

Summary
Elucidating how homing endonucleases undergo changes in recognition site specificity will facilitate efforts to engineer proteins for gene therapy applications. I-SceI is a monomeric homing endonuclease that recognizes and cleaves within an 18-base-pair (bp) target. It tolerates limited degeneracy in its target sequence, including substitution of a C/G+4 base-pair for the wild-type A/T+4 base-pair. Libraries encoding randomized amino acids at I-SceI residue positions that contact or are proximal to A/T+4 were used in conjunction with a bacterial one-hybrid system to select I-SceI derivatives that bind to recognition sites containing either the A/T+4 or C/G+4 base-pairs. As expected, isolates encoding wild-type residues at the randomized positions were selected using either target sequence. All I-SceI proteins isolated using the C/G+4 recognition site included small side chain substitutions at G100, and either contained (K86R/G100T, K86R/G100S and K86R/G100C) or lacked (G100A, G100T) a K86R substitution. Interestingly, the binding affinities of the selected variants for the wild-type A/T+4 target are 4–11-fold lower than that of wild-type I-SceI, whereas those for the C/G+4 target are similar. The increased specificity of the mutant proteins is also evident in binding experiments in vivo. These differences in binding affinities account for the observed ~36-fold difference in target preference between the K86R/G100T and wild-type proteins in DNA cleavage assays. An X-ray crystal structure of the K86R/G100T mutant protein bound to a DNA duplex containing the C/G+4 substitution suggests how sequence specificity of a homing enzyme can increase. This biochemical and structural analysis defines one pathway by which site specificity is augmented for a homing endonuclease.
Keywords: homing enzyme, protein-DNA interactions, protein engineering, specificity, directed evolution
Introduction
Homing endonuclease genes (HEGs) are mobile DNA elements that propagate throughout a population by catalyzing a gene conversion process termed homing 1; 2. Homing requires the activity of a site-specific homing endonuclease encoded by the HEG that introduces a double-strand break into a cognate allele that lacks the sequence. The mobile DNA sequence is copied into the cleaved allele via homologous recombination when it is used as a template during double-strand break repair. Homing endonucleases genes are found associated with self-splicing RNA introns, as part of inteins that self-splice at the protein level, or as free-standing elements. They are pervasive in nature, occurring in organisms from all phyla. Several families of HEGs have evolved, the largest being the LAGLIDADG family that is defined on the basis of conserved sequence and structural motifs.
A “life cycle” has been proposed for HEGs in which the elements are introduced into a naive species by horizontal transmission, where they are subsequently inherited vertically and propagated to other individuals by homing 3; 4; 5. Once fixed within the population, HEGs degenerate in the absence of positive selection, and it has been suggested that they are eventually deleted. For HEGs that are part of inteins, the endonucleolytic activities that initiate homing contribute little to host survival and appear to degenerate prior to the intein protein splicing activities, which are required for maximal host fitness by generating functional host proteins6. Once the HEG and the associated intron and intein have been entirely deleted from the genome, subsequent rounds of HEG invasion may re-introduce the mobile element. Alternative models based on stochastic simulations of populations indicate that HEGs persist even in the absence of inter-species horizontal transfer 7. The complex evolutionary pathway of HEGs requires that they encode homing endonucleases with seemingly paradoxical properties; not only possessing the specificity to initiate homing by cleaving single genomic target sites, but also being sufficiently flexible to recognize diverged target sites in related species that contain nucleotide polymorphisms in order to promote cross-species invasion and long-term persistence of the element 8. The molecular basis for homing endonuclease specificity has been revealed by X-ray crystal structures of LAGLIDADG endonucleases bound to their recognition sequences that indicate that these enzymes utilize only a subset of the total number of possible protein-DNA interactions, with some base-pairs making no contact to the protein 8; 9; 10; 11; 12; 13; 14; 15. As a result, homing endonucleases tolerate limited base-pair degeneracy within their recognition sites.
Homing endonucleases have been the subject of intense study because they can greatly stimulate DNA repair at defined loci in gene therapy protocols 16. When a homing endonuclease is delivered to and expressed in cells and generates a double strand break proximal to a genetic mutation that causes disease, repair of the gene can occur by homologous recombination if a non-mutant template DNA is also present. A major challenge to exploit this method has been in developing the means to engineer homing enzymes with the requisite specificities needed to cleave complex genomes at particular loci. A variety of approaches have been applied to alter LAGLIDADG homing endonuclease specificity that involve screening or selecting variants from mutagenic libraries based on DNA cleavage or recombination activities 17; 18; 19; 20; 21; 22; 23; 24; 25, and structure-based computational design 26; 27. Previously, we applied a two-hybrid method to select variants of the PI-SceI intein from a combinatorial library with altered DNA binding specificity 28. In PI-SceI, one domain contains the active sites that catalyze the double strand scission reaction, while a second, separate domain catalyzes protein splicing, and residues in both domains establish base-specific contacts to the DNA. These studies demonstrated that PI-SceI variants with altered DNA binding specificity exhibited a parallel shift in their DNA cleavage specificity. However, this method could not be used to identify altered interactions mediated by the endonuclease domain because DNA binding of this domain is dependent on prior binding by the protein splicing domain 29; 30. Furthermore, crystals of PI-SceI bound to its DNA recognition sequence are difficult to produce and only low resolution X-ray crystallographic structures of the complex are available 10, preventing detailed analysis of the altered binding interactions. In contrast, the DNA binding and DNA cleavage determinants of the intron-encoded I-SceI homing endonuclease are both situated within the same domains and high resolution X-ray crystal structures of the I-SceI/DNA complex are available 11; 13. Here, we report the selection and characterization of I-SceI variants from combinatorial libraries with altered specificity. Interestingly, the specificity of the mutant I-SceI proteins for a variant recognition site is increased as a result of lower affinity for the wild-type sequence. Examination of the X-ray crystal structure of one of the mutant proteins provides insight into the types of atomic adjustments that occur as homing endonucleases evolve increased target site specificity.
RESULTS
Preparation of I-SceI expression libraries and DNA substrates
The 2.25 Å X-ray crystal structure of the I-SceI/DNA complex reveals that residues within β-strands 3 and 4 of β-sheet 1 interact with base-pairs +1 to +7 in the major groove of the DNA. Recognition is the result of extensive water-mediated contacts, backbone phosphate interactions and direct contacts to nitrogenous bases (Figure 1a) 11. Sequence specificity profiling in vitro 22; 31 as well as the X-ray crystal structure suggested that some of these base-specific contacts are critical recognition determinants that may be amenable to alteration. Moreover, the absence of interactions between the hairpin turn and the remainder of the protein indicates that the residues are not essential for stabilizing the protein tertiary structure 11 nor do they comprise part of the two overlapping endonucleolytic active sites. We focused on the interactions between the β-hairpin turn and nucleotides +3 and +4 (Fig 1a, 1b, 1c). The wild-type C/G+3 base-pair is essential for function, whereas either A/T+4, found in the wild-type site, or C/G+4, are active, indicating that limited sequence variation is permissible at position +422; 31. Glu61 directly contacts C+3 while also making water-mediated contacts to A+2 and A+4 (Figure 1c) 11. The complementary G+3 base is contacted by Arg88, which also makes a water-mediated contact to A+2. At position +4, the Lys86 terminal amino (NZ) group directly hydrogen bonds to the T+4 O4 in addition to making a water mediated contact to C+5. The complementary base A+4 is indirectly contacted by Gln59 and Glu61.
Figure 1.

A) A ribbon representation of the 2.25 Å X-ray crystal structure of I-SceI bound to a 24 base-pair duplex containing the 18-bp recognition sequence (1R7M 11). Highlighted within the protein secondary structure elements (colored gray) are the two LAGLIDADG α-helices (colored orange) and β-sheet 1 (colored magenta). The nucleotides of the top (colored green) and bottom (colored blue) DNA strands are depicted in a stick representation. The positions of the scissile phosphodiester bonds within the DNA backbone are indicated (colored orange). B) Schematic representation of the 18-bp I-SceI recognition sequence showing the cleavage reaction products in blue and green. Bases are numbered from the center of the 4-bp overhang generated by DNA cleavage. Base-specific and water-mediated (indicated by blue spheres) contacts made to DNA by Gln59, Glu61, Lys86 and Arg88 are indicated. Base-pairs C/G+3 and A/T+4 are indicated (colored violet). C) A detailed view of a region of the I-SceI/DNA complex structure showing the locations of Gln59, Glu61, Lys86, Arg88 and Gly100 (colored gray) relative to the C/G+3 (colored salmon) and A/T+4 (colored green) base-pairs (depicted as sticks). The protein (cyan) and DNA strand (green and blue) backbones are represented with ribbons. Hydrogen bonds are indicated by dashed lines, and water molecules are represented by spheres (colored red).
I-SceI expression libraries containing randomized codons at defined positions were created to study the interactions made by wild-type I-SceI to positions +3 and +4 and to attempt to alter these interactions. A phagemid library (5var) was constructed in which the codons for residues Gln59, Glu61, Lys86, and Arg88, which contact base-pairs +3 and/or +4, and for residue Gly100, which is proximal to the DNA, were randomized. Only the codons for Glu61, Arg88 and Gly100 were randomized in the 3var plasmid library. Each library is designed to express variants with any of the twenty amino acids at the randomized positions.
The set of all possible mutant DNA substrates was generated to select I-SceI derivatives with shifted or altered specificity. Six mutant DNA targets that included transversion and transition mutations were created by randomizing nucleotide positions +3 or +4 (Figure 1b). This set is also used to assay the extent of substrate selectivity of the isolated I-SceI variants.
Selection of I-SceI mutants using a bacterial one-hybrid system
We applied a bacterial one-hybrid selection system to isolate I-SceI derivatives from the randomized libraries with shifted or altered specificity (Figure 2). This strategy is a simplified version of a two-hybrid approach32 that was used to isolate altered binding variants of the PI-SceI homing enzyme 28. The one-hybrid system consists of two components: a plasmid (pACLαI-SceI (D145A)-f1) that expresses an I-SceI derivative fused to the Escherichia coli RNA polymerase α subunit and a single-copy F′ episome that contains the I-SceI recognition sequence. The I-SceI fusion protein includes a D145A substitution that eliminates DNA strand scission without affecting DNA binding33. The I-SceI target lies upstream of a weak lac promoter on the F′ factor that drives expression of the selectable HIS3 gene. When the I-SceI fusion protein fails to bind to the I-SceI target due to mutations that disrupt binding, auxotrophic his- reporter strains fail to grow on minimal media lacking supplemental histidine. If an I-SceI fusion protein binds to the recognition sequence, the close proximity of the fused RNA polymerase subunit to the promoter increases transcription of the HIS3 gene, thereby allowing colony growth on minimal media. The stringency of the selection is controlled by varying the concentration of 3-aminotriazole, a competitive inhibitor of His3, in the growth medium.
Figure 2.

One-hybrid selection strategy for I-SceI variants with altered DNA binding specificity. A) An I-SceI derivative fused to the E. coli RNA polymerase α subunit does not bind to the target DNA sequence and does not recruit RNA polymerase to the weak lac promoter. Low levels of His3 expression prevents rapid growth of bacterial colonies on His-selective media in the presence of 3-AT. B) I-SceI derivatives within the randomized libraries that have high affinity for the target sequence localize RNA polymerase to the promoter, thereby activating transcription to yield rapidly growing bacterial colonies.
We first tested whether wild-type fusion proteins could be recovered from the libraries in a selection for binding to the wild-type recognition site. This experiment would also reveal whether any other amino acid residues effect DNA binding to the cognate site (Table 1, Supplemental Table 1). Of the six possible codon combinations that encode the wild-type residues, four and five different combinations were isolated from the 3var and 5var libraries, respectively, and one to three independent isolates of each were obtained. Thus, the libraries contain wild-type sequences, but some of the different wild-type codon combinations were not recovered, either because the libraries lacked these codon arrangements or because an insufficient number of candidates were analyzed. Three isolates from the 3var library and five from the 5var library contained wild-type amino acids at the randomized positions that contact the DNA and a G100A substitution. Since no candidate survivor colonies were obtained using targets containing substitutions at position +3, no further attempt was made to alter specificity at this position. When the C/G+4 recognition sequence was used in the selection, isolates that encoded the wild-type residues at the randomized positions were obtained from both the 5var (three of six possible codon combinations) and 3var (four of six possible codon combinations) libraries. In addition, six variants were selected that contained a G100A substitution. It is evident, therefore, that insertion of an alanine at position 100 permits binding to both the wild-type and C/G+4 recognition sites.
Table 1.
Amino acid residue identities of selected I-SceI variants
| Target Site | Isolate | Library |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 var |
5 var |
||||||||||
| Amino Acid Position | |||||||||||
| A/T+4 (WT) | 61 | 88 | 100 | a# | 59 | 61 | 86 | 88 | 100 | a# | |
| WT | Glu | Arg | Gly | 6 | Gln | Glu | Lys | Arg | Gly | 10 | |
| G100A | Glu | Arg | Ala | 3 | Gln | Glu | Lys | Arg | Ala | 5 | |
| C/G+4 | WT | Glu | Arg | Gly | 6 | Gln | Glu | Lys | Arg | Gly | 6 |
| G100T | Glu | Arg | Thr | 5 | |||||||
| G100A | Gln | Glu | Lys | Arg | Ala | 6 | |||||
| K86R/G100T | Gln | Glu | Arg | Arg | Thr | 3 | |||||
| K86R/G100C | Gln | Glu | Arg | Arg | Cys | 4 | |||||
| K86R/G100S | Gln | Glu | Arg | Arg | Ser | 5 | |||||
The number of isolates containing each combination of residues is indicated.
In the selection that used the C/G+4 recognition sequence, five isolates were obtained from the 3var library that contained threonine at position 100 (G100T). Double mutants containing a K86R substitution plus either a threonine, a cysteine or a serine substitution at position 100 were isolated from the 5var library. These results suggest that small amino side chains are tolerated at position 100 and that either of the two long chain, basic amino acids function at position 86. Interestingly, among 59 selected isolates on two different targets, none had any substitutions at the other three randomized positions, Gln59, Glu61 and Arg88, suggesting that the side chains that mediate DNA contacts at these positions cannot be replaced with other residues.
Substitutions at randomized amino acids in yeast I-SceI homologues
It is apparent that some LAGLIDADG homing enzymes utilize different protein-DNA contacts to recognize identical target sites 34. We surveyed the sequences of I-SceI homologues to determine whether the amino acid substitutions obtained by our selection occur in other yeasts (Figure 3). The I-SceI homologues from Kluyveromyces thermotolerans and Zygosaccharomyces bisporus are predicted to be functional enzymes 3, and neither contains amino acid substitutions at the positions that were randomized in this report. However, in the I-SceI homologue from the distantly related yeast Pichia canadensis, which is 55% identical to I-SceI, Lys86 is substituted with tyrosine, and Gly100 is substituted with arginine. The inferred target sites for the K.thermotolerans and P.canadensis homologues are identical to that of I-SceI (data not shown), but sequence information is not available for Z. bisporus. Thus, naturally occurring homologues of homing endonucleases have evolved amino acid mutations at some of the same positions where substitutions have been introduced by directed evolution. Similar observations have been made for the Aspergillus nidulans I-AniI homing endonuclease, but in that case, it was found that amino acid substitutions obtained by directed evolution that increased DNA cleavage activity were identical to those present in I-AniI homologues from other fungal species 35.
Figure 3.

Alignment of the amino acid sequence of I-SceI from S. cerevisiae with those of I-SceI homologues from Kluyveromyces thermotolerans, Zygosaccharomyces bisporus and Pichia canadensis. The positions of five residues randomized in the study are numbered in red. Residues identical in all homologues are highlighted in dark gray, and residues identical in I-SceI and in at least one other homologue are highlighted in light gray. Non-identical, conserved substitutions common in all four homologues are labeled with the symbol “:”. ClustalW2 was used to perform alignments and to define conserved substitutions55.
In vivo binding analysis of selected and designed mutants
The one-hybrid selection strategy was used to qualitatively assay DNA binding activity in vivo. Control reporter strains that lack the I-SceI fusion protein or that express a non-functional protein do not grow on the selective media while those that produce the wild-type I-SceI protein or active selected variants produce large colonies after 60 hours of growth. Table 2 shows that strains that express the wild-type I-SceI fusion protein and harbor either the wild-type (A/T+4) or the C/G+4 target sites grew equally well, suggesting that the protein binds similarly to both targets. By contrast, no growth was observed when the wild-type I-SceI fusion protein was expressed in strains containing the G/C+4 or T/A+4 targets. Wild-type I-SceI may be unable to bind the T/A+4 or G/C+4 substituted sites because neither the O4 of T+4 nor the exocyclic N6 of A+4 can accept a hydrogen bond from the side chain amino group of Lys86. These results are consistent with in vitro experiments that determined the target specificity of the protein22; 31. Table 2 shows that expression of engineered Q59A, Q59E, E61A, E61Q, K86A, and R88A mutant proteins does not permit growth of the reporter strain, suggesting that these proteins bind poorly or not at all to the different DNA substrates. These results are consistent with the role that Gln59, Glu61, Lys86 and Arg88 play in directly contacting DNA bases.
Table 2.
In vivo growth phenotypes of I-SceI variant proteins
| I-SceI | DNA recognition site |
||
|---|---|---|---|
| A/T+4 (WT) | C/G+4 | Other sitesa | |
| WT (K86/G100) | +++b | +++ | - |
| Q59A | - | - | - |
| Q59E | - | - | - |
| E61A | - | - | - |
| E61Q | - | - | - |
| K86A | - | - | - |
| R88A | - | - | - |
| R88K | - | - | - |
| K86R/G100T | - | +++ | - |
| K86R/G100C | - | +++ | - |
| K86R/G100S | - | +++ | - |
| G100T | + | +++ | - |
| G100A | +++ | +++ | - |
| K86R | ++ | ++ | - |
| G100C | - | + | - |
| G100S | - | - | - |
| G100V | - | - | - |
The DNA recognition sites that were assayed included G/C+3, A/T+3, T/A+3, T/A+4, and G/C+4.
Growth phenotypes of bacteria transformed with pACLαI-SceI(D145A) derivatives that express the indicated I-SceI variant proteins and containing the indicated I-SceI recognition sequence on the single copy F′-episome. The transformants were grown on selective mediate for 60 hours at 37° C and the growth phenotypes were determined by averaging the measured diameters of 15 colonies from each of three experimental trials. Growth phenotypes: +++, colonies >2 mm; ++, colonies < 2 mm and > 1 mm; +, colonies < 1 mm; -, no growth.
Large colonies resulted when the K86R/G100T, K86R/G100C and K86R/G100S derivatives were expressed in the C/G+4 reporter strain on which they were selected, but none resulted when the strains contained the wild-type site (A/T+4) (Table 2). Expression of the G100T variant resulted in the formation of large colonies in strains containing the C/G+4 site and small colonies in the strain containing the wild-type site. The lack of growth or slow growth of strains containing the wild-type site was unexpected because it suggested that the K86R/G100T, K86R/G100C, K86R/G100S and G100T proteins bound poorly to the A/T+4 site despite the fact that there was no selection against binding to it. Like the wild-type I-SceI fusion protein, none of the variants bound to the G/C+4 or T/A+4 sites in vivo. We dissected the role of the K86R substitution in the double mutants by assaying an engineered K86R protein for DNA binding in vivo. When the protein was expressed in reporter strains containing either the A/T+4 or C/G+4 substrates, the resulting colonies were smaller than those observed when the wild-type protein was expressed, suggesting that the arginine-substituted protein binds less tightly to these substrates than wild-type I-SceI.
The roles of the small, polar side chains identified at position 100 were assessed using the in vivo DNA binding assay. Expression of the G100A variant in strains containing either the A/T+4 or C/G+4 targets resulted in colonies that were similar in size to those expressing the wild-type protein. By contrast, expression of the G100S protein yielded no growth in either the A/T+4 or C/G+4 strains, and expression of the G100C variant in the C/G+4 strain resulted in small colonies. Combination of the G100S and G100C mutations with the K86R mutation resulted in more robust growth. No colonies expressing the Val100 protein were observed under any conditions. Finally, we determined that lysine cannot substitute for arginine at position 88 by showing that reporter strains expressing an I-SceI fusion protein containing a R88K substitution do not grow on either the A/T+4 or C/G+4 recognition sites.
In vitro DNA binding affinities of I-SceI derivatives
Equilibrium dissociation constants (Kd) were determined using a filter binding assay (Table 3). Wild-type I-SceI enzyme exhibited tight binding to both the wild-type and C/G+4 mutant targets under in vitro conditions with binding constants of approximately 9 nM and 20 nM, respectively, but it binds weakly to the non-specific target (Kd>400nM) (Table 3). The Kd value for wild-type I-SceI and its canonical target is similar to that reported previously by our group and others 22; 33; 36. The double mutant proteins, K86R/G100T, K86R/G100C and K86R/G100S, and the single mutant G100T protein that were each selected for binding to the C/G+4 substrate in vivo, bind in vitro to that target as tightly as the wild-type protein (Kd values ranging from 7 nM to 23 nM). However, the in vitro assays indicate that binding of these single and double mutant proteins to the wild-type recognition site is reduced, with decreases in affinity ranging from 4- to 11-fold. Thus, these results confirm the unexpected finding that the double mutants bind poorly to the wild-type site, indicating that they are less flexible than wild-type I-SceI in binding different substrates.
Table 3.
Thermodynamic parameters of I-SceI variants
| I-SceI protein |
KD (nM) |
||
|---|---|---|---|
| A/T+4 (WT)a | C/G+4 | Non-specificb | |
| WT(K86/G100) | 9.0±3 | 20±4.6 | 420±140 |
| K86R/G100T | 96±16 | 8.2±0.86 | 440±82 |
| K86R/G100C | 74±12 | 7.5±2.3 | ND |
| K86R/G100S | 83±27 | 23±4.3 | ND |
| G100T | 39±13 | 10±1.5 | 420±91 |
| K86R | 35±5 | 39±10 | ND |
| G100C | 110±30 | 34±8.8 | ND |
| G100S | 150±26 | 32±10 | ND |
| G100V | 240±51 | 230±27 | ND |
ND, not determined.
Equilibrium dissociation constants (mean ± SD, n=3) were obtained by direct measurement by a filter-binding assay.
The sequence of the non-specific DNA was derived by randomizing the order of the base-pairs comprising the wild-type recognition sequence (see Materials and Methods).
We compared the binding affinities of the K86R/G100T double mutant with those of the G100T and K86R single mutants to evaluate the contribution of these mutations to binding. None of the mutations has a marked effect on the binding affinity for the C/G+4 target. However, both mutations decrease the binding affinity of the protein to the wild-type recognition site approximately four-fold relative to the wild-type protein (39 nM (G100T) and 35 nM (K86R) compared with 9 nM (WT)). Combining the two mutations in the K86R/G100T double mutant results in an even larger decrease in binding (Kd = 96 nM) that is approximately equal to the combined reduction in binding contributed by each single mutant. The data suggest that both the K86R and G100T mutations deleteriously affect binding to the wild-type substrate.
Substitution with cysteine and serine at position 100 is more deleterious to I-SceI binding to the wild-type site than substitution with threonine (Kd values of 110 nM and 150 nM for the cysteine and serine substitutions, respectively, compared with 39 nM for the threonine mutation (Table 3)). This finding is consistent with the fact that cells expressing the G100C and G100S proteins yield no colonies in the in vivo assay while those expressing the G100T protein yield small colonies (Table 2). Substitution of the glycine hydrogen at position 100 with the hydrophobic side chain of valine decreases binding to both the wild-type and mutant targets by at least an order of magnitude, relative to wild-type I-SceI. Thus, side chain size and polarity at position 100 influence DNA binding activity both in vivo and in vitro. Proteins containing a hydrogen (G100) or a methyl group (G100A) are active, but addition of a sulfhydryl (G100C) or hydroxyl (G100S) group reduces the DNA binding activity. Addition of a methyl group to the serine side chain (G100T) increases activity, but replacement of the hydroxyl with a methyl group (G100V) eliminates activity.
There is a correlation between the in vivo and in vitro binding assays (Tables 2 and 3). For proteins in which the Kd values for a target site were greater than 40 nM, expression of the proteins in the one-hybrid growth assay yielded no colonies. Conversely, if the Kd values were less than 25 nM, expression of the protein in vivo resulted in similarly sized bacterial colonies as those observed for strains expressing wild-type I-SceI in reporter strains containing the wild-type recognition sequence. When the equilibrium dissociation constant determined in vitro was between 25 nM and 40 nM, no strict correlation existed between the assays.
Determination of substrate preferences of wild-type, G100T and K86R/G100T I-SceI proteins using competitive cleavage assays
The one-hybrid strategy selects variants on the basis of their DNA binding rather than their DNA cleavage activity, so it is uncertain whether selected proteins that exhibit shifted DNA binding specificities also acquire new cleavage profiles. Therefore, substrate cleavage preferences were determined by competitive DNA cleavage assays using I-SceI constructs in which catalytic residue Asp145 was present. Wild-type I-SceI prefers the canonical I-SceI target approximately five-fold compared to the C/G+4 target (Figure 4a and 4d). In contrast, the G100T protein does not show a marked preference for either target (Figure 4b). The K86R/G100T variant displays a switch in substrate specificity relative to the wild-type protein since it preferentially cleaves the C/G+4 target 7-fold more effectively than the wild-type target (Figure 4c and 4d). When the relative cleavage efficiencies of all three proteins are compared, it is apparent that they all cleave the C/G+4 target similarly since there is <2-fold difference in C50 values. In marked contrast, the G100T protein cleaves the wild-type target approximately 13-fold less efficiently than the wild-type protein and the K86R/G100T protein cleaves it 29-fold less efficiently. This loss of activity towards the wild-type target by the double mutant protein results in a 36-fold switch in overall target preference relative to the wild-type protein.
Figure 4.

Competitive DNA cleavage assay by wild-type I-SceI and mutant derivatives. A) Two linearized plasmid substrates containing either the wild-type I-SceI recognition sequence (A/T+4) or the C/G+4-substituted site (10 nM) were incubated for 20 min at 25° C with different concentrations of wild-type I-SceI (0, 5 nM, 10 nM, 20 nM, 40 nM, 80 nM, 160 nM, 320 nM, or 640 nM). The uncleaved DNA substrates were separated from the cleavage products by agarose gel electrophoresis, the gels were stained with ethidium bromide and the amounts of each band were quantitated from digital images. B) As in panel A) using the G100T I-SceI derivative. C) As in panel A) using the K86R/G100T protein. D) Average C50 values from duplicate DNA cleavage experiments.
These findings indicate that, in general, there is a correlation between the protein/DNA binding affinities and the cleavage efficiencies. The equilibrium dissociation constants of the wild-type, G100T and K86R/G100T proteins for the C/G+4 target are similar, as are their C50 values for DNA cleavage of the substrate (Table 3 and Figure 4d). The differences in binding of the wild-type site by the three proteins, with the wild-type protein binding most tightly and the double mutant binding most weakly, mirrored the cleavage activity profiles of the three proteins. The moderate decrease in cleavage activity of the wild-type substrate by the G100T variant, relative to wild-type I-SceI, and the severe decrease in activity by the K86R/G100T highlights the additive negative effect of the G100T and K86R mutations. The consequence of having both mutations in I-SceI is a significant switch in substrate preference.
Crystallographic analysis of wild-type and K86R/G100T I-SceI bound to the C/G+4 recognition sequence
The X-ray crystal structure of wild-type I-SceI protein bound to the C/G+4 recognition site was determined at a resolution of 2.3Å in order to elucidate how this protein interacts with the non-cognate sequence (Table 4 and Supplemental Figure 1). The RMSD value of the superposed structures of wild-type I-SceI bound to the A/T+4 and C/G+4 recognition sequences is 0.41Å. Overall, there are few differences between the structures except at the position of the substituted nucleotide where it is apparent that Lys86 hydrogen bonds to the O6 of G+4 (3.27 Å) in the non-cognate site structure rather than to O4 of T+4 (Figure 5). The presence of few modeled water molecules in this structure precludes conclusions to be drawn about the role of solvent in the protein-DNA interaction.
Table 4.
Data collection and refinement statistics
| Protein-DNA substrate | ||
|---|---|---|
| WT-C/G+4 | K86R/G100T-C/G+4 | |
| Space group | C 2 | C 2 |
| Cell dimensions: | ||
| a, b, c (Å) | 129.2,49.9, 63.4 | 131.7, 50.5, 62.9 |
| α, β, γ (°) | 90, 107.5, 90 | 90, 108.5, 90 |
| Wavelength (Å) | .97872 | .97869 |
| Rsym (%)a | 5.1(10.3)b | 5.7(40.1)b |
| I/σ | 28.8(5.4)b | 38.3(5.7)b |
| Redundancy | 3.4(3.6)b | 7.4(7.2)b |
| Completeness (%) | 91.5(96.9)b | 98.7(98.0)b |
| Resolution (Å) | 30.8–2.3 (2.36–2.30) | 31.7–2.5 (2.57–2.50)b |
| No. of reflections used in refinement | 14747 | 12597 |
| No of reflections in test set | 1133 | 1033 |
| Rwork(%)/Rfree(%) | 24.6/29.4 | 23.4/28.2 |
| No. of atoms: | 2876 | 2857 |
| Protein | 1853 | 1859 |
| DNA | 984 | 984 |
| Ion | 3 | 3 |
| Solvent | 36 | 11 |
| Ramachandran plot: | ||
| Preferred regions (%) | 95.93 | 95.02 |
| Allowed regions (%) | 4.07 | 4.07 |
| Outliers(%) | 0 | 0.9 |
| Bond length r.m.s.d(Å) | 0.006 | 0.005 |
| Bond angle r.m.s.d(deg) | 1.14 | 1.01 |
| Average B-factor | 43.9 | 67.5 |
| Coordinate error by Luzzati plot(Å) | .49 | .56 |
Rsym=ΣhΣi|Ii(h)-<I(h)>-|/=ΣhΣiI(h).
Values for high-resolution shell in parentheses.
Figure 5.

Views of wild-type I-SceI bound to a 24-bp duplex containing the C/G+4 substitution within the I-SceI recognition sequence. The two views are rotated 180° about the indicated axis and show the locations of Gln59, Glu61, Lys86, Arg88 and Gly100 (colored gray and by element) relative to the C/G+3 (colored salmon and by element) and C/G+4 (colored green and by element) base-pairs (all depicted as sticks). The protein (colored cyan) and DNA strand (green and blue) backbones are represented with ribbons. Hydrogen bonds are indicated by dashed lines.
Our biochemical analyses indicated that the interaction between the K86R/G100T protein and the C/G+4 target site was as strong as that between the wild-type protein and the cognate sequence, but also showed that the double mutant protein had low affinity for the wild-type A/T+4 site. We determined the co-crystal structure of the double mutant protein bound to the C/G+4 recognition sequence at 2.5 Å resolution to provide insight into these observations (Table 4 and Supplemental Figure 2). The protein directly contacts the nucleotide at position +4 of the mutant target (Figure 6). Specifically, the guanidino group of Arg86 makes bidentate contacts to the O6 and to N7 (2.7 Å) of G+4 (3.6 Å) in the K86R/G100T-C/G+4 structure. The threonine substitution at position 100 is distant from the DNA and does not make direct DNA contacts. Furthermore, analysis of the wild-type-A/T+4 structure indicates the presence of a pocket formed by Glu61, Gly100, His84, Arg88 and the DNA in which Lys86 makes a hydrogen bond to O4 at T+4 (Figure 7a). Gly100 does not interfere with any of the adjacent side-chains forming this pocket. However in the K86R/G100T-C/G+4 structure, the presence of the Thr100 side chain in the pocket 4.0 Å from the Cγ of Arg86 (Figure 7b) may constrain the number of rotamers that Arg86 can adopt, resulting in an orientation that is favorable to bind guanidine but not thymidine at nucleotide position +4. This indirect effect of Thr100 may explain how this residue affects DNA binding, but other mechanisms of action cannot be ruled out. The structures of the K86R/G100C and K86R/G100S complexes have not been determined, but a similar role for the cysteine or serine in these mutant proteins is suggested.
Figure 6.

Views of the K86R/G100T double mutant protein bound to a 24-bp duplex containing the C/G+4 substitution in the I-SceI recognition sequence. The two views are rotated 180° about the indicated axis. The side chains of Gln59, Glu61, Arg86, Arg88 and Thr100 (colored gray and by element) and the C/G+3 (colored salmon and by element) and C/G+4 (colored green and by element) base-pairs are represented by sticks. The protein (colored cyan) and DNA strand (green and blue) backbones are depicted as ribbons. Hydrogen bonds are indicated by dashed lines.
Figure 7.
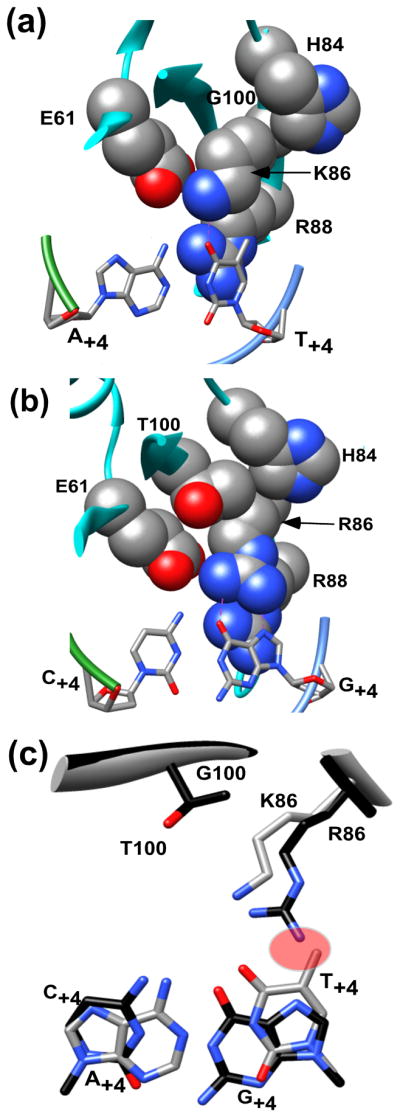
A) A detailed view of amino acid residues Glu61, His84, Lys86, Arg88, and Gly100 from the wild-type structure (1R7M) depicted as space filling representations in order to highlight the space surrounding Lys86 (depicted as stick and colored gray and by element). The protein and the two DNA strand backbones are depicted as ribbon representations (colored cyan and blue and green, respectively) and the DNA bases are depicted as sticks (colored gray and by element) B) Detailed view of residues Glu61, His84, Arg86, Arg88 and Thr100 depicted in space filling representations that indicates the limited space surrounding the Arg86 side chain due to the presence of Thr100. The protein and DNA backbones are represented as in Panel A.C) Superposition of Gly100, Lys86 and A/T+4 residues from the 2.25Å wild-type I-SceI-A/T+4 co-crystal structure (1R7M,; colored gray and by element) and the Thr100, Arg100 and C/G+4 residues from the 2.5Å K86R/G100T-C/G+4 structure (colored black and by element) described in this work. A predicted steric clash is highlighted in red.
Efforts to obtain co-crystals of the K86R/G100T mutant bound to the wild-type target were unsuccessful, perhaps because of the low binding affinity between the macromolecules. To gain insight into why the double mutant protein binds poorly to the site, the wild-type I-SceI/DNA complex structure was superposed on the K86R/G100T-C/G+4 structure, yielding an alignment with a Cα RMSD value of 0.36 Å. Interestingly, the superposed structures indicated a severe steric clash between the guanidino group of Arg86 and the T+4 C5 methyl group (distance=1.0Å; Figure 7c). The exclusion of favorable rotamers in the presence of Thr100 in addition to the steric clash between Arg86 and the DNA may account for the significant loss of affinity by the double mutant for the wild-type A/T+4 target.
DISCUSSION
Proteins that have been engineered in the laboratory to interact with alternative DNA sequences provide insight into how DNA binding proteins naturally evolve specificity for target sites. Here, we combined directed evolution and rational design methods to isolate variants of the I-SceI homing endonuclease that displayed greater specificity than the wild-type enzyme for a particular DNA sequence. I-SceI cleaves DNA sequences containing either the cognate A/T+4 base-pair or a non-cognate C/G+4 base-pair within this region, and the crystal structure indicates that in the protein-DNA complex, the Lys86 amino group makes direct contacts to T+4 while Glu61 and Gln59 side chain make indirect water-mediated contacts to A+4. We applied a bacterial one-hybrid method to select variants from combinatorial plasmid or phagemid libraries that bind to substrates containing base substitutions at position +4. An unexpected finding was that the variants selected on the C/G+4 substrate, unlike wild-type I-SceI, bind poorly to the wild-type A/T+4 substrate. This is explained at the molecular level by the introduction of amino acid side chains in the mutant proteins that limit the number of possible rotamers available to amino acids side chains that contact the DNA.
A major goal of several research groups has been to engineer homing endonucleases with any desired target site specificities to use in gene therapy strategies 37. These efforts have entailed screening or selecting altered specificity enzymes from rationally designed combinatorial libraries based on DNA binding and/or cleavage activities, and generating proteins using computational design 21; 22; 24; 26; 27; 28; 38; 39; 40. Our previous application of a bacterial two-hybrid method to select altered specificity variants of PI-SceI was restricted to modifying the protein-DNA interactions made by the protein splicing domain, whose establishment are a prerequisite for DNA binding by the endonuclease domain28. A modified one-hybrid E.coli version of the method was used here to select altered specificity variants of I-SceI. Variants of the His-Cys box I-PpoI homing endonuclease with changed specificity were isolated using a similar method 41. The extent of the specificity shift observed in the selected I-SceI variants was similar to those evolved in PI-SceI, and in neither case were proteins generated that recognized a single target with high specificity. For reasons that are unclear, fewer variants with different specificities were identified in this study than were obtained in the selection using PI-SceI. This may indicate that there is tight coupling in I-SceI between adjacent contacts within the β-sheet 1/DNA interface, and that small perturbations introduced by amino acid substitutions disrupt all protein binding. Incorporation of a stringent negative selection or combination of the selection methods with computational design might be necessary to achieve the levels of specificity that are required to prevent deleterious ectopic cleavage.
Two other efforts to engineer altered specificity variants of I-SceI targeted the extreme ends of the I-SceI/DNA interface, with one focusing on a short 27 residue amino-terminal subdomain that contacts the DNA minor groove between nucleotides +8 through +11 and the other addressing the interaction between a short loop that contacts nucleotide −7 22; 24. The latter study succeeded in engineering a mutant protein that exhibited a 17-fold greater preference for a non-cognate substrate containing four nucleotide substitutions relative to the wild-type substrate 24, while the former study evolved a protein that preferred a singly-substituted target sequence nearly 4-fold 22. These methods selected variants based on the DNA cleavage activities rather than DNA binding affinities, as done here, but the magnitudes of the shifts in cleavage specificity of the selected enzymes were similar. It is also noteworthy that one of the previous studies included a negative selection against cleavage of the wild-type substrate in order to obtain variants with altered, rather than broadened, specificity 22. An unexpected result of our study was that even in the absence of a negative selection, homing enzymes were obtained that acquire higher specificity for a mutant target sequence.
The in vivo and in vitro assay results indicate that there is a correlation between the in vivo growth phenotypes and the in vitro equilibrium dissociation constants, suggesting that the balance of the different in vivo parameters, including the concentrations of I-SceI, DNA target site, non-specific DNA targets and the His3 competitor, 3-aminotriazole (3-AT), permits bacterial growth to reflect in vivo I-SceI binding. Furthermore, as noted above, we observed a correlation between the in vitro DNA binding affinities and DNA cleavage efficiencies. When altered specificity variants are isolated based on DNA cleavage activity, the observed differences between survival rates measured in vivo for wild-type and mutant proteins appears to be much greater than the differences in cleavage efficiencies measured in vitro, suggesting that the conditions present in vivo result in greater levels of specificity than those detected in vitro 22. We cannot conclude that there is a difference in in vivo and in vitro DNA binding specificities because we have not measured the DNA binding affinities in vivo.
I-SceI recognition and cleavage of sites containing different nucleotides at position +4 can be understood in terms of homing endonuclease structure and function. The several X-ray crystal structures of LAGLIDADG homing enzymes bound to their DNA substrates reveal that the proteins use only a small subset of the total number of available hydrogen bond contacts to effect specific recognition. I-SceI makes 27 hydrogen bonds to the DNA bases within its 18-bp recognition sequence, corresponding to only 1.5 hydrogen bonds per base-pair 11, which is nearly as low as the single contact per base-pair observed for I-CreI 42. This subsaturation of available contacts contrasts with the much larger number of interactions per base-pair made by restriction enzymes, which recognize large numbers of short sequences with high specificity in order to destroy the genomes of invading bacteriophage without risking the integrity of the host chromosome42. By contrast, homing endonucleases must only be able to cleave a single genomic site in order to initiate homing, which they accomplish by utilizing long target sequences. However, they must also be able to facilitate HEG lateral transmission into related recognition sequences of other species, which they accomplish by tolerating sequence divergence. Given this role, it might be expected that homing endonucleases encoded by HEGs that have recently invaded a new host genome would exhibit broadened specificity, but that subsequent selection would increase their specificity for the cognate site in order to reduce their cleavage of ectopic sites that would deleteriously affect host fitness. 23; 43. Therefore, many homing endonucleases may not have evolved optimal specificity for their cognate sites, and further improvement in DNA binding, target site specificity, and/or DNA cleavage activities may be possible 44. The pliable nature of LAGLIDADG enzymes has permitted the DNA cleavage activities of the intein-derived PI-SceI and the intron-encoded I-AniI homing endonucleases to be increased by introduction of mutations into the enzymes or by nucleotide replacement within the target sites 29; 35; 44; 45. Evolution of increased specificity by I-SceI is demonstrated here since substitution of two amino acids in wild-type I-SceI, which cleaves either of two substrates similarly, yields a mutant derivative that significantly prefers to cleave only one of the sequences. The structure of the K86R/G100T structure suggests that intramolecular steric occlusion in the protein constrains the specificity of the protein by limiting the flexibility of Arg86. This mode of action may be a general strategy that limits substrate promiscuity in homing endonucleases. Side chain constraint by hydrogen bonding also plays a key role in determining specificity. Removal of a PI-SceI glutamic acid side chain that hydrogen bonded and constrained an arginine making a base-specific DNA contact resulted in significant specificity broadening 28.
Taken together, the results of this study demonstrate that it is possible to augment the specificity of I-SceI even in the absence of negative selection pressure. This offers the possibility that site-specific enzymes can be engineered in the future from progenitors more flexible in their recognition of DNA substrates.
MATERIALS AND METHODS
Media and bacterial strains
Luria-Bertani medium (LB), LB agar, YEG agar plates, SOC medium, histidine-selective NM medium and NM agar plates were prepared according to published protocols46. Liquid media and plates were supplemented with one or more of the following antibiotics and reagents: ampicillin (Fisher Scientific, Pittsburgh, PA) (Ap, 100 μg/ml), chloramphenicol (Sigma-Aldrich, St. Louis, MO) (Cm, 30 μg/ml), kanamycin (Sigma-Aldrich, St. Louis, MO) (Kn, 50 μg/ml), tetracycline (Tc, 15 μg/ml), isopropyl-β-D-thiogalactopyranoside (Indofine Chemical Co., Hillborough, NJ), (IPTG, 250 μM), D, L–p-Cl-phenylalanine (Sigma-Aldrich), (2mg/ml) and 3-aminotriazole (Acros Organics, Morris Plains, NJ) (3-AT, 20 mM or 40 mM).
Escherichia coli strain XL-1 blue (Stratagene, La Jolla, CA) (recA endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F′ proAB lacIqZΔM15 Tn10 (tetr)]) was used to construct plasmid and phagemid libraries. CSH100 was used to construct strains containing different episome-encoded I-SceI substrates. Strain KJ1C (F−, ΔhisB463, Δ(gpt-proAB-arg-lac)XIII, zaj::Tn10), which does not produce the hisB gene product, was used with CSH100 (F′ lacproA +, B +(lacIq lacPL8)/araD(gpt-lac)5) in a conjugation method to generate reporter strains containing the F′-episome-based DNA targets. Strain BL21 (DE3) (F−ompT gal[dcm] [lon] hsdSB (rB−mB−; an E.coli B strain) with DE3 contains a λ prophage that expresses the bacteriophage T7 RNA polymerase and was used to express wild-type I-SceI and I-SceI derivative proteins. Electro-competent cells for all strains were prepared using established protocols 47. CSH100 and KJ1C were kindly provided by Drs. Keith Joung and Carl Pabo.
Construction of randomized plasmid expression libraries and subcloning of designed I-SceI derivatives
Plasmid pACLαI-SceI(D145A)-f1 expresses a fusion protein consisting of the first 248 residues of the RNA polymerase α subunit connected by a 23-amino acid linker (AAADYKDDDDKFRTGSKTPPHRS) to 241 residues of a catalytically inactive I-SceI homing endonuclease containing a His6 carboxyl-terminal tail. This plasmid was made by replacing the coding sequence of Gal4 in plasmid pAClαGal4 32 with that of the I-SceI homing endonuclease by inserting an 810 base-pair (bp) PCR product into the NotI and AvrII restriction sites. In addition, a unique silent NsiI site was inserted into the sequence and a HindIII site was removed to facilitate subsequent library construction. The presence of a D145A mutation in the I-SceI coding region abolishes catalytic activity but does not affect DNA binding. Plasmid pACLαI-SceI(D145A)-f1 was created by insertion of a 306-bp PCR product containing the f1 origin of replication sequence from pBluescript SK+ (Stratagene, La Jolla, CA) into the AatII and EcoO109I sites of pACLα-I-SceI (D145A). To facilitate efficient cloning of I-SceI mutants and libraries, and to reduce contamination of libraries with sequences encoding wild-type I-SceI, a 803-bp non-coding stuffer fragment derived from plasmid pF11-stuffer 28 was inserted into the NsiI and SfoI sites of the I-SceI coding sequence in pACLαI-SceI(D145A)-f1 to generate pACLαI-SceI(D145A)-f1-stuffer.
Two pACLαI-SceI (D145A)-f1-stuffer-based plasmid libraries, termed 3var and 5var, were generated by randomizing either the codons for I-SceI residues E61, R88 and G100 or those for residues Q59, E61, K86, R88 and G100, respectively, using a mixed base NNK codon. Degenerate oligonucleotides from Integrated DNA Technologies, Inc. (Coralville, IA) (3var library: 5′-GA TCC TGG GTG ATG CAT ACA TCA GAT CTC GTG ATG AAG GTA AAA CCT ACT GTA TGC AGT TCN NKT GGA AAA ACA AAG CAT AC-3′ and 5′-CA GTT TGT TGA AAG CTT GGT GTT TGA AAG TCT GGG CMN NCC AGG TGA TTA CCA GGT TAC CCA GGT GGT TAA CMN NTT CTT TTT TGT GCG GCG GGG ACA G-3′; 5var library: 5′-TGG GTG ATG CAT ACA TCA GAT CTC GTG ATG AAG GTA AAA CCT ACT GTA TGN NKT TCN NKT GGA AAA ACA AAG CAT AC-3′ and 5′-TGT TGA AAG CTT GGT GTT TGA AAG TCT GGG CMN NCC AGG TGA TTA CCA GGT TAC CCA GGT GGT TAA CMN NTT CMN NTT TGT GCG GCG GGG ACA G-3′) were used in PCR to create two sets of 189-bp fragments randomized at specified positions that were inserted into the NsiI and HindIII sites of pACLαI-SceI(D145A)-f1-stuffer. The resulting plasmid libraries were electroporated into XL-1 Blue cells and grown for 1 hour at 37 °C in SOC media. The 3var and 5var libraries yielded 6.2 × 106 and 4.2 × 108 tetracycline-resistant and chloramphenicol-resistant colonies, respectively. The predicted codon complexities for the 3var and 5var libraries were estimated to be 3.3 × 104 and 3.4 × 107, respectively. The cells were amplified for 1 hour at 37° C, harvested by centrifugation and diluted in SOC media supplemented with 15% glycerol before being frozen at −80° C. Plasmid library DNA was purified from thawed cells.
A phagemid library was generated from the 5var plasmid library to maximize coverage of the library. Cells harboring the 5var plasmid library were thawed, diluted five-fold in SOC media and infected with M13 K07 helper phage (New England Biolabs, Inc., Ipswich, MA) as described previously 28. The titer of the resulting 5var phagemid library was ~4.2 × 107 transducing units/μl of stock.
Various DNA fragments that contained the sequences of engineered I-SceI mutants were generated using appropriate DNA primers and PCR. These were inserted into the pACLαI-SceI(D145A)-f1-stuffer plasmid using the NsiI and HindIII restriction sites.
Construction of reporter strains to use in the one-hybrid selection
Reporter plasmids were constructed that express the selectable HIS3 gene product from a weak lac promoter located downstream of either the 18-bp wild-type or mutant I-SceI recognition sequences. Duplex oligonucleotide cassettes containing the wild-type I-SceI recognition sequence (5′-AATTACGCTAGGGATAACAGGGTAATAC-3′ and 5′-GGCCGTATTACCCTGTTATCCCTAGCGT-3) or recognition sequences with A/T+3, T/A+3 or G/C+3 substitutions for C/G+3 or with C/G+4, T/A+4 or G/C+4 substitutions for A/T+4 were inserted into the EcoRI and NotI sites of pF11-stuffer-HIS3-aadA-pheS 28 to generate seven reporter constructs. The I-SceI target sequences were confirmed by DNA sequencing.
The portions of the plasmids including the I-SceI recognition sites, the downstream lac promoters and the HIS3 reporter gene were transferred to single-copy F′-episomes using an homologous recombination and conjugation strategy as described previously 28. Briefly, the target site and reporter gene sequences located on the pF11-stuffer-HIS3-aadA-pheS derivatives were allowed to recombine from the plasmids onto F′-episomes, which were then isolated from the parental plasmids by their transfer to strain KJ1C by conjugation.
One hybrid selection of DNA binding activity in Escherichia coli
Selection of I-SceI variants that bind to specific recognition sequences was effected using a bacterial one-hybrid strategy that was adapted from a previously described two-hybrid protocol 32. Here, cells that express RNA polymerase α-subunit/I-SceI fusion proteins capable of binding to a I-SceI recognition sequence located upstream of a weak lac promoter and the HIS3 gene can be selected since these are able to grow on media lacking histidine. The 3var plasmid library DNA was transformed into tetracycline- and kanamycin-resistant, electrocompetent cells of each of the seven KJ1C reporter strains. The transformation efficiency was ~106 transformants/μg DNA. The transformed cells were grown for 1 hour in SOC media, centrifuged and washed three times in NM/Tc/Kn/Cm/IPTG media and then grown on NM/Tc/Kn/Cm/IPTG plates containing 20 mM 3-AT for 60 hours at 37° C. A reporter strain containing the wild-type target and a plasmid that expresses the wild-type RNA polymerase α-subunit/I-SceI fusion protein was grown in parallel to serve as a positive control, and a reporter strain transformed with an empty pACL vector was used as a negative control.
The 5var phagemid library was transduced into each of the seven reporter strains as described previously. A 5 μL aliquot of infected reporter strain cells was grown for 24 hours at 37° C on LB/Tc/Kn media to yield a total of ~109 cells. The number of transductants was estimated to be ~107 by determining the number of cells that grew on LB/Tc/Kn/Cm media. The infected reporter strains were grown on selective NM/Tc/Kn/Cm/IPTG media containing 20 mM 3-AT for 60 hours at 37° C. DNA from isolates that grew on selective plates was extracted and sequenced. To confirm the phenotypes of the selected plasmids or phagemids, the isolated DNA was transformed back into the naïve reporter strain on which the isolate was selected and grown for 60 hours at 37 °C on selective NM/Tc/Kn/Cm/IPTG plates containing 20 mM or 40mM 3-AT.
In vivo binding assays using the one-hybrid method
E.coli KJ1C strains containing F′-episome constructs with either the wild-type or one of the six mutant I-SceI recognition sequences were transformed with plasmid pACLαI-SceI(D145A) derivatives that express I-SceI proteins containing either wild-type or selected mutant residues at positions 59, 61, 86, 88 and 100. These strain constructs were grown on NM/Tc/Kn/Cm/IPTG media containing 20 mM 3-AT for 72 hours at 37° C. The diameters of 15 colonies, from each of three experimental trials, were averaged and scored after 60 hours of growth.
Construction of protein expression plasmids and protein purification
In order to characterize the DNA binding and cleavage activities of the selected I-SceI proteins, it was necessary to remove the RNA polymerase component of the fusion proteins and to replace alanine-145, which had been inserted to prevent DNA cleavage during the selection, with aspartic acid. DNA fragments encoding the I-SceI portion of the fusion proteins were generated by PCR, and were inserted into the NdeI and BamHI sites of pET15b (EMD4Biosciences, Gibbstown, NJ), yielding constructs that expressed I-SceI proteins containing aspartic acid at position 145, and a carboxyl-terminal His6 tag. The proteins were expressed in E. coli strain BL21 (DE3) cells and purified according to an established protocol 45 Proteins used in crystallization studies were expressed without a His6 tag and were purified as described. All proteins were estimated to be >97% purified by SDS-PAGE analysis.
Filter binding protocol
Binding affinities of wild-type and mutant I-SceI proteins for three radiolabeled duplexes were determined by filter hybridization. DNA duplexes were created by annealing a synthetic 54-mer (5′-aacgaataaaagttacgcTAGGGATAACAGGGTAATatagcgaaagagtagatatt-3′), and a 49-mer (5′-caatatctactctttcgctatATTACCCTGTTATCCCTAgcgtaacttt-3′) that contained either the 18-bp wild-type I-Sce recognition sequence (shown in capitals), the C/G+4 mutation, or a randomized 18-bp non-specific sequence (5′-CTGTGGTAAATAAGGGAA -3′). The construction of the DNA binding substrates and the nitrocellulose filter binding assays were conducted as described previously 33. DNA binding buffer included 2 mM CaCl2 in all experiments. Calcium can replace magnesium at the I-SceI active sites, but does not permit DNA cleavage. Binding experiments were performed in triplicate. Curve fitting using Kaleidagraph software (Synergy Software, Inc, Reading, PA) was performed to generate binding curves and to obtain equilibrium dissociation constants.
Competitive DNA cleavage assay
DNA fragments containing the wild-type (2520-bp) or C/G+4 mutant (2006-bp) I-SceI recognition sequences were generated by PCR from pF11-stuffer-HIS3-aadA-PheS derivatives to use as substrates in competitive DNA cleavage assays. Purified wild-type, G100T and K86R/G100T I-SceI proteins were mixed with 10 nM of each of the two DNA substrates, the reaction was initiated by addition of MgCl2 (15 mM) and the mixtures were incubated in cleavage buffer (10 mM Tris-HCl (pH8.8), 1 mM DTT, 0.1 mg/ml BSA) for 20 mins at 25° C. The reaction samples were halted by the addition of EDTA (100 mM). The reaction products were resolved by electrophoresis on a 1% agarose gel and visualized by staining with ethidium bromide (100mM). Gel images were produced using the EDAS 290 imager and software (Kodak, Rochester, NY) and C50 values were calculated from curves generated from duplicate experiments using KaleidaGraph software.
Crystallization, data collection and refinement
A protein-DNA solution containing either wild-type or K86R/G100T I-SceI proteins was co-crystallized with 24-mer DNA duplexes containing either the wild-type (Top oligo:5-CACGCTAGGGATAACAGGGTAATAC-3′, Bottom oligo:5′ GGTATTACCCTGTTATCCCTAGCGT-3′) or C/G+4 mutant targets (Top oligo: 5′-CACGCTAGGGATAACCGGGTAATAC-3′, Bottom oligo:5-GGTATTACCCGGTTATCCCTAGCGT-3 DNA duplexes were prepared by resuspending the oligonucleotides in a buffer containing 20mM Tris (pH8.0), 100 mM KCl and 2mM EDTA and annealing the samples by using a slow-cool protocol described in a previous publication 11. The DNA:protein ratio during incubation was 2 parts DNA duplex:1 part protein. Equal volumes (2 μl) of the protein-DNA samples and the precipitant solution (14%–24% PEG 400, 20mM CaCl2, 1 mM DTT and 0.1 M sodium acetate pH 4.7) were mixed and suspended as hanging drops over reservoirs containing 1–2 ml of the precipitant solution. The co-crystals were allowed to grow for three weeks at 4° C. The crystals had average dimensions of 0.3 mm × 0.2 mm × 0.125 mm. Crystals were transferred in a single step into cryo-protectants containing 24–34 % PEG-400 and 5% glycerol, soaked for 1–2 hours and flash-frozen in liquid nitrogen.
The data sets were collected on the LS-CAT beam-line at the Argonne Photon Source. The data was processed and merged using the HKL2000 software suite and indexed in space group C2 48. Molecular replacement, using the Phaser program 49 implemented within the CCP4 software suite 50, was conducted using the 1R7M crystal structure with solvent and metals removed as a search model. Iterative manual rebuilding using Coot51, the CCP4 suite (Refmac552 and ARP/wARP53 programs) and Phenix54 were used to refine the structures at 2.3Å and 2.5Å resolutions.
Protein Data Bank accession numbers
Coordinates and structure factors have been deposited in the Protein Data bank with accession numbers 3OOL for the wild-type-C/G+4 structure and 3OOR for the K86R/G100T-C/G+4 structure.
Supplementary Material
Supplemental Figure 1: The composite omit map (1σ; blue) of the WT-C/G+4 structure depicting two amino positions, Lys86 and Gly100 (colored cyan and by element), and the substituted C/G+4 base-pair (colored magenta and green and by element) represented as sticks.
Supplemental Figure 2: The composite omit map (1σ; blue) of the K86R/G100T-C/G+4 (1σ; blue) depicting the two substitutions at Arg86 and Thr100 (colored cyan and by element) and the substituted base-pair C/G+4 (colored magenta and green and by element) represented as sticks.
Acknowledgments
We thank Weijia Luo and Jennifer L. Meyers for technical assistance, the personnel at the LS-CAT beamline (Advanced Photon Source) for assistance in data collection, Dr. Long Li for assistance with data collection and discussions on refinement and Dr. T.J. Kappock for helpful discussions on the project. This work was supported by grants from the National Institutes of Health (GM 070553) and the National Science Foundation (MCB-0321550) to F.S.G. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (Grant 085P1000817).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gimble FS. Invasion of a multitude of genetic niches by homing endonuclease genes. FEMS Microbiol Letters. 2000;185:99–107. doi: 10.1111/j.1574-6968.2000.tb09046.x. [DOI] [PubMed] [Google Scholar]
- 2.Stoddard BL. Homing endonuclease structure and function. Q Rev Biophys. 2005;38:49–95. doi: 10.1017/S0033583505004063. [DOI] [PubMed] [Google Scholar]
- 3.Goddard MR, Burt A. Recurrent invasion and extinction of a selfish gene. Proc Natl Acad Sci U S A. 1999;96:13880–13885. doi: 10.1073/pnas.96.24.13880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haugen P, Wikmark OG, Vader A, Coucheron DH, Sjottem E, Johansen SD. The recent transfer of a homing endonuclease gene. Nucleic Acids Re s. 2005;33:2734–41. doi: 10.1093/nar/gki564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gogarten JP, Hilario E. Inteins, introns, and homing endonucleases: recent revelations about the life cycle of parasitic genetic elements. BMC Evol Bio l. 2006;6:94. doi: 10.1186/1471-2148-6-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gimble FS. Degeneration of a homing endonuclease and its target sequence in a wild yeast strain. Nucleic Acids Res. 2001;29:4215–4223. doi: 10.1093/nar/29.20.4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yahara K, Fukuyo M, Sasaki A, Kobayashi I. Evolutionary maintenance of selfish homing endonuclease genes in the absence of horizontal transfer. Proc Natl Acad Sci U S A. 2009;106:18861–6. doi: 10.1073/pnas.0908404106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chevalier B, Turmel M, Lemieux C, Monnat RJ, Stoddard BL. Flexible DNA target site recognition by divergent homing endonuclease isoschizomers I-CreI and I-MsoI. J Mol Biol. 2003;329:253–269. doi: 10.1016/s0022-2836(03)00447-9. [DOI] [PubMed] [Google Scholar]
- 9.Jurica MS, Monnat RJ, Jr, Stoddard BL. DNA recognition and cleavage by the LAGLIDADG homing endonuclease I-CreI. Mol Cell. 1998;2:469–476. doi: 10.1016/s1097-2765(00)80146-x. [DOI] [PubMed] [Google Scholar]
- 10.Moure CM, Gimble FS, Quiocho FA. Crystal structure of the intein homing endonuclease PI-SceI bound to its recognition sequence. Nat Struct Biol. 2002;9:764–770. doi: 10.1038/nsb840. [DOI] [PubMed] [Google Scholar]
- 11.Moure CM, Gimble FS, Quiocho FA. The crystal structure of the gene targeting homing endonuclease I-SceI reveals the origins of its target site specificity. J Mol Biol. 2003;334:685–95. doi: 10.1016/j.jmb.2003.09.068. [DOI] [PubMed] [Google Scholar]
- 12.Spiegel PC, Chevalier B, Sussman D, Turmel M, Lemieux C, Stoddard BL. The structure of I-CeuI homing endonuclease: evolving asymmetric DNA recognition from a symmetric protein scaffold. Structure. 2006;14:869–880. doi: 10.1016/j.str.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 13.Moure CM, Gimble FS, Quiocho FA. Crystal structures of I-SceI complexed to nicked DNA substrates: snapshots of intermediates along the DNA cleavage reaction pathway. Nucleic Acids Res. 2008;36:3287–3296. doi: 10.1093/nar/gkn178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nomura N, Nomura Y, Sussman D, Klein D, Stoddard BL. Recognition of a common rDNA target site in archaea and eukarya by analogous LAGLIDADG and His-Cys box homing endonucleases. Nucleic Acids Res. 2008;36:6988–98. doi: 10.1093/nar/gkn846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaiser BK, Clifton MC, Shen BW, Stoddard BL. The structure of a bacterial DUF199/WhiA protein: domestication of an invasive endonuclease. Structure. 2009;17:1368–76. doi: 10.1016/j.str.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paques F, Duchateau P. Meganucleases and DNA double-strand break-induced recombination: perspectives for gene therapy. Curr Gene Ther. 2007;7:49–66. doi: 10.2174/156652307779940216. [DOI] [PubMed] [Google Scholar]
- 17.Seligman LM, Chisholm KM, Chevalier BS, Chadsey MS, Edwards ST, Savage JH, Veillet AL. Mutations altering the cleavage specificity of a homing endonuclease. Nucleic Acids Res. 2002;30:3870–3879. doi: 10.1093/nar/gkf495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Epinat JC, Arnould S, Chames P, Rochaix P, Desfontaines D, Puzin C, Patin A, Zanghellini A, Paques F, Lacroix E. A novel engineered meganuclease induces homologous recombination in yeast and mammalian cells. Nucleic Acids Res. 2003;31:2952–2962. doi: 10.1093/nar/gkg375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chames P, Epinat JC, Guillier S, Patin A, Lacroix E, Paques F. In vivo selection of engineered homing endonucleases using double-strand break induced homologous recombination. Nucleic Acids Res. 2005;33:e178. doi: 10.1093/nar/gni175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Z, Zhao H. A highly sensitive selection method for directed evolution of homing endonucleases. Nucleic Acids Res. 2005;33:e154. doi: 10.1093/nar/gni148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arnould S, Chames P, Perez C, Lacroix E, Duclert A, Epinat JC, Stricher F, Petit AS, Patin A, Guillier S, Rolland S, Prieto J, Blanco FJ, Bravo J, Montoya G, Serrano L, Duchateau P, Paques F. Engineering of large numbers of highly specific homing endonucleases that induce recombination on novel DNA targets. J Mol Biol. 2006;355:443–58. doi: 10.1016/j.jmb.2005.10.065. [DOI] [PubMed] [Google Scholar]
- 22.Doyon JB, Pattanayak V, Meyer CB, Liu DR. Directed evolution and substrate specificity profile of homing endonuclease I-SceI. J Am Chem Soc. 2006;128:2477–84. doi: 10.1021/ja057519l. [DOI] [PubMed] [Google Scholar]
- 23.Rosen LE, Morrison HA, Masri S, Brown MJ, Springstubb B, Sussman D, Stoddard BL, Seligman LM. Homing endonuclease I-CreI derivatives with novel DNA target specificities. Nucleic Acids Res. 2006;34:4791–800. doi: 10.1093/nar/gkl645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Z, Wen F, Sun N, Zhao H. Directed evolution of homing endonuclease I-SceI with altered sequence specificity. Protein Eng Des Sel. 2009;22:249–256. doi: 10.1093/protein/gzp001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jarjour J, West-Foyle H, Certo MT, Hubert CG, Doyle L, Getz MM, Stoddard BL, Scharenberg AM. High-resolution profiling of homing endonuclease binding and catalytic specificity using yeast surface display. Nucleic Acids Res. 2009;37:6871–80. doi: 10.1093/nar/gkp726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ashworth J, Havranek JJ, Duarte CM, Sussman D, Monnat RJ, Jr, Stoddard BL, Baker D. Computational redesign of endonuclease DNA binding and cleavage specificity. Nature. 2006;441:656–9. doi: 10.1038/nature04818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ashworth J, Taylor GK, Havranek JJ, Quadri SA, Stoddard BL, Baker D. Computational reprogramming of homing endonuclease specificity at multiple adjacent base pairs. Nucleic Acids Res. 2010 doi: 10.1093/nar/gkq283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gimble FS, Moure CM, Posey KL. Assessing the plasticity of DNA target site recognition of the PI-SceI homing endonuclease using a bacterial two-hybrid selection system. J Mol Biol. 2003;334:993–1008. doi: 10.1016/j.jmb.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 29.Gimble FS, Wang J. Substrate recognition and induced DNA distortion by the PI-SceI endonuclease, an enzyme generated by protein splicing. J Mol Biol. 1996;263:163–180. doi: 10.1006/jmbi.1996.0567. [DOI] [PubMed] [Google Scholar]
- 30.Grindl W, Wende W, Pingoud V, Pingoud A. The protein splicing domain of the homing endonuclease PI-SceI is responsible for specific DNA binding. Nucleic Acids Res. 1998;26:1857–1862. doi: 10.1093/nar/26.8.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Colleaux L, D’Auriol L, Galibert F, Dujon B. Recognition and cleavage site of the intron-encoded omega transposase. Proc Natl Acad Sci USA. 1988;85:6022–6026. doi: 10.1073/pnas.85.16.6022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Joung JK, Ramm EI, Pabo CO. A bacterial two-hybrid selection system for studying protein-DNA and protein-protein interactions. Proc Natl Acad Sci U S A. 2000;97:7382–7387. doi: 10.1073/pnas.110149297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niu Y, Tenney K, Li H, Gimble FS. Engineering variants of the I-SceI homing endonuclease with strand-specific and site-specific DNA-nicking activity. J Mol Biol. 2008;382:188–202. doi: 10.1016/j.jmb.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lucas P, Otis C, Mercier JP, Turmel M, Lemieux C. Rapid evolution of the DNA binding site in LAGLIDADG homing endonucleases. Nucleic Acids Res. 2001;29:960–969. doi: 10.1093/nar/29.4.960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takeuchi R, Certo M, Caprara MG, Scharenberg AM, Stoddard BL. Optimization of in vivo activity of a bifunctional homing endonuclease and maturase reverses evolutionary degradation. Nucleic Acids Res. 2009;37:877–90. doi: 10.1093/nar/gkn1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beylot B, Spassky A. Chemical probing shows that the intron-encoded endonuclease I-SceI distorts DNA through binding in monomeric form to its homing site. J Biol Chem. 2001;276:25243–53. doi: 10.1074/jbc.M101200200. [DOI] [PubMed] [Google Scholar]
- 37.Galetto R, Duchateau P, Paques F. Targeted approaches for gene therapy and the emergence of engineered meganucleases. Expert Opin Biol Ther. 2009;9:1289–303. doi: 10.1517/14712590903213669. [DOI] [PubMed] [Google Scholar]
- 38.Sussman D, Chadsey M, Fauce S, Engel A, Bruett A, Monnat R, Jr, Stoddard BL, Seligman LM. Isolation and characterization of new homing endonuclease specificities at individual target site positions. J Mol Biol. 2004;342:31–41. doi: 10.1016/j.jmb.2004.07.031. [DOI] [PubMed] [Google Scholar]
- 39.Smith J, Grizot S, Arnould S, Duclert A, Epinat JC, Chames P, Prieto J, Redondo P, Blanco FJ, Bravo J, Montoya G, Paques F, Duchateau P. A combinatorial approach to create artificial homing endonucleases cleaving chosen sequences. Nucleic Acids Res. 2006;34:e149. doi: 10.1093/nar/gkl720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thyme SB, Jarjour J, Takeuchi R, Havranek JJ, Ashworth J, Scharenberg AM, Stoddard BL, Baker D. Exploitation of binding energy for catalysis and design. Nature. 2009;461:1300–4. doi: 10.1038/nature08508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eklund JL, Ulge UY, Eastberg J, Monnat RJ., Jr Altered target site specificity variants of the I-PpoI His-Cys box homing endonuclease. Nucleic Acids Res. 2007;35:5839–50. doi: 10.1093/nar/gkm624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chevalier BS, Stoddard BL. Homing endonucleases: structural and functional insight into the catalysts of intron/intein mobility. Nucleic Acids Res. 2001;29:3757–3774. doi: 10.1093/nar/29.18.3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koufopanou V, Goddard MR, Burt A. Adaptation for horizontal transfer in a homing endonuclease. Mol Biol Evol. 2002;19:239–246. doi: 10.1093/oxfordjournals.molbev.a004077. [DOI] [PubMed] [Google Scholar]
- 44.Scalley-Kim M, McConnell-Smith A, Stoddard BL. Coevolution of a homing endonuclease and its host target sequence. J Mol Biol. 2007;372:1305–19. doi: 10.1016/j.jmb.2007.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He Z, Crist M, Yen H-C, Duan X, Quiocho FA, Gimble FS. Amino acid residues in both the protein splicing and endonuclease domains of the PI-SceI intein mediate DNA binding. J Biol Chem. 1998;273:4607–4615. doi: 10.1074/jbc.273.8.4607. [DOI] [PubMed] [Google Scholar]
- 46.Serebriiskii IG, Fang R, Latypova E, Hopkins R, Vinson C, Joung JK, Golemis EA. A combined yeast/bacteria two-hybrid system: development and evaluation. Mol Cell Proteomics. 2005;4:819–26. doi: 10.1074/mcp.T500005-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maniatis T, Fritsch EF, Sambrook J. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, N.Y: 1982. [Google Scholar]
- 48.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 49.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Project CC. The CCP4 suite: programs for protein crystallography. Acta Crystallogr A. 1994;4:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 51.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr D Biol Crystallogr. 2004;60:2184–95. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- 53.Cohen SX, Ben Jelloul M, Long F, Vagin A, Knipscheer P, Lebbink J, Sixma TK, Lamzin VS, Murshudov GN, Perrakis A. ARP/wARP and molecular replacement: the next generation. Acta Crystallogr D Biol Crystallogr. 2008;64:49–60. doi: 10.1107/S0907444907047580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: The composite omit map (1σ; blue) of the WT-C/G+4 structure depicting two amino positions, Lys86 and Gly100 (colored cyan and by element), and the substituted C/G+4 base-pair (colored magenta and green and by element) represented as sticks.
Supplemental Figure 2: The composite omit map (1σ; blue) of the K86R/G100T-C/G+4 (1σ; blue) depicting the two substitutions at Arg86 and Thr100 (colored cyan and by element) and the substituted base-pair C/G+4 (colored magenta and green and by element) represented as sticks.