

Abstract
Computational simulations of the conversion from the normal cellular prion (PrPc) to the scrapie prion (PrPSc) are usually based on the structures determined by NMR because of the difficulties in crystallizing prion protein. Due to insufficient experimental restraints, a biologically critical loop region in PrPc (residues 167–171), which is a potential binding site for Protein X, is under-determined in most mammalian species. Here, we show that by adding information about distance constraints derived from a database of high-resolution protein structures, this under-determined loop as well as other secondary structural elements of the E200K variant of human prion protein (hPrPc), a disease-related isoform, can be refined into more realistic structures in the structural ensemble with improved quality and increased accuracy. In particular, the ensemble becomes more compact after the refinement and the percentage of residues in the most favourable region of the Ramachandran diagram is increased to about 90% in the refined structures from the 80 to 85% range in the previously reported structures. Our results not only provide significantly improved structures of the prion protein and hence would facilitate insights into its conversion in the spongiform encephalopathies, but also demonstrate the strong potential for using databases of known protein structures for structure determination and refinement.
Keywords: Protein structure refinement, NMR structure determination, database-derived distance constraints, prion structure refinement
1. Introduction
Spongiform encephalopathies, or prion diseases, are a group of neurodegenerative diseases in mammalian species characterized by a progressive vacuolation of brain tissue, amyloid protein deposits, and astrogliosis [1]. Specific examples of the diseases include scrapie in sheep, transmissible mink encephalopathy in mink (TME), chronic wasting disease in mule deer (CWD), bovine spongiform encephalopathy (BSE) in cows, and Gerstmann-Sträussler-Scheinker disease (GSS), fatal familial insolmnia (FFI), kuru, and Alpers syndrome in humans [2–3]. The pathogenesis of the diseases is associated with accumulation of the infectious “scrapie” form of prion protein (PrPSc) in brain tissue, which is transformed from its normal cellular form (PrPc) [2–5]. The two forms of prion protein are distinct in many aspects. For example, PrPc is α-helix-rich, soluble, and highly sensitive to proteinase K digestion, whereas PrPSc is β-sheet-rich, insoluble, and resistant to proteinase K digestion [6–11]. The striking difference in secondary structures implies a major conformational transition from PrPc to PrPSc, which has been considered as a key process involved in the pathogenesis of prion diseases. However, this so-called ‘prion-protein-only’ hypothesis is still not fully validated, and the mechanism of the conformational conversion is still unclear. One of the key obstacles for understanding the details of the prion conformational conversion is that the PrPSc sample is hard to purify for biochemical and structural characterization. Moreover, the normal, cellular isoform of PrP (PrPc) is also difficult for high-resolution spectroscopic or crystallographic study [12], especially for certain biologically critical loop regions (see below). Thus, high-quality prion structures are urgently needed to provide better insights into its transition process.
The E200K variant of the human prion protein (denoted as E200K hereafter) contains a point mutation from negative-charged glutamic acid (E) to positive-charged lysine (K) at the 200th codon. This mutation is linked to the human familiar prion diseases [13]. However, this charge-switching mutation that may alter surface electrostatic potential seems not bring dramatic changes in 3-D structures. Comparison between the E200K NMR structures [14] and other wild-type prion NMR [15–16] and X-ray [17–18] structures from different species shows that they agree very well in several stable secondary elements: Helix 1 (residues 144–153), Helix 2 (residues 172–194), and Helix 3 (residues 200–227), and two β-sheets (residues 129–131 and 161–163). However, they differ significantly at two loop regions, Loop 1 (residues 167–171) and Loop 2 (residue 195–199). The Loop 1 region is biologically critical because it is shown to act as a binding site for “Protein X,” which might function as a mediator for the transition from PrPc to PrPSc [19–23]. Whether the E200K mutation can alter the conformations of this loop region still remains unclear.
Nevertheless, the Loop 1 is under-determined in E200K. There are only a few experimental NOE (Nuclear Overhauser Effect) constraints available in this region (see Table 1), much fewer than other regions of the same protein. The analysis of PROCHECK [24–25] also showed that none of the residues in Loop 1 fall within the most favored regions of the Ramachandran plot [26] (see Results). Thus, alternative constraints that are derived from structural analysis of high resolution X-ray structure may be well suited for this particular case.
Table I.
Experimental Restraints of E200K
| Experimental Restraints of Loop 1 (residue 167–171) | ||||
|---|---|---|---|---|
| Residue | NOE† | Torsion | H-bond | J-coupling‡ |
| 167 | 3 | 0 | 0 | 0 |
| 168 | 3 | 0 | 0 | 0 |
| 169 | 0 | 0 | 0 | 0 |
| 170 | 1 | 0 | 0 | 0 |
| 171 | 21 | 1 | 2 | 0 |
| Experimental Restraints of Loop 2 (residue 195–199) | ||||
| Residue | NOE | Torsion | H-bond | J-coupling |
| 195 | 17 | 0 | 0 | 0 |
| 196 | 45 | 0 | 0 | 1 |
| 197 | 34 | 0 | 0 | 0 |
| 198 | 76 | 0 | 2 | 1 |
| 199 | 32 | 1 | 0 | 1 |
| NOE | Torsion | H-bond | J-coupling | |
| Total | 3157 | 177 | 96 | 44 |
| Per Res. | 29.8 | 1.7 | 0.9 | 0.4 |
Note: Total number of Residue is 106.
Total distance restraints,
J HNHA-coupling constants
The enhancement of NMR structures can be achieved by adding theoretical conformational constraints such as dihedral angles [27] and inter-atomic distances [28] based on statistical analysis of databases of high-resolution protein structures. In particular, it has been shown that inter-atomic distance constraints can improve NMR structures with increased precision and accuracy [28], and can function in an equivalent way as some experimental NMR restraints such as torsion angle restraints, without compromising the quality of the NMR structures [28]. Moreover, these distance constraints impose literally no extra cost on NMR structure refinement [28–29].
In this work, we used a selected set of inter-atomic distance constraints between heavy atoms as additional constraints to refine the E200K variant of human prion protein [14]. Our results showed that the critical Loop 1 region as well as some other secondary elements of E200K can be significantly improved in terms of the precision and accuracy and the Ramachandran plots of the structures by using database-derived distance constraints. The improved structures provide additional structural information that can be taken as a better starting point for the studies of prion protein conversion in the spongiform encephalopathies. To the best of our knowledge, this is the first evidence that the distance constraints derived from a structural database can be used to optimize the under-determined regions of biologically important proteins.
2. Methods
Inter-atomic distances can be categorized according to the specific atom pairs, residue pairs, and sequential residual separations. Different types of distances are subject to different statistical distributions in the structural databases, which have been employed to construct various statistical potentials for contact determination, inverse folding, structure alignment, X-ray structure refinement, etc [30–35]. A set of X-ray structures (~3000) with similarity of 90% or less and resolutions of 2.0 Ǻ or better have been collected from the Protein Data Bank [36] (PDB) and used to obtain the statistical distributions of distances of these different types. To generate a minimal amount of required data, the distances between two heavy atoms N, Cα, C′, Cβ, and O in two residues either sequentially adjacent or separated by one residue have been considered, although they could be extended to other more general types. The distribution of each type of distances was calculated by counting the number of occurrences of the distances within a set of distance intervals. Two distance distributions are given as examples in Fig. 1, one for the distances between the C atom of ARG at position i and the Oof ILE at neighboring position i + 1 (Fig. 1(a)) and the other for the distances between the Cβ atom of ALA at position i and the N atom of LEU at the next-nearest neighbor position i + 2 (Fig. 1(b)). Two hundred distance intervals are specified in the horizontal direction. The length of each interval is equal to 0.1 Å. The ordinate values show the frequencies of the distances in the corresponding distance intervals. The means μ and standard deviations σ of the distributions have been used to specify the range constraints for the corresponding distances to be between μ − 2σ and μ + 2σ.
Fig. 1.

Sample distance distribution functions. (a) Distances between the C atom of ARG at position i and the O atom ILE at position i + 1. (b) Distances between the Cβ atom of ALA at position i and the N atom of LEU at position i + 2.
The NMR experimental data of E200K (residues 125–228) (Table 1) was downloaded from BioMagResBank [37] (BMRB). The structure was then refined using NMR experimental constraints plus additional database-derived distance constraints. The standard torsion angle dynamic simulated annealing protocol implemented in CNS [38] was used for the refinement.
3. Results
A.. Refining NMR structures of the disease-related hPrPc (E200K)
The NMR structure of the E200K variant of the human prion protein was refined by adding the database-derived distance constraints. Two ensembles of 50 accepted structures were collected: one was obtained with experimental constraints (denoted as <E200K>NMR) and the other with experimental data plus database-derived distance constraints (denoted as <E200K>NMR+D). The statistics of resulting ensembles in terms of the agreement with the experimental constraints, optimal covalent geometry, and the local and global potential energy are given in Table 2–4.
Table II.
Average RMSD from Experimental Restraints
| †<E200K>NMR | ‡<E200K>NMR | †<E200K>NMR+D | ‡<E200K>NMR+D | |
|---|---|---|---|---|
| Distances (Ǻ) | 0.0046 ± 0.0018 | 0.0030 | 0.0047 ± 0.0016 | 0.0040 |
| Angles (degrees) | 0.1664 ± 0.0368 | 0.1540 | 0.1589 ± 0.0340 | 0.1380 |
| J-couplings (Hz) | 0.3787 ± 0.0951 | 0.2470 | 0.2105 ± 0.0186 | 0.2550 |
Average RMSD ± standard deviations for the ensemble of structures,
RMSD for the lowest energy structure in the ensemble
Table IV.
Potential Energy of Different Types
| †<E200K>NMR | ‡<E200K>NMR | †<E200K>NMR+D | ‡<E200K>NMR+D | |
|---|---|---|---|---|
| Total Energy | 104.16 ± 24.80 | 82.08 | 102.31 ± 23.09 | 86.30 |
| Bonds | 3.30 ± 1.11 | 2.45 | 3.54 ± 1.58 | 2.70 |
| Bond-Angles | 46.78 ± 6.67 | 42.53 | 46.11 ± 4.92 | 44.11 |
| Improper-Angles | 6.78 ± 1.54 | 5.80 | 6.49 ± 1.21 | 6.53 |
| Van der Waals | 34.44 ± 9.44 | 26.23 | 31.81 ± 6.97 | 26.29 |
| NOE | 5.85 ± 4.80 | 2.11 | 6.97 ± 6.57 | 3.60 |
| Dihedral-Angles | 0.31 ± 0.14 | 0.26 | 0.28 ± 0.14 | 0.21 |
Average energy ± standard deviations for the ensemble of structures,
energy for the lowest energy structure in the ensemble (kcal/mol)
The quality analysis by PROCHECK on the average and energy-minimized structures1 and the lowest energy structures of the ensembles showed a significantly higher percentage (89.6%) of residues in most favorable regions of the Ramachandran plot of the structures in <E200K>NMR+D compared to the 85.4% of these residues in <E200K>NMR (Table 5). Note that the latter percentage (85.4%) is consistent with what was reported by Zhang et al. for their experimental structures (85.7%) [14]. This increase in the percentage of residues in the most favorable regions indicated an improvement of the structural conformations coming from the use of the database-derived distance constraints.
Table V.
Percentage of Residues in Different Ramachandran Plot Regions
| †<E200K>NMR | ‡<E200K>NMR | †<E200K>NMR+D | ‡<E200K>NMR+D | |
|---|---|---|---|---|
| Most favorable | 85.40% | 84.40% | 89.60% | 88.50% |
| Additional allowed | 14.60% | 14.60% | 10.40% | 11.50% |
| Generously allowed | 0.00% | 0.00% | 0.00% | 0.00% |
| Disallowed | 0.00% | 1.00% | 0.00% | 0.00% |
Average percentage of residues in different (ϕ,ψ)-regions for the average and energy-minimized structure in the ensemble,
the percentage of residues in different (ϕ,ψ)-regions for the lowest energy structure in the ensemble
B. Comparison between the refined E200K structures and the wild-type hPrPc NMR and X-Ray structures
The residue-residue comparison of the average and energy-minimized structures of <E200K>NMR+D and <E200K>NMR with the experimentally-determined NMR and X-ray structures of wild-type PrPc was conducted. The residual RMSD values between the refined E200K structures and wild-type structures 1QM0, 1HJM, 1UW3, and 1I4M were plotted in Fig. 2 (see Fig. 2 legends for details).
Fig. 2.
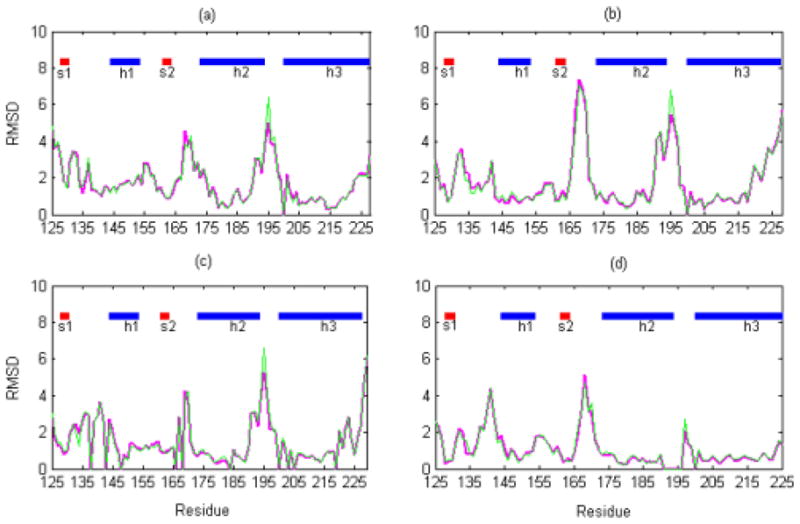
Residue-residue comparison between the refined E200K and wild-type NMR and X-ray structures. The graphs show the residue RMSD values (with the backbone atoms, N, Cα, C′, and O) for the average and energy-minimized structures of <E200K>NMR+D (magenta line) and <E200K>NMR (green line) compared with the structures of (a) the wild-type hPrPc NMR structure (1QM0) at mildly acidic condition (pH 4.5); (b) the wild-type hPrPc NMR structure (1HJM) at neutral condition (pH 7.0); (c) the wild-type shPrPc X-ray structure (1UW3) in a monomeric form; (d) the wild-type hPrPc X-ray structure (1I4M) in a dimeric form. The secondary structures are indicated along the top of each part of the figure with h representing alpha helix and s beta sheet.
The residue RMSD values of Loop 1 and 2 in all these plots are all significantly higher compared to other regions of the structure (> 4 Ǻ), indicating that the loop regions are more flexible compared to the helical and sheet regions. The <E200K>NMR+D structure is closer to the hPrPc X-ray structure (1I4M) at Loop 1 with smaller residue RMSD values compared with <E200K>NMR (in Fig. 2(d)). This indicates that the database-derived distance constraints indeed ‘improve’ the loop conformations. As to Loop 2, the residue RMSD values in the <E200K>NMR+D structure are consistently smaller than those in <E200K>NMR structure (in Fig. 2(a–d)), showing that the database-derived distance constraints make the loop more consistent with the experimental NMR and X-ray structures.
In addition to the under-determined loop regions, differences between <E200K>NMR+D and <E200K>NMR were observed in well-defined helical regions (Helix 2 and Helix 3) (Figure 3). The dimeric hPrPc X-ray structure was used as a reference structure for this comparison. The residual RMSD values for <E200K>NMR+D are slightly smaller than those for <E200K>NMR in the N terminal of Helix 2 (residues 172–190) and Helix 3 (residues 201–228), showing that the helical regions of <E200K>NMR+D are closer to the corresponding X-ray structure compared with those of <E200K>NMR (Fig. 3(a), 3(c)). As to the region between the β-sheet and Helix 2 (residues 191–199), the <E200K>NMR+D structure is closer to the X-ray structure compared with <E200K>NMR (Fig. 3(b)). Overall, the <E200K>NMR+D structure has been ‘improved’ in both the under-determined loop regions and the well-defined helix regions.
Fig. 3.
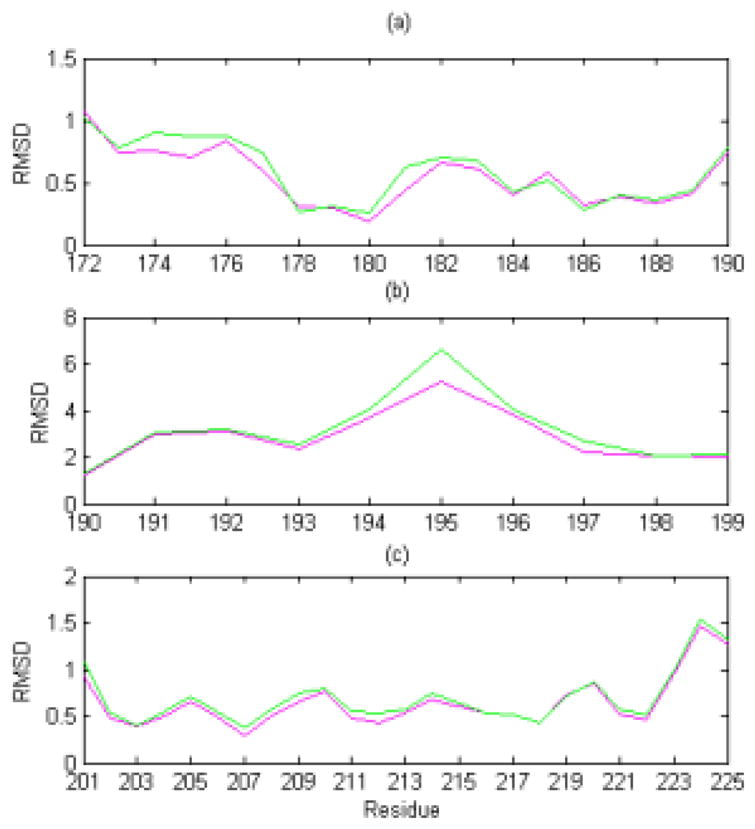
Detailed residue RMSD plots for helix and loop regions. The graphs show the detailed residue RMSD values (of the backbone atoms, N, Cα, C′, and O) for the average and energy-minimized structures of <E200K>NMR+D (magenta line) and <E200K>NMR (green line) at (a) N-terminal of Helix 2 (residues 172–190) when compared with the hPrPc X-ray structure; (b) C-terminal of Helix 2 and the loop between Helix 2 and Helix 3 (residues 191–199) compared with the hPrPc X-ray structure; and (c) Helix 3 (residues 201–228) when compared with the hPrPc X-ray structure.
C. Conformational analysis of the refined E200K loop regions
To further elucidate the improvements of the under-determined loop regions of E200K, the sequential ϕ and ψ angles for each residue between <E200K>NMR and <E200K>NMR+D were compared (Fig. 4) and displayed in Ramachandran plots (Fig. 5). As shown in Fig. 4, the angles ϕ and ψ of <E200K>NMR+D (magenta square) and <E200K> NMR (green square) were extremely close to one another, except in Loop 1 and 2. In the Loop 1 of <E200K>NMR, the residues lie far outside the most favourable regions of the Ramachandran plot (Fig. 5(a)). However, most of these residues have moved into the most favourable regions of Ramachandran plot (Fig. 5(b)) in the <E200K>NMR+D structure. We obtained a similar result for the Loop 2 (data not shown).
Fig. 4.

The ϕ and ψ angles of the refined E200K structures. The angles of <E200K>NMR+D are represented by magenta squares and for <E200K>NMR by green squares.
Fig. 5.

Ramachandran plots of <E200K>NMR and <E200K>NMR+D. (a) Ramachandran plot showing the values of (ϕ, ψ) angles of the average and energy-minimized structure of <E200K>NMR; (b) Ramachandran plot of <E200K>NMR+D. Only the residues of Loop 1 are shown. In (a), most of the residues in Loop 1 are found outside the most favourable (red) regions, while in (b), most of these residues lie within the most favourable regions.
D. Backbone and side-chain packing analysis of the refined E200K loop regions
To illustrate the difference of backbones and side-chains in the loop regions between the <E200K>NMR+D and <E200K>NMR structures, we superimposed the backbones of the whole structure (Fig. 6(a)) and of the loop structures (Fig. 6(a)–6(e). Apparently, the Loop 1 of <E200K>NMR+D differ significantly from the same region of <E200K>NMR (Fig. 6(b)), implying that the database-derived distance constraints affect the backbone conformations in regions where experimental restraints are insufficient (Table 1). The conformations of Loop 2 in both structures appear quite similar except at residue GLY195 (Fig. 6(c)). It has been shown that the conformations of glycine and neighbouring residues can be improved by using database-derived distance constraints [28–29]. Examination of side-chain packing in the two loop regions showed that overall conformations of side-chains are quite similar between <E200K>NMR+D and <E200K>NMR (Fig. 6(d–e)). No change in either hydrogen bonds or salt bridges was observed. It suggests that the database-derived distance constraints do not affect the side-chain packing, which is not so surprising since only the constraints between backbone heavy atoms were utilized.
Fig. 6.

Superimposition of tertiary structures. Tertiary structures of the average and energy-minimized structures of <E200K>NMR+D (small magenta cylinders) and <E200K>NMR (small green cylinders) are superimposed in (a) the backbone of the whole protein; (b) the backbone of Loop 1; (c) the backbone of Loop 2; (d) the backbone and side-chain of Loop 1; (e) the backbone and side-chain of Loop 2.
4. Discussion
Prion diseases can be sporadic (spontaneous), inherited, or transmitted by infectious agents. Some prion diseases in humans, such as the familiar CJD, FFI, and GSS, are inherited and linked to mutations in the PrPc-coding gene, PRNP. More than 20 mutations in this gene have been associated with prion diseases [39]. The mutation E200K, where a glutamic acid is substituted with a lysine at residue 200, is a major cause of familiar CJD [40]. The tertiary structure of the variant E200K of human PrPc is almost identical to the wild-type prion [14]. This mutation changes not only the surface potential and charge distribution of the protein, which might affect its interaction with Protein X or other cellular components and the conversion from PrPc to PrPSc [14], but also the stability of prion protein [41]. On the other hand, this mutation by itself cannot lead to the conversion, which could require additional modification of the protein and additional cofactors [42]. The possible involvement of Protein X in the pathogenic process of the familiar CJD makes the structure of the binding sites between the mutated prion (E200K variant) and Protein X, which include the loop regions encompassing residues 167–171, urgently needed. However, the lack of experimental constraints in this critical region makes the structures poorly defined [14–16]. Here, we have employed distance constraints that are derived from a database of high-resolution protein structures and significantly improved the conformation of the loop region. With the database-derived distance constraints, both loop regions (residues 167–171 and 195–199) in <E200K>NMR+D showed more generally acceptable conformations. The comparisons between the generated structures and the experimentally determined structures of the prion protein confirmed a convergence between the structures, which implies that an increase in the accuracy of the poorly determined loop structures is possible with database-derived distance constraints. The improved structures may provide a better structural understanding of prion proteins and hence facilitate insights into its conversion in the spongiform encephalopathies.
Table III.
Average RMSD from Idealized Geometries
| †<E200K>NMR | ‡<E200K>NMR | †<E200K>NMR+D | ‡<E200K>NMR+D | |
|---|---|---|---|---|
| Bond lengths ((Ǻ) | 0.0014 ± 0.0002 | 0.0012 | 0.0014 ± 0.0002 | 0.0012 |
| Bond Angles (°) | 0.3128 ± 0.0212 | 0.2990 | 0.3108 ± 0.0153 | 0.3040 |
| Improper-Angles (°) | 0.2148 ± 0.0236 | 0.2000 | 0.2105 ± 0.0186 | 0.2120 |
Average RMSD ± standard deviations for the ensemble of structures,
RMSD for the lowest energy structure in the ensemble
Acknowledgments
The work is partially supported by the research funds provided by the Department of Mathematics, the Graduate Program on Bioinformatics and Computational Biology, and the Lawrence Baker Center for Bioinformatics and Biological Statistics at Iowa State University and the NIH/NIGMS grant R01GM081680.
Biographies
Feng Cui received a B.S. degree in Medicine from Hunan School of Medicine, China in 1995, a M.S. degree in Biology from Trument State University in 2000, and a PhD degree in Bioinformatics and Computational Biology from Iowa State University in 2005. He is currently a research fellow in the Laboratory of Cell Biology at the National Cancer Institute, the National Institutes of Health.
Kriti Mukhopadhyay received a B.S. degree in Mathematics from and a M.S. degree in Applied Mathematics from Iowa State University in 2005. She is currently working in industry and planning to come back to school to pursue a PhD degree in Applied Mathematics.
Won-Bin Young received his B.S. and M.S. degrees in Animal Science from National Taiwan University in 1985 and 1990, respectively, and his PhD degree in Molecular, Cellular, and Developmental Biology from Iowa State University in 1999. He did postdoctoral research in the Bio-therapeutics Development Lab. at the Beth Israel Deaconess Center, Harvard University from 1999 to 2000. He then joined the Newlink Genetics as a senior scientist. He is currently a research scientist in the Pittsburg NMR Center for Biomedical Research at the Carnegie Mellon University.
Robert Jernigan received his B.S. degree in Chemistry from California Institute of Technology in 1963 and his Ph.D. degree in Physical Chemistry from Stanford University in 1968. He did postdoctoral research at University of California at San Diego from 1968 to 1970. He joined NIH from 1970 to 2002 and was the Deputy Chief and Chief of the Laboratory of Experimental and Computational Biology and Section of Molecular Structures of NCI. He is currently professor of biochemistry, biophysics, and molecular biology of Iowa State University and also the Director of the ISU Baker Center for Bioinformatics and Biological Statistics.
Zhijun Wu received his Bachelor degree in Computer Science from Huazhong University of Science and Technology, China, and his Ph.D. degree in Computational Mathematics from Rice University, United States, in 1982 and 1991, respectively. He did postdoctoral research at Cornell University and Argonne National Lab from 1991 to 1997. He is currently a full professor in the Department of Mathematics of Iowa State University and also a joint faculty member of the ISU Program on Bioinformatics and Computational Biology.
Footnotes
It is obtained by minimizing the total energy of the structure using the CNS energy minimization routine with the averaged structure of the ensemble as the starting structure.
References
- 1.Brown P, Gajdusek DC. The human spongiform encephalopathies: kuru, Creutzfeldt-Jakob disease, and the Gerstmann-Straussler-Scheinker syndrome. Curr Top Microbiol Immunol. 1991;171:1–20. doi: 10.1007/978-3-642-76540-7_1. [DOI] [PubMed] [Google Scholar]
- 2.Horwich AL, Weissman JS. Deadly conformations – protein misfolding in prion disease. Cell. 1997;89:499–510. doi: 10.1016/s0092-8674(00)80232-9. [DOI] [PubMed] [Google Scholar]
- 3.Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 5.Stahl N, Baldwin MA, Teplow DB, Hood L, Gibson BW, Burlingame AL, Prusiner SB. Structural studies of the scrapie prion protein using mass spectrometry and amino acid sequencing. Biochemistry. 1993;32:1991–2002. doi: 10.1021/bi00059a016. [DOI] [PubMed] [Google Scholar]
- 6.Meyer RK, McKinley MP, Bowman KA, Braunfeld MB, Barry RA, Prusiner SB. Separation and properties of cellular and scrapie prion proteins. Proc Natl Acad Sci USA. 1986;83:2310–2314. doi: 10.1073/pnas.83.8.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oesch B, Westaway D, Walchli M, Mckinley MP, Kent SB, Aebersold R, Barry RA, Tempst P, Teplow DB, Hood LE, et al. A cellular gene encodes scrapie PrP 27–30 protein. Cell. 1985;40:735–746. doi: 10.1016/0092-8674(85)90333-2. [DOI] [PubMed] [Google Scholar]
- 8.Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci USA. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caughey BW, Dong A, Bhat KS, Ernst D, Hayes SF, Caughey WS. Secondary structure analysis of the scrapie-associated protein PrP 27–30 in water by infrared spectroscopy. Biochemistry. 1991;30:7672–7680. doi: 10.1021/bi00245a003. [DOI] [PubMed] [Google Scholar]
- 10.Safar J, Roller PP, Gajdusek DC, Gibbs CJ., Jr Thermal stability and conformational transitions of scrapie amyloid (prion) protein correlate with infectivity. Protein Sci. 1993;2:2206–2216. doi: 10.1002/pro.5560021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wüthrich K. NMR structure of the mouse prion protein domain PrP (121–321) Nature. 1996;382:180–182. doi: 10.1038/382180a0. [DOI] [PubMed] [Google Scholar]
- 12.Rudd PM, Wormald MR, Wing DR, Prusiner SB, Dwek RA. Prion glycoprotein: structure, dynamics, and roles for the sugars. Biochemistry. 2001;40:3759–3766. doi: 10.1021/bi002625f. [DOI] [PubMed] [Google Scholar]
- 13.Apetri AC, Surewicz K, Surewicz WK. The effect of disease-associated mutations on the folding pathway of human prion protein. J Biol Chem. 2004;279(17):18008–18014. doi: 10.1074/jbc.M313581200. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, Swietnicki W, Zagorski MG, Surewicz WK, Sönnichsen FD. Solution structure of the E200K variant of human prion protein. J Biol Chem. 2000;275:33650–33654. doi: 10.1074/jbc.C000483200. [DOI] [PubMed] [Google Scholar]
- 15.Zahn R, Liu A, Lührs T, Riek R, von Schroetter C, Garcia FL, Billeter M, Calzolai L, Wider G, Wüthrich K. NMR solution structure of the human prion protein. Proc Natl Acad Sci USA. 2000;97:145–150. doi: 10.1073/pnas.97.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calzolai L, Zahn R. Influence of pH on NMR structure and stability of the human prion protein globular domain. J Biol Chem. 2003;278:35592–35596. doi: 10.1074/jbc.M303005200. [DOI] [PubMed] [Google Scholar]
- 17.Knaus K, Morillas M, Swietnicki W, Malone M, Surewicz WK, Yee VC. Crystal structure of the human prion protein reveals a mechanism for oligomerization. Nat Struct Biol. 2001;8:770–774. doi: 10.1038/nsb0901-770. [DOI] [PubMed] [Google Scholar]
- 18.Haire LF, Whyte SM, Vasisht N, Gill AC, Verma C, Dodson EJ, Dodson GG, Bayley PM. The crystal structure of the globular domain of sheep prion protein. J Mol Biol. 2004;336:1175–1183. doi: 10.1016/j.jmb.2003.12.059. [DOI] [PubMed] [Google Scholar]
- 19.Telling GC, Scott M, Hsiao KK, Foster D, Yang SL, Torchia M, Sidle KC, Collinge J, DeArmond SJ, Prusiner SB. Transmission of Creutzfeldt-Jakob disease from humans to transgenic mice expressing chimeric human-mouse prion protein. Proc Natl Acad Sci USA. 1994;91:9936–9940. doi: 10.1073/pnas.91.21.9936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE, DeArmond SJ, Prusiner SB. Prion propagation in mice expressing human and chimeric PrP transgenes implicates that interaction of cellular PrP with another protein. Cell. 1995;83:79–90. doi: 10.1016/0092-8674(95)90236-8. [DOI] [PubMed] [Google Scholar]
- 21.Billeter M, Riek R, Wider G, Hornemann S, Glockshuber R, Wüthrick K. Prion protein NMR structure and species barrier for prion diseases. Proc Natl Acad Sci USA. 1997;94:7281–7285. doi: 10.1073/pnas.94.14.7281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaneko K, Zulianello L, Scott M, Cooper CM, Wallace AC, James TL, Cohen FE, Prusiner SB. Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc Natl Acad Sci USA. 1997;94:10069–10074. doi: 10.1073/pnas.94.19.10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gossert AD, Bonjour S, Lysek DA, Fiorito F, Wuthrich K. Prion protein NMR structures of elk and of mouse/elk hybrids. Proc Natl Acad Sci USA. 2005;102:646–650. doi: 10.1073/pnas.0409008102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 25.Morris AL, MacArthur MW, Hutchinson EG, Thornton JM. Stereochemical quality of protein structure coordinates. Proteins. 1992;12:345–364. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]
- 26.Ramachandran GN, Sasisekharan V. Conformation of polypeptides and proteins. Adv Protein Chem. 1968;23:283–437. doi: 10.1016/s0065-3233(08)60402-7. [DOI] [PubMed] [Google Scholar]
- 27.Clore GM. Improving the quality of NMR and crystallographic protein structures by means of a conformational database potential derived from structure databases. Protein Sci. 1996;5:1067–1080. doi: 10.1002/pro.5560050609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cui F, Jernigan RL, Wu Z. Refinement of NMR-determined protein structures with database-derived distance constraints. J of Bioinformatics and Computational Biology. 2005;3:1315–1331. doi: 10.1142/s0219720005001582. [DOI] [PubMed] [Google Scholar]
- 29.Cui F, Jernigan RL, Wu Z. Knowledge-Based versus Experimentally-Acquired Distance and Angle Constraints for NMR Structure Refinement. J of Bioinformatics and Computational Biology. 2007 doi: 10.1142/s0219720008003448. submitted. [DOI] [PubMed] [Google Scholar]
- 30.Miyazawa S, Jernigan RL. Estimation of effective inter-residue contact energies from protein crystal structures: quasi-chemical approximation. Macromolecules. 1985;18:534–552. [Google Scholar]
- 31.Miyazawa S, Jernigan RL. Residue-residue potentials with a favourable contact pair term and an unfavourable high packing density term for simulation and threading. J Mol Biol. 1996;256:623–644. doi: 10.1006/jmbi.1996.0114. [DOI] [PubMed] [Google Scholar]
- 32.Sippl MJ. Calculation of conformational ensembles from potentials of mean force. J Mol Biol. 1990;213:859–883. doi: 10.1016/s0022-2836(05)80269-4. [DOI] [PubMed] [Google Scholar]
- 33.Sippl MJ, Weitckus S. Detection of native-like models for amino acid sequence of unknown three-dimensional structure in a database of known protein conformations. Proteins. 1992;13:258–271. doi: 10.1002/prot.340130308. [DOI] [PubMed] [Google Scholar]
- 34.Rojnuckarin A, Subramaniam S. Knowledge-based potentials for protein structure. Proteins. 1999;36:54–67. doi: 10.1002/(sici)1097-0134(19990701)36:1<54::aid-prot5>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 35.Wall ME, Subramaniam S, Phillips GN., Jr Protein structure determination using a database of inter-atomic distance probabilities. Protein Sci. 1999;8:2720–2727. doi: 10.1110/ps.8.12.2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov LN, Bourne PE. The Protein Data Bank. Nuc Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doreleijers JF, Mading S, Maziuk D, Sojourner K, Yin L, Zhu J, Makley JL, Ulrich EL. BioMagResBank database with sets of experimental NMR constraints corresponding to the structures of over 1400 biomolecules deposited in the Protein Data Bank. J Biomol NMR. 2003;26:139–146. doi: 10.1023/a:1023514106644. [DOI] [PubMed] [Google Scholar]
- 38.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang S, Kuszewski J, Nilges N, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR System: A new software suite for macromolecular structure determination. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 39.Goldfarb LG, Brown P, Cervenakova L, Gajdusek DC. Genetic analysis of Creutzfeldt-Jakob disease and related disorders. Phil Trans Roy Soc Lond B Biol Sci. 1994;343:379–384. doi: 10.1098/rstb.1994.0032. [DOI] [PubMed] [Google Scholar]
- 40.Hsiao K, Meiner Z, Kahana E, Cass C, Kahana I, Avrahami D, Scarlato G, Abramsky O, Prusiner SB, Gabizon R. Mutation of the prion protein in Libyan Jews with Creutzfeldt-Jakob disease. N Engl J Med. 1991;324:1091–1097. doi: 10.1056/NEJM199104183241604. [DOI] [PubMed] [Google Scholar]
- 41.Levy Y, Becker OM. Conformational polymorphism of wild-type and mutant prion proteins: Energy landscape analysis. Proteins. 2002;47(4):458–468. doi: 10.1002/prot.10095. [DOI] [PubMed] [Google Scholar]
- 42.Capellari S, Parchi P, Russo CM, Sanford J, Sy MS, Gambetti P, Petersen RB. Effect of the E200K mutation on prion protein metabolism. Amer J Pathol. 2000;157:613–622. doi: 10.1016/S0002-9440(10)64572-5. [DOI] [PMC free article] [PubMed] [Google Scholar]