nucleophilicity owing to the presence of a hydrogen bond between the alcohol oxygen atom and the carbamate hydrogen atom,(28) and while the reaction of 16 proceeds readily with “armed” per-TMS donor 15, the corresponding less-armed per-benzylated glycosyl iodide is unreactive to this class of acceptor.24a,24b These observations serve to highlight how sensitive is the reactivity of these glycosyl iodide donors to small changes in the sugar substitution pattern.
Scheme 4. Attempted More Convergent Synthesis of α-Galactoside 22.

In summary, a short and convenient route to 6-N-derivatized galactosyl ceramides has been developed, which allows late-stage variation of both the N-acyl chain of the sphingosine unit and the substituent introduced at the 6-position of the sugar. Azides 23 and 24 are also primed for further functionalization via Click Chemistry. Future work will focus on using such derivatization strategies in the synthesis and biological evaluation of novel 6-N-derivatized glycolipids.
Experimental Section
General Procedure for Selective 6-Desilylation (Alcohols 21 and 26)
AcOH (3.00 mmol) was added to a solution of galactoside 20 or 13 (1.57 mmol) in acetone (3.5 mL) and MeOH (4.7 mL) at 0 °C. The reaction mixture was allowed to warm to rt. After 8 h, the reaction was quenched by the addition of NaHCO3 (5.95 mmol) and then filtered. After concentration of the filtrate under reduced pressure, the resulting oil was purified by flash column chromatography (EtOAc in hexane) to afford alcohol 21 (from 20) or 26 (from 13) as a colorless oil.
General Procedure for the Mitsunobu Reaction (Azides 22 and 25)
PPh3 (1.86 mmol), DIAD (1.86 mmol), and DPPA (1.86 mmol) were added sequentially to a cooled solution of alcohol 21 or 26 (0.90 mmol) in THF (20 mL). The mixture was allowed to warm to rt and then stirred overnight. Concentration under reduced pressure followed by purification of the residue by flash column chromatography (EtOAc in hexane) afforded azide 22 (from 21) or 25 (from 26) as a colorless oil.
Pentaol 9
TFA (0.50 mL, 6.6 mmol) was added dropwise over 5 min to a solution of azide 22 (200 mg, 0.19 mmol) in CH2Cl2 (5 mL) at rt. After 30 min, the reaction mixture was concentrated under reduced pressure to afford pentaol 9 as a colorless oil (115 mg, quant.): [α]20D +12.4 (c 0.5, CDCl3:CD3OD, 2:1); νmax(film) (cm−1) 3282s br (O−H), 2114s (N3), 1696 m (C=O); 1H NMR (300 MHz, CDCl3:CD3OD, 2:1) 0.85 (t, J = 6.0 Hz, 3H), 1.18−1.39 (stack, 22H), 1.40−1.71 (stack, 4H), 1.44 (s, 9H), 3.26 (A of ABX, JA−B = 12.6 Hz, JA−X = 4.9 Hz, 1H), 3.51−3.64 (stack, 3H), 3.65−3.98 (stack, 7H), 4.89 (d, J = 3.3 Hz, 1H); 13C NMR (100 MHz, CDCl3:CD3OD, 2:1) 14.3 (CH3), 23.2 (CH2), 26.4 (CH2), 28.6 (CH3), [29.9, 30.2, 32.4, 32.8 (CH2, resonance overlap)], 51.7 (CH), 51.8 (CH2), 68.3 (CH2), 69.3 (CH), 70.4 (CH), 70.6 (2 × CH, resonance overlap), 72.5 (CH), 75.3 (CH), 80.1 (C), 156.8 (C); MS (TOF ES+) m/z 627.3 ([M + Na]+, 100%); HRMS (TOF ES+) calcd for C29H56N4O9Na [M + Na]+ 627.3945, found 627.3956.
Amine 10 from Azide 22
TFA (1.0 mL, 13.2 mmol) was added dropwise over 5 min to azide 22 (400 mg, 0.38 mmol) at rt. After 30 min, the reaction mixture was concentrated under reduced pressure. The resulting colorless oil was used in the next step without further purification (192 mg, quant.).
From Azide 9
TFA (0.50 mL, 6.6 mmol) was added dropwise over 5 min to azide 9 (114 mg, 0.19 mmol) at rt. After 30 min, the reaction mixture was concentrated under reduced pressure. The resulting colorless oil was used in the next step without further purification (96 mg, quant.).
Acknowledgments
G.S.B. acknowledges support in the form of a Personal Research Chair from Mr. James Bardrick, Royal Society Wolfson Research Merit Award, and as a former Lister Institute-Jenner Research Fellow; and P.J.J. acknowledges The Wellcome Trust (084923/B/08/Z) for funding. The NMR spectrometers used in this research were funded in part through Birmingham Science City: Innovative Uses for Advanced Materials in the Modern World (West Midlands Centre for Advanced Materials Project 2), with support from Advantage West Midlands and part-funded by the European Regional Development Fund.
Supporting Information Available
Experimental procedures, characterization, and 1H NMR and 13C NMR spectra for products 6, 8−10, 13, 16, 18, and 20−25. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Kawano T.; Cui J.; Koezuka Y.; Toura I.; Kaneko Y.; Motoki K.; Ueno H.; Nakagawa R.; Sato H.; Kondo E.; Koseki H.; Taniguchi M. Science 1997, 278, 1626–1629. [DOI] [PubMed] [Google Scholar]
- Crowe N.; Uldrich A. P.; Kyparissoudis K.; Hammond K. J. L.; Hayakawa Y.; Sidobre S.; Keating R.; Kronenberg M.; Smyth M. J.; Godfrey D. I. J. Immunol. 2003, 171, 4020–4027. [DOI] [PubMed] [Google Scholar]
- Burdin N.; Brossay L.; Kronenberg M. Eur. J. Immunol. 1999, 29, 2014–2025. [DOI] [PubMed] [Google Scholar]
- Carnaud C.; Lee D.; Donnars O.; Park S. H.; Beavis A.; Koezuka Y.; Bendelac A. J. Immunol. 1999, 163, 4647–4650. [PubMed] [Google Scholar]
- Taniguchi M.; Harada M.; Kojo S.; Nakayama T.; Wakao H. Annu. Rev. Immnunol. 2003, 21, 483–513. [DOI] [PubMed] [Google Scholar]
- Godfrey D. I.; MacDonald H. R.; Kronenberg M.; Smyth M. J.; Van Kaer L. Nat. Rev. Immunol. 2004, 4, 231–237. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Aseguinolaza G.; Van Kaer L.; Bergmann C. C.; Wilson J. M.; Schmieg J.; Kronenberg M.; Nakayama T.; Taniguchi M.; Koezuka Y.; Tsuji M. J. Exp. Med. 2002, 195, 617–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto K.; Miyake S.; Yamamura T. Nature 2001, 413, 531–534. [DOI] [PubMed] [Google Scholar]
- Chiba A.; Oki S.; Miyamoto K.; Hashimoto H.; Yamamura T.; Miyake S. Arthritis Rheum. 2004, 50, 305–313. [DOI] [PubMed] [Google Scholar]
- a Oki S.; Chiba A.; Yamamura T.; Miyake S. J. Clin. Invest. 2004, 113, 1631–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yu K. O. A.; Porcelli S. A. Immunol. Lett. 2005, 100, 42–55. [DOI] [PubMed] [Google Scholar]
- a Savage P. B.; Teyton L.; Bendelac A. Chem. Soc. Rev. 2006, 35, 771–779. [DOI] [PubMed] [Google Scholar]; b Wu D.; Fujioa M.; Wong C.-H. Bioorg. Med. Chem. 2008, 16, 1073–1083 and references cited therein.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff R. D.; Gao Y.; Mattner J.; Zhou D.; Yin N.; Cantu C.; Teyton L. III; Bendelac A; Savage P. B. J. Am. Chem. Soc. 2004, 126, 13602–13603. [DOI] [PubMed] [Google Scholar]
- Oki S.; Tomi C.; Yamamura T; Miyake S. Int. Immunol. 2005, 17, 1619–1629. [DOI] [PubMed] [Google Scholar]
- a Yu K. O. A.; Im J. S.; Molano A.; Dutronc Y.; Illarionov P. A.; Forestier C.; Fujiwara N.; Arias I.; Miyake S.; Yamamura T.; Chang Y. T.; Besra G. S.; Porcelli S. A. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 3383–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Im J. S.; Arora P.; Bricard G.; Molano A.; Venkataswamy M. M.; Baine I.; Jerud E. S.; Goldberg M. F.; Baena A.; Yu K. O. A.; Ndonye R. M.; Howell A. R.; Yuan W. M.; Cresswell P.; Chang Y. T.; Illarionov P. A.; Besra G. S.; Porcelli S. A. Immunity 2009, 30, 888–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Schmieg J.; Yang G.; Franck R. W.; Tsuji M. J. Exp. Med. 2003, 198, 1631–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yang G.; Schmeig J.; Tsuji M.; Franck R. W. Angew. Chem., Int. Ed. 2004, 43, 3818–3822. [DOI] [PubMed] [Google Scholar]
- a Trappeniers M.; Van Beneden K.; Decruy T.; Hillaert U.; Linclau B.; Elewaut D.; Van Calenbergh S. J. Am. Chem. Soc. 2008, 130, 16468–16469. [DOI] [PubMed] [Google Scholar]; b Liu Y.; Goff R. D.; Zhou D.; Mattner J.; Sullivan B. A.; Khurana A.; Cantu C. III; Ravkov E. V.; Ibegbu C. C.; Altman J. D.; Teyton L.; Bendelac A.; Savage P. B. J. Immunol. Methods 2006, 312, 34–39. [DOI] [PubMed] [Google Scholar]; c Xia C.; Zhang W.; Zhang Y.; Woodward R. L.; Wang J.; Wang P. G. Tetrahedron 2009, 65, 6390–6395. [Google Scholar]; d Zhou X. T.; Forestier C.; Goff R. D.; Li C.; Teyton L.; Bendelac A.; Savage P. B. Org. Lett. 2002, 4, 1267–1270. [DOI] [PubMed] [Google Scholar]
- Koch M.; Stronge V. S.; Shepherd D.; Gadola S. D.; Mathew B.; Ritter G.; Fersht A. R.; Besra G. S.; Schmidt R. R.; Jones E. Y.; Cerundolo V. Nat. Immunol. 2005, 6, 819–826. [DOI] [PubMed] [Google Scholar]
- Borg N. A.; Wun K. S.; Kjer-Nielson L.; Wilce M. C.; Pellicci D. G.; Koh R.; Besra G. S.; Bharadwaj M.; Godfrey D. I.; McCluskey J.; Rossjohn J. Nature 2007, 448, 44–49. [DOI] [PubMed] [Google Scholar]
- Prigozy T. I.; Naidenko O.; Qazba P.; Elewaut D.; Brossay L.; Khurana A.; Natori T.; Koezuka Y.; Kulkarni A.; Kronenberg M. Science 2001, 291, 664–667. [DOI] [PubMed] [Google Scholar]
- Fernández C.; Nieto O.; Rivas E.; Montenegro G.; Fontenla J. A.; Fernández-Mayoralas A. Carbohydr. Res. 2000, 327, 353–365. [DOI] [PubMed] [Google Scholar]
- a Mootoo D. R.; Konradsson P.; Udodong U.; Fraser-Reid B. J. Am. Chem. Soc. 1988, 110, 5583–5584. [Google Scholar]; b Hashimoto S.; Sakamoto H.; Honda T.; Abe H.; Nakamura S.; Ikegami S. Tetrahedron Lett. 1997, 38, 8969–8972. [Google Scholar]; c Crich D.; Li M. Org. Lett. 2007, 9, 4115–4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toubiana R.; Das B. C.; Defaye J.; Mompon B.; Toubiana M. J. Carbohydr. Res. 1975, 44, 308–312. [DOI] [PubMed] [Google Scholar]
- Mormeneo D.; Casas J.; Llebaria A.; Delgado A. Org. Biomol. Chem. 2007, 5, 3769–3777. [DOI] [PubMed] [Google Scholar]
- a Du W.; Kulkarni S. S.; Gervay-Hague J. Chem. Commun. 2007, 2336–2338. [DOI] [PubMed] [Google Scholar]; b Du W.; Gervay-Hague J. Org. Lett. 2005, 7, 2063–2065. [DOI] [PubMed] [Google Scholar]; c Schombs M.; Park F. E.; Du W.; Kulkarni S. S.; Gervay-Hague J. J. Org. Chem. 2010, 75, 4891–4898. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Jervis P. J.; Veerapen N.; Bricard G.; Cox L. R.; Porcelli S. A.; Besra G. S. Bioorg. Med. Chem. Lett. 2010, 20, 3475–3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson P.-O.; Chen Y.; Belfrage A. K.; Blackman M. J.; Kvarnström I.; Jansson K.; Vrang L.; Hamelink E.; Hallberg A.; Rosenquist Å.; Samuelsson B. J. Med. Chem. 2004, 47, 3353–3366. [DOI] [PubMed] [Google Scholar]
- Worthington R. J.; Bell N. M.; Wong R.; Micklefield J. Org. Biomol. Chem. 2008, 6, 92–103. [DOI] [PubMed] [Google Scholar]
- a Li Z.; Gildersleeve J. C. Tetrahedron Lett. 2007, 48, 559–562. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Li Z.; Gildersleeve J. C. J. Am. Chem. Soc. 2006, 128, 11612–11619. [DOI] [PubMed] [Google Scholar]
- a Polt R.; Szabo L.; Treiberg J.; Li Y.; Hruby V. J. J. Am. Chem. Soc. 1992, 114, 10249–10258. [Google Scholar]; b Schmidt R. R.; Zimmermann P. Angew. Chem., Int. Ed. Engl. 1986, 25, 725–726. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.