

Abstract
Objectives
To correlate the genotype of von Hippel-Lindau (VHL) mutations and the clinical characteristics of associated retinal hemangioblastomas (RCHs).
Design
Cross-sectional and molecular genetic study.
Patients and Methods
Of 890 VHL patients (327 pedigrees), 335 had ocular involvement in the form of RCHs. Statistical analysis was employed to correlate the structure of the mutated VHL protein to ocular phenotype.
Results
Three genotype categories, amino-acid substitutions, protein-truncating mutations, and complete deletion of the VHL protein, were defined in all patients. Prevalence of RCHs was lowest in complete deletions (14.5%, overall prevalence = 37.1%). Genotypic category had no effect on the unilaterality or bilaterality of ocular disease, or number and extent of peripheral RCHs. The prevalence of RCH at the juxtapapillary location was lower in protein truncations compared to amino-acid substitutions. Complete deletions had the highest mean visual acuity (84.7 letters) compared to amino-acid substitutions (54.9 letters) or protein truncating mutations (51.7 letters).
Conclusions
Patients with complete deletion of the VHL protein have the lowest prevalence of ocular disease and the most favorable visual outcome.
Clinical relevance
VHL mutation genotype may be used to predict the prevalence and outcome of ocular VHL disease and to guide ophthalmic follow-up.
INTRODUCTION
Von Hippel-Lindau disease (Online Mendelian Inheritance in Man [OMIM] 193300) is a relatively uncommon (1 in 36,000 live births), multi-system, dominantly-inherited cancer syndrome that predisposes affected persons to the development of benign and malignant tumors in the retina and central nervous system, kidney, pancreas, adrenal gland, epididymis and broad ligament, and endolymphatic sac 1. In the retina, VHL disease may result in the formation of benign, solitary or multiple retinal capillary hemangioblastomas (RCHs) that have a globular, reddish-pink appearance, with prominent, dilated feeding and draining vessels 2. Often slow-growing, RCHs in VHL, through their exudative and/or tractional secondary effects, can result in vision loss, structural disruption of the retina and the globe, and loss of the eye 3. Treatment, especially at an early stage for a small RCH, may limit vision loss 4,5. Because RCHs may progress or develop de novo over time, annual ophthalmic evaluation is advised for all patients with VHL disease.
VHL disease results from dominantly inherited, germline mutations in a ubiquitously-expressed tumor-suppressor gene located on chromosome 3p26 6. Patients mostly inherit one mutated copy from an affected parent from the germline and then subsequently develop a second mutation somatically in susceptible tissues. Cells having both a germline and a somatic mutation then become tumorous according to Knudson’s two-hit hypothesis 7. The disease is highly penetrant and almost all patients having one germline VHL mutation develop disease characteristics in the course of a lifetime 8.
With the identification of the VHL gene in 1993, germline mutations can now be identified almost in all VHL pedigrees 9. Encoding a novel protein, the mechanism by which mutations in VHL gene results in the range of phenotypes seen in VHL disease is still incompletely understood 10. There is much variability in the nature of documented mutations (ranging from single amino-acid substitutions to the complete deletion of the gene), as well as in the manifestations of the disease phenotype 11. Genotype-phenotype correlations relating the nature of mutations to the tissue distribution of disease have been performed and have generated some hypotheses about how the VHL protein function may function 12,13. However, large-scale correlation studies focusing on ocular manifestations remain few in number 14,15.
Using the largest population of patients collected to-date, we aim 1) to characterize the germline mutations found in this large population, 2) to measure the prevalence of RCHs in VHL disease generally and specifically for each genotype category, 3) to establish genotype-phenotype correlations between genotypic class and the phenotypic features of ocular VHL disease, and 4) to establish genotype-phenotype correlations between genotypic class and visual function. These findings may help us better understand how VHL mutations influence the occurrence and progression of RCHs in the eye and assist clinicians in employing genotypic information for the prognosis and monitoring of ocular disease in patients affected with VHL.
PATIENTS AND METHODS
Patient Selection and Ascertainment of VHL disease
All patients referred to the study were enrolled into a study protocol at the National Cancer Institute (NCI, National Institute of Health, Bethesda, Maryland) from 1988 to 2005. Patients were systemically evaluated at this single center with history and physical examination, laboratory evaluation, and radiographical (CT or MRI) studies of the abdomen, pelvis, brain, and spine. Patients were also evaluated with a complete ophthalmological evaluation with slit lamp examination, indirect fundoscopy, and, when clinically indicated, fluorescein angiography.
A total of 890 patients were found to meet clinical diagnostic criteria of VHL 16 based on genetic testing, physical exam, imaging studies, or pathology and were admitted to the study. Of these, 335 patients were confirmed to have a history or present evidence of retinal angiomatosis in at least one eye.
Patients were followed longitudinally, and treatments, if necessary, were delivered. Owing to the cross-sectional nature of this study, each patient’s most recent visit was designated as the study visit.
Genotype analysis
Analysis for mutations in the VHL gene was carried out on peripheral blood samples from at least one member of each kindred as previously described 9. Mutations were classified into one of the following groups: missense, partial deletions, complete deletions, splice mutations, amino-acid deletions (microdeletions in multiples of 3), and nonsense mutations. These were further classified into 3 main genotypic categories according to their expected effect on protein structure: missense mutations were classified as single “amino-acid substitutions”; partial deletions, frameshift, and nonsense mutations as “protein-truncating mutations”; and complete deletions of the coding sequence were designated as “complete deletions”. Amino-acid mutations and splice mutations were not assigned to any sub-group as their effect on the structure of the VHL protein is uncertain.
Assessment of Retinal Phenotype
Patients with retinal angiomatosis were interviewed on whether they have had a previous history of ocular involvement from VHL, date of onset of eye findings, and prior therapy. Ocular examination included a best-corrected visual acuity, intraocular pressure measurements, slit-lamp examination, and indirect ophthalmoscopic fundoscopy. Both eyes in each patient were accounted for, including normal eyes with no VHL ocular lesions,, eyes that have been enucleated due to complications of ocular angiomatosis, and phthisical or pre-phthisical eyes for which a view of the posterior pole was often unavailable. For eyes that can be evaluated by funduscopy, the location of RCHs were noted (as juxtapapillary, macular, peripheral), the number of RCHs was counted, and the extent of retinal involvement recorded (as extending to greater or less than 1 quadrant of the retina). The location of tumors was described as either “optic nerve tumors” if they were located on or touching the optic nerve, “macular” if they were located within the vascular arcades, and “peripheral” if they were located peripheral to the arcades and twice the fovea-to-disk distance from the optic nerve.
Statistical analysis
Age-adjusted logistic regression and analysis of variance was used to assess the relationship of the germline mutations and age, visual function, and the clinical phenotypes of the patients included in this analysis. Because outcomes occurred in familial clusters of varying sizes, multiple outputation 17 was applied for all comparisons to account for intra-family correlation as well as for size of family. For the outputation technique it was determined that 2000 resamplings within family were optimal for drawing inferences from these data.
Analysis of variance was performed to evaluate the differences in age and the differences in mean visual acuity among the three genotypic mutation classes. The clinical phenotypes analyzed using logistic regression were: presence or absence of RCH, laterality of angiomatosis, presence of severe ocular involvement, location of retinal angioma, number of peripheral RCH, and extent of peripheral involvement. Given that the mean age was found to be significantly different among the three mutation classes, all analyses were adjusted for age.
Prevalence rates were calculated by dividing the number of patients with a phenotype by the total number of patients.
RESULTS
Genotype analysis
Germline mutations in the VHL gene can be characterized in 873 (98.1%) out of all 890 patients (with and without retinal angiomatosis) included in the study. In the remaining cases, germline mutations in the VHL were not found. The distributions of patients in each mutation category according to DNA mutation and each category according to effect on protein structure are shown in Table 1.
Table 1.
Genotype analysis of all patients with clinically definite VHL disease (with and without ocular angiomatosis).
| Germline mutation according to effect on DNA sequence | Number of patients (% of total) | Mutational category according to effect on protein structure (genotypic mutation category) | Number of patients (% of total) | Number of pedigrees (% of total) |
|---|---|---|---|---|
| Missense | 418 (47.0) | Amino-acid substitution | 418 (47.0) | 138 (42.2) |
| Partial deletions | 218 (24.5) | Protein truncation | 354 (39.8) | 128 (39.1) |
| Nonsense | 70 (7.9) | |||
| Frameshift | 66 (7.4) | |||
| Complete deletions | 62 (7.0) | Complete deletion of protein | 62 (7.0) | 23 (7.0) |
| Splice mutations | 21 (2.4) | Undefined effect | 38 (4.3) | 22 (6.7) |
| Amino acid deletions | 17 (1.9) | |||
| Unknown | 18 (2.0) | Unknown | 18 (2.0) | 16 (4.9) |
| Total | 890 (100) | Total | 890 (100) | 327 (100) |
Mutations were grouped according to their predicted effect on the structure of the VHL protein. All missense mutations were expected to result in a single amino acid substitution. Partial deletions, nonsense mutations, and frameshift mutations were grouped together as resulting in protein truncating mutations. Complete deletion of the coding region of the VHL gene was expected to result in a complete deletion of the protein product. Splice mutations, microdeletions in multiples of 3 bases, and undetected mutations had an undefined predicted effect on protein structure.
Of the entire group of patients, 834 (93.7%) patients have mutations that have known effects on the VHL protein and can be placed in one of the 3 genotypic mutation categories of amino-acid substitution, protein truncation, or complete deletion. In the remainder of the study, genotype-phenotype correlations will be performed between these 3 genotypic mutation categories and the patients’ demographic, ocular, and visual function phenotypes.
Genotypic class and the prevalence of retinal angiomatosis
Table 2 provides a breakdown of all 834 patients in the 3 major genotypic classes according to the presence and absence of retinal angiomatosis. Approximately one third of patients (310 out of 834 patients, 37.1%) in these categories have a history or clinical evidence of retinal angiomatosis in at least one eye. This compares quite similarly to the overall prevalence of retinal angiomatosis (37.6%; 335 out of 890) when all patients in the study regardless of genotype are considered.
Table 2.
Prevalence of Retinal Angiomatosis by VHL Mutational Class
| Type of Mutational Class | Presence of Retinal Angiomatosis (in at least one eye) | |||
|---|---|---|---|---|
| Yes | No | Total | Prevalence of Retinal Angiomatosis* | |
| Amino-acid Substitution | 159 | 259 | 418 | 38.0% |
| Protein Truncation | 142 | 212 | 354 | 40.1% |
| Complete Deletion | 9 | 53 | 62 | 14.5%** |
| Total | 310 | 524 | 834 | 37.1% |
| Comparison | Odds ratio (95% confidence limits) for rate of RCH present vs. RCH absent ** | P value |
|---|---|---|
| Amino-acid substitution vs. Protein Truncation | 1.01(0.67-1.54) | 0.9453 |
| Amino-acid substitution vs. Complete deletion | 5.85 (2.51-13.65) | <.0001 |
| Protein Truncation vs. Complete deletion | 5.77 (2.45-13.59) | <.0001 |
Calculated as number of patients with retinal angiomatosis divided by total number of patients in each subset.
Analyses were adjusted for age, gender, and familial relationships.
We examined the question of whether the prevalence of retinal angiomatosis was influenced by mutational class. Results shown in Table 2 and Figure 1 demonstrate that the prevalence of RCHs is significantly lower in mutations resulting in a complete deletion of the VHL protein compared to mutations resulting in amino-acid substitutions or protein truncations. The odds ratio of having RCHs versus not having RCHs was about 5 to 6 times greater for amino-acid substitutions and protein truncations compared to complete deletions, indicating a clear influence of mutational genotype on RCH prevalence.
Figure 1.
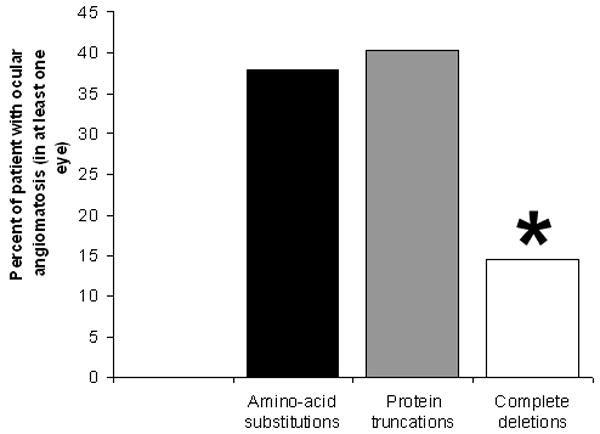
Prevalences of retinal angiomatosis in VHL patients in 3 genotypic mutational categories of single amino-acid substitutions, protein-truncating mutations, and complete deletion of VHL protein. Asterisk (*) indicates that the prevalence for complete deletions were significantly lower than those for amino-acid substitutions and protein truncations (p<0.0001).
Genotypic class and demographics of patients with ocular angiomatosis
The demographic features (age, gender, race) of patients in the 3 genotypic mutation classes (n = 310) are listed in Table 3. We examined the possible correlation between genotypic class and the demographical categories of age, gender, and race in this group of patients. For gender, there was no evidence to reject the null hypothesis that all genotypes had a similar prevalence (p > 0.50 for all pairwise comparisons). For age, the cross-sectional ages (with 95% confidence limits) for the 3 major genotype classes are shown in Figure 2. The difference in the mean ages of patients with amino-acid mutations (39.2 years) and protein truncations (34.6 years) was found to be statistically significant (p = 0.0026). The mean age of complete deletions (38.3 years) was not significantly different from those in the other categories (p= 0.34-0.85). Because the racial distribution of the patient population was predominantly white (91.6%, Table 3), a statistical analysis on the basis of racial category was not performed.
Table 3.
Demographics of 310 patients with ocular angiomatosis with documented mutations in the 3 major genotypic classes (amino-acid substitutions, protein truncations, and complete deletions).
| Age | Mean age ± standard deviation = 37.1 ± 13.9 years |
| Range = 8.6–84.3 years | |
| Gender | Males = 142; Females = 168; M : F ratio = 1 : 1.18 |
| Race (self-described) | White = 284; Hispanic = 15; Asian = 6; Black = 5 |
Figure 2.
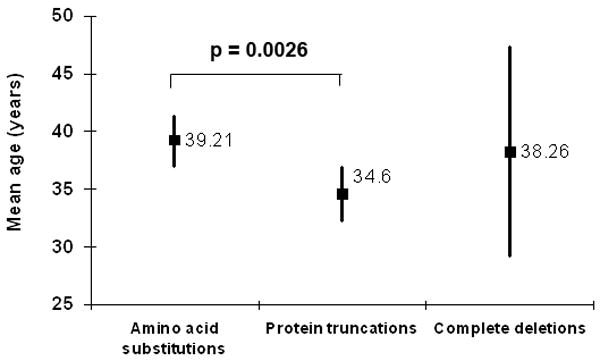
Mean ages of VHL patients with ocular angiomatosis according to genotypic categories (n = 310 patients). Square symbols and adjacent notation indicate mean age in each category. Vertical error bars indicate 95% confidence limits. Mean age for amino acid substitutions is significantly higher than for protein truncations (asterisk *).
Genotypic class and ocular phenotype in eyes with retinal angiomatosis
The ocular phenotype of the same 310 patients in the 3 major genotypic classes were documented and categorized according to separate phenotypic features as shown in Table 4. These phenotypic features illustrate the nature, extent, and severity of retinal angiomatosis in affected eyes.
Table 4.
Ocular phenotypes of VHL patients with ocular angiomatosis in the 3 genotypic categories
| Phenotypic category | Number of patients in each phenotypic subcategory (percentage in parentheses) | Definitions for each phenotypic category |
|---|---|---|
| Laterality of angiomatosis | With only 1 eye involved = 127 (41.0) | History or evidence of retinal hemangioblastomas |
| With both eyes involved = 183 (59.0) | ||
| Presence of severe ocular involvement | Without severe ocular involvement in either eye = 246 (79.3) | Eyes that have been enucleated or structurally disrupted due to complications from ocular angiomatosis such that a fundus view was not possible were judged as having “severe ocular involvement” |
| At least one eye with severe ocular involvement = 64 (20.7) | ||
| Location of retinal hemangioblastomas | Without optic nerve involvement in either eye = 249 (80.3) | “Optic nerve involvement” was judged to be present when 1 or more retinal hemangioblastomas were located in the juxtapapillary region (located either directly on the optic nerve or less than one disc diameter from the optic nerve). |
| At least one eye with documented optic nerve involvement = 61 (19.7) | ||
| Number of peripheral retinal hemangioblastomas (RCH) | Fewer than 3 RCHs in either eye = 153 (56.9) | For eyes with RCHs located in the periphery, the individual RCHs were counted. Peripheral RCHs referred to those located beyond the vascular arcades and twice the foveal disk distance from the optic nerve. |
| At least 3 RCHs in either eye = 116 (43.1) | ||
| At least 5 RCHs in either eye = 60 (22.3) | ||
| Extent of peripheral retina involvement | < 1 quadrant of peripheral retinal involvement in either eye = 185 (68.8) | For eyes with RCHs located in the periphery, the extent of peripheral retina involved by RCHs was estimated. |
| > 1 quadrant of peripheral retinal involvement in either eye = 84 (31.2) | ||
Approximately 6 out of 10 patients in this subset had ocular angiomatosis in both eyes; the remainder had only 1 eye affected. The prevalences of bilateral involvement in the 3 genotypic classes, amino-acid substitutions, protein truncations, and complete deletions, were similar (Table 5) and the odds of having bilateral involvement vs. unilateral involvement did not differ statistically from each other (p > 0.60 for all pairwise comparisons still correct). Therefore, genotypic class appeared not to influence whether one or both eyes were involved with angiomatosis.
Table 5.
Ocular phenotype of patients with ocular angiomatosis in each of 3 genotypic mutation classes.
| Phenotypic category | Genotype mutation class |
||||
|---|---|---|---|---|---|
| Amino-Acid Substitution | Protein Truncation | Complete Deletion | Total patients | ||
| Laterality of angiomatosis | With only 1 eye involved | 64 (40.3) | 59 (41.6) | 4 (44.4) | 310 |
| With both eyes involved | 95 (59.8) | 83 (58.5) | 5 (55.6) | ||
| Presence of severe ocular involvement | No severe ocular involvement in either eye | 128(80.5) | 109 (76.8) | 9(100) | 310 |
| At least one eye with severe ocular involvement | 31(19.5) | 33 (23.2) | 0(0) | ||
| Location of retinal angioma | No optic nerve involvement in either eye | 120 (75.5) | 122 (85.9) | 7(77.8) | 310 |
| At least one eye with documented optic nerve involvement | 39 (24.5) | 20 (14.1) | 2 (22.2) | ||
| Number of peripheral retinal hemangioblastomas (RCH) | Fewer than 3 RCHs in either eye | 73 (54.1) | 77 (60.6) | 3 (42.9) | 269 |
| At least 3 RCHs in either eye | 62 (45.9) | 50 (39.4) | 4 (57.1) | ||
| Fewer than 5 RCHs in either eye | 103 (76.3) | 99 (78.0) | 7 (100) | 269 | |
| At least 5 RCHs in either eye | 32 (23.7) | 28 (22.0) | 0 (0) | ||
| Extent of peripheral retina involvement | < 1 quadrant of peripheral retinal involvement in either eye | 92 (68.2) | 88 (69.3) | 5 (71.4) | 269 |
| > 1 quadrant of peripheral retinal involvement in either eye | 43 (31.8) | 39 (30.7) | 2 (28.6) | ||
Both eyes in each patient were examined and accounted for. A number of eyes had been enucleated secondary to complications of severe ocular angiomatosis. A further number had undergone structural disruption in the form of total retinal detachment, massive subretinal exudation, and phthisical changes that prevented visualization of the posterior pole. These eyes were collectively termed as having “severe ocular involvement” to distinguish them from eyes in which the posterior pole was intact and individual RCHs visualized and counted. These eyes had poor (lower than 20/160) or no vision. About 1 in 5 patients had one or both eyes that were severely affected. Genotype-correlation analysis did not produce evidence to reject the null hypothesis that all genotypes have a similar rate of “severe involvement” in at least one eye (p = 0.60 for amino-acid substitution vs. protein truncation). However, it should be noted that for complete deletions, none of the 9 patients in this genotypic category had severely affected eyes (Table 5). Owing to the small number of patients in this genotypic class, the statistical significance of this rate cannot be established.
Retinal hemangioblastomas were found to be located in two fundus locations; in the juxtapapillary area (termed “optic nerve involvement”) and in the peripheral retina. The predilection of RCHs to occur in these locations and avoid the macular retina is unknown. The location of RCHs is of clinical and functional significance, as unlike peripheral RCHs, juxtapapillary RCHs often cannot be safely treated using commonly available treatment modalities of laser photocoagulation and cryotherapy 18. The growth and exudative behavior of juxtapapillary lesions, left unchecked, may in some cases lead to vision loss and structural disruption to the posterior pole more frequently than do peripheral lesions. Approximately 1 in 5 patients having at least one eye affected by retinal angiomatosis have a juxtapapillary tumor in at least 1 eye. On the level of the individual eye, about 1 in 6 eyes affected by ocular angiomatosis have a juxtapapillary tumor (Wong et al., unpublished data). We examined the influence of genotypic class on the location of RCHs. The rate of having a juxtapapillary tumor was lowest in protein truncations (14.1%) and this was statistically lower than the rate in amino-acid substitutions (24.5%, p = 0.0070), but not distinct from the rate in complete deletions (22.2%, p = 0.19) (Figure 3).
Figure 3.
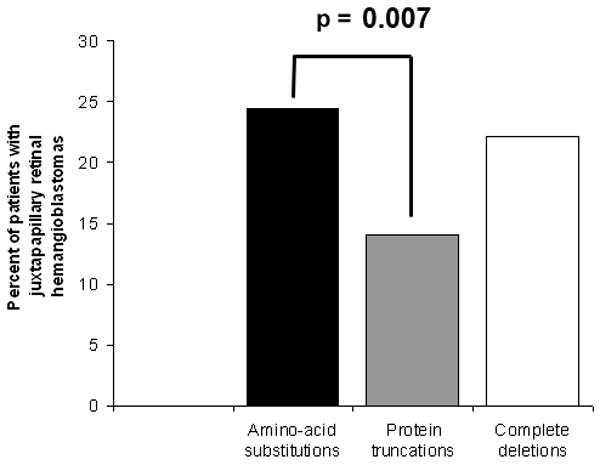
Prevalence of retinal angiomatosis at a juxtapapillary location (in at least one eye) in each of the 3 genotypic categories. The prevalence of juxtapapillary RCHs is significantly lower for protein truncations than for amino-acid substitutions.
Retinal hemangioblastomas located in the peripheral retina were found in a majority of patients in at least one eye (n = 269). In these eyes, individual hemangioblastomas could be counted and the extent of peripheral retinal involvement scored. We examined if genotypic class influenced the number of peripheral RCHs or the extent of peripheral involvement in this group of patients. We correlated genotypic class to the proportion of patients having at least 3 or at least 5 RCHs in either eye (Table 5). There were no statistically significant differences in the genotypes in regard to the number or extent of peripheral lesions (p>0.30 in all comparisons).
Genotypic class and visual acuity
Visual acuities in letters on the ETDRS chart in both eyes were separately evaluated for patients in the 3 genotypic categories (eyes that have been enucleated were given a score of 0). The mean visual acuities in the better- and worse-seeing eyes of all 310 patients were correlated separately with genotype category (Figure 4) and adjusted for age and familial correlation. In the better-seeing eye, the mean acuities in all 3 genotypic categories were relatively similar and did not vary statistically from each other (p>0.1 in all comparisons). In the worse-seeing eye, patients with complete deletions had a statistically higher visual acuity (84.7 letters) than patients with amino-acid substitutions (54.9 letters, p = 0.01) or deletions (51.7 letters, p = 0.01).
Figure 4.
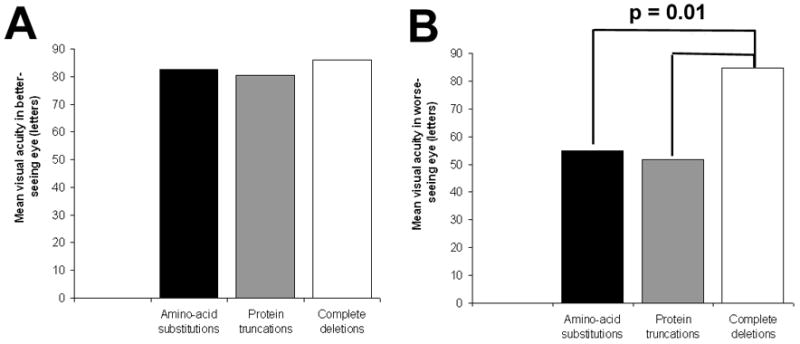
Mean visual acuities in each of 3 genotypic categories. A. Mean visual acuity in the better-seeing eye. There were no detectable significant differences between the means (p > 0.10 for all 3 pair-wise comparisons). B. Mean visual acuity in the worse-seeing eye. Mean visual acuity for complete deletions were significantly higher than those for amino-acid substitutions and protein truncations (p = 0.01).
COMMENT
The findings from the present study draw from the largest group of patients thus far assembled in the literature. These patients have been referred to the study as a result of systemic findings of VHL disease rather than ocular findings or complaints. They have also evaluated systemically in a single center and germline genotype information had been obtained in almost all patients. The nature of ascertainment of the patients, the systemic clinical diagnosis of VHL, and the comprehensive genotype analysis of patients provide a comprehensive evaluation of ocular angiomatosis in VHL disease.
The prevalence of retinal angiomatosis in our study population was 37.1%, slightly lower than the other previously reported prevalences of 49 to 68% for ocular involvement 14,19,20. Our study focuses only on hemangioblastomas in the retina and excludes tumors found in the retrobulbar optic nerve and also non-angiomatous retinal lesions that appear as retinal neovascularization (Chew, EY. unpublished data). Our method of ascertainment also does not draw primarily from patients referred from eye centers with ocular findings or complaints but from patients with systemic VHL disease. As such, the lower measured prevalence in this study may reflect more accurately on the occurrence of retinal hemangioblastomas in all VHL patients as a whole.
We have chosen to analyze genotype mutations in the VHL gene according to their effect on protein structure. The 3 genotypic classes of amino-acid mutations, protein-truncating mutations, and complete deletions provide one comparison for how ocular phenotype may differ when only a single residue on the protein is altered to when only part or none of the protein is present. These genotypic classes have also been previously employed for genotype-phenotype correlations for ocular disease 14,15 and for other systemic associations 21. Because members in the same pedigree share not only the same genotype class but also many other genes that may affect ocular phenotype, our statistical analyses of genotype-phenotype correlations takes into account familial relations (Corbin et al., unpublished data). The aims for defining these correlations in this study are to uncover possible mechanisms by which VHL mutations may result in retinal disease and to provide ways in which clinicians can use genotypic information for the prognosis and monitoring of eye disease.
Our analyses indicate that the prevalence of RCHs in VHL patients is significantly lower for complete deletions than for the other genotypes. The reason why the complete absence of VHL protein from one allele results in less frequent eye disease than a focally mutated or a truncated protein is not clear. It is possible that adjacent sequences flanking the VHL gene also lost in large deletions may be important for the production of RCHs. Alternatively, the complete loss of one VHL allele in all cells produces compensatory mechanisms in the retina that partially protects it from the formation of RCHs.
Genotype category was also found to correlate slightly but significantly with age. Single amino-acid substitutions were found in a slightly older distribution of patients than protein truncations. As the overall prevalence of RCH is not different between the 2 categories, it is possible that protein-truncating mutations, perhaps resulting in lower levels of residual VHL function, may cause the RCH phenotype to emerge at a slightly younger age.
On the whole, genotype category exerted little effect on the overall phenotype of RCHs. Previous studies 14,15 have also not found many differences between genotype and different aspects of ocular phenotype. In our study, genotypic category did not significantly influence whether one or both eyes were affected by RCHs, how many peripheral RCHs were found, and how much area of the retina was occupied. However, we did find that protein truncating mutations resulting in a lower prevalence of juxtapapillary lesions. The mechanism underlying this trend and the functional significance of this finding is not clear.
Genotype category was not found to have a significant effect on the better-seeing eye of affected patients. Many better-seeing eyes of affected patients lack any RCH involvement and most had good vision (in the 20/20 to 20/40 range). However, all the worse-seeing eyes of affected patients had RCHs and some had significantly compromised vision. In these eyes, amino-acid substitutions and protein truncations have a significantly lower level of visual acuity compared to complete deletions. One reason may be that although these eyes did not differ significantly in RCH number or area involved by genotype, the natural history of RCHs in complete deletions may be more favorable than in the other classes (i.e. have a less exudative behavior or result in fewer tractional complications), resulting in a better preservation of visual function.
In our study, patients with complete deletions as a whole develop RCHs less frequently, and even for patients who do, the overall extent of visual morbidity is lower than for the other genotypic categories. The genetic and cellular mechanisms underlying this difference may arise from additional deleterious gain-of-function properties that are absent in complete deletions but present in the other mutations. Alternatively, the complete absence of protein or flanking genetic regions may produce compensatory mechanisms that ameliorate the ocular phenotype.
In the present age of genetic diagnosis and counseling, ophthalmologists are often consulted when a patient is found to have a germline mutation in the VHL gene of a certain genotypic category 23. The present study provides some data for helping clinicians counsel on the risk of developing ocular angiomatosis and the resulting visual morbidity for each genotypic category. Although at present multiple screening protocols exist for the surveillance for patient with or at risk for VHL disease 19,20,23, future protocols may be formulated that take into account the prognostic information provided by genotype analysis.
Acknowledgments
This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, Division of Epidemiological and Clinical Research, National Eye Institute.
References
- 1.Lonser RR, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet. 2003;361:2059–67. doi: 10.1016/S0140-6736(03)13643-4. [DOI] [PubMed] [Google Scholar]
- 2.Singh AD, Shields CL, Shields JA. von Hippel-Lindau disease. Surv Ophthalmol. 2001;46:117–42. doi: 10.1016/s0039-6257(01)00245-4. [DOI] [PubMed] [Google Scholar]
- 3.Vail D. Angiomatosis retinae, eleven years after diathermy coagulation. Am J Ophthalmol. 1958;46:525–34. doi: 10.1016/0002-9394(58)91132-2. [DOI] [PubMed] [Google Scholar]
- 4.Annesley WH, Jr, Leonard BC, Shields JA, Tasman WS. Fifteen year review of treated cases of retinal angiomatosis. Trans Sect Ophthalmol Am Acad Ophthalmol Otolaryngol. 1977;83:446–53. [PubMed] [Google Scholar]
- 5.Blodi CF, Russell SR, Pulido JS, Folk JC. Direct and feeder vessel photocoagulation of retinal angiomas with dye yellow laser. Ophthalmology. 1990;97:791–5. doi: 10.1016/s0161-6420(90)32509-5. [DOI] [PubMed] [Google Scholar]
- 6.Latif F, Tory K, Gnarra J, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317–20. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 7.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820–3. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maher ER, Iselius L, Yates JR, et al. Von Hippel-Lindau disease: a genetic study. J Med Genet. 1991;28:443–7. doi: 10.1136/jmg.28.7.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stolle C, Glenn G, Zbar B, et al. Improved detection of germline mutations in the von Hippel-Lindau disease tumor suppressor gene. Hum Mutat. 1998;12:417–23. doi: 10.1002/(SICI)1098-1004(1998)12:6<417::AID-HUMU8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 10.Follmann D, Proschan M, Leifer E. Multiple outputation: inference for complex clustered data by averaging analyses from independent data. Biometrics. 2003;59:420–9. doi: 10.1111/1541-0420.00049. [DOI] [PubMed] [Google Scholar]
- 11.Barry RE, Krek W. The von Hippel-Lindau tumour suppressor: a multi-faceted inhibitor of tumourigenesis. Trends Mol Med. 2004;10:466–72. doi: 10.1016/j.molmed.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 12.Zbar B, Kishida T, Chen F, et al. Germline mutations in the Von Hippel-Lindau disease (VHL) gene in families from North America, Europe, and Japan. Hum Mutat. 1996;8:348–57. doi: 10.1002/(SICI)1098-1004(1996)8:4<348::AID-HUMU8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 13.Crossey PA, Richards FM, Foster K, et al. Identification of intragenic mutations in the von Hippel-Lindau disease tumour suppressor gene and correlation with disease phenotype. Hum Mol Genet. 1994;3:1303–8. doi: 10.1093/hmg/3.8.1303. [DOI] [PubMed] [Google Scholar]
- 14.Chen F, Kishida T, Yao M, et al. Germline mutations in the von Hippel-Lindau disease tumor suppressor gene: correlations with phenotype. Hum Mutat. 1995;5:66–75. doi: 10.1002/humu.1380050109. [DOI] [PubMed] [Google Scholar]
- 15.Webster AR, Maher ER, Moore AT. Clinical characteristics of ocular angiomatosis in von Hippel-Lindau disease and correlation with germline mutation. Arch Ophthalmol. 1999;117:371–8. doi: 10.1001/archopht.117.3.371. [DOI] [PubMed] [Google Scholar]
- 16.Dollfus H, Massin P, Taupin P, et al. Retinal hemangioblastoma in von Hippel-Lindau disease: a clinical and molecular study. Invest Ophthalmol Vis Sci. 2002;43:3067–74. [PubMed] [Google Scholar]
- 17.Glenn G, Choyke P, Zbar B, Linehan W. von Hippel-Lindau disease: clinical review and molecular genetics. Problems in Urology. 1990;4:312–30. [Google Scholar]
- 18.McCabe CM, Flynn HW, Jr, Shields CL, et al. Juxtapapillary capillary hemangiomas. Clinical features and visual acuity outcomes. Ophthalmology. 2000;107:2240–8. doi: 10.1016/s0161-6420(00)00422-x. [DOI] [PubMed] [Google Scholar]
- 19.Lamiell JM, Salazar FG, Hsia YE. von Hippel-Lindau disease affecting 43 members of a single kindred. Medicine. 1989;68:1–29. doi: 10.1097/00005792-198901000-00001. [DOI] [PubMed] [Google Scholar]
- 20.Maher ER, Yates JR, Harries R. Clinical features and natural history of von Hippel-Lindau disease. Q J Med. 1990;77:1151–63. doi: 10.1093/qjmed/77.2.1151. [DOI] [PubMed] [Google Scholar]
- 21.Friedrich CA. Genotype-phenotype correlation in von Hippel-Lindau syndrome. Hum Mol Genet. 2001;10:763–7. doi: 10.1093/hmg/10.7.763. [DOI] [PubMed] [Google Scholar]
- 22.Moore AT, Maher ER, Rosen P, Gregor Z, Bird AC. Ophthalmological screening for von Hippel-Lindau disease. Eye. 1991;5:723–8. doi: 10.1038/eye.1991.133. [DOI] [PubMed] [Google Scholar]
- 23.Choyke PL, Glenn GM, Walther MM, Patronas NJ, Linehan WM, Zbar B. von Hippel-Lindau disease: genetic, clinical, and imaging features. Radiology. 1995;194:629–42. doi: 10.1148/radiology.194.3.7862955. [DOI] [PubMed] [Google Scholar]