

Abstract
Endometriosis is a common gynaecological disease associated with pelvic pain and sub-fertility. We conducted a genome-wide association (GWA) study in 3,194 surgically confirmed endometriosis cases and 7,060 controls from Australia and the UK. Polygenic predictive modelling showed significantly increased genetic loading among 1,364 cases with moderate-severe endometriosis. The strongest association signal was on 7p15.2 (rs12700667) for ‘all’ endometriosis (P = 2.6 × 10−7, OR = 1.22 (1.13-1.32)) and for moderate-severe disease (P = 1.5 × 10−9 (OR = 1.38 (1.24-1.53)). We replicated rs12700667 in an independent US cohort of 2,392 self-reported surgically confirmed endometriosis cases and 2,271 controls (P = 1.2 × 10−3, OR = 1.17 (1.06-1.28)), resulting in a genome-wide significant P-value of 1.4 × 10−9 (OR = 1.20 (1.13-1.27)) for ‘all’ endometriosis in our combined datasets of 5,586 cases and 9,331 controls. SNP rs12700667 is located in an inter-genic region upstream of plausible candidate genes NFE2L3 and HOXA10.
Endometriosis [MIM 131200] is a disease affecting 6-10% of women of reproductive age1 with significant annual health costs2 and health burden for individuals3,4. Common symptoms include chronic pelvic pain, severe dysmenorrhea (painful periods), and sub-fertility. The causes of endometriosis remain uncertain despite over 50 years of hypothesis-driven research. Disease severity is classified using the revised American Fertility Society (rAFS) system5, assigning patients to one of four stages (I–IV, minimal-severe) based on lesion size and associated pelvic adhesions. However, it remains unclear whether the disease progresses through these stages, and it has been suggested that small lesions (stages I-II) represent an epiphenomenon rather than a disease entity6. Endometriosis risk is influenced by genetic factors7-14, with heritability estimated around 51%9.
We genotyped 3,194 unrelated cases with surgically confirmed endometriosis, recruited by the International Endogene Consortium, IEC (QIMR, Australia dataset = 2,270; Oxford, UK dataset = 924)15, using the Illumina Human670Quad Beadarray (Online Methods). Disease stage was assessed from surgical records using the rAFS classification system5,15 and grouped into two phenotypes: stage A (stage I-II or some ovarian disease plus a few adhesions; N = 1,686, 52.7%), and stage B (stage III–IV disease; N = 1,364, 42.7%), or unknown (N = 144, 4.6%) (Supplementary Table 1). Illumina Human610Quad control genotypes for QIMR cases were available for 1,870 individuals in an adolescent twin study16,17. For Oxford cases, Illumina Human1M-Duo genotypes for 5,190 UK population controls were obtained from the Wellcome Trust Case Control Consortium (WTCCC2). Although endometriosis affects women, Australian and UK control sets included men to maximise power of association detection on autosomal chromosomes (Online Methods). No significant autosomal allele frequency differences were detected between male and female control samples (Supplementary Fig. 1), indicating that association signals would not be influenced by differing female/male ratio in the cases and controls.
Studies to date have established that endometriosis is heritable, but have not addressed genetic burden for different disease stages. We used the GWA data to assess genetic loading in cases in two complementary ways. Using a novel method18 we estimated the proportion of variation in case-control status that can be explained by considering all SNPs simultaneously through inference of distant relatedness from marker data and comparing it to case-control status (Online Methods). The proportion of variation in case-control status explained by the GWA data was highly significant for both “all” and stage B endometriosis (Table 1; Supplementary Table 2). The estimate for stage B (0.34, SE: 0.04) was significantly higher than for stage A (0.15, SE: 0.04 (Table 1)).
Table 1.
Estimates of proportion of variation due to common genetic variants for “all” endometriosis and stage A or B disease using genome-wide SNP data from cases and controls
| Phenotypes | Cases | Controls | Proportion of variation (SE) |
P valuea |
|---|---|---|---|---|
| All endometriosis | 3154 | 6981 | 0.27 (0.04) | 4.4 × 10−16 |
| Stage B | 1347 | 6981 | 0.34 (0.04) | 4.4 × 10−16 |
| Stage A | 1666 | 6981 | 0.15 (0.04) | 2.6 × 10−4 |
Proportion of variation and associated P values for the likelihood ratio test were estimated using a linear mixed model incorporating 203,826 SNPs from the GWA panel after additional QC. Case and control numbers are slightly lower than for the GWA analyses due to the stricter QC measures (Online Methods). Stage A and stage B estimates of the variance explained are significantly different from each other (P = 1.8 × 10−3, using a two sample t-test which is conservative since the control samples are the same). Results were verified by prediction of individual genetic risk using QIMR and Oxford as alternate “discovery” and “target” datasets (Supplementary Table 2).
We also assessed the genetic loading of the different stages using a “prediction” approach (Online Methods)19 in which we used the Oxford data as a “discovery” set to identify increasingly large SNP sets ranked on significance of association (“allele specific scores”), and used these scores to predict disease status in “target” samples from QIMR. The discovery and target sets were then reversed (Supplementary Fig. 2). Oxford “all” endometriosis predicted endometriosis in the QIMR sample, with the smallest P value (8.4 × 10−6) obtained for a score set including ~75% of SNPs (Fig. 1). This result was highly significant, although the proportion of variance explained was small (maximum Nagelkerke R2 of 0.007; 0.7% of the variance). For stage B cases the proportion of variance explained by most score sets was higher (e.g. the score set including the ~20% most associated SNPs (P = 3.5 × 10−7) explained 1.3% of the variance, consistent with a greater (polygenic) genetic loading for stage B disease.
Figure 1.
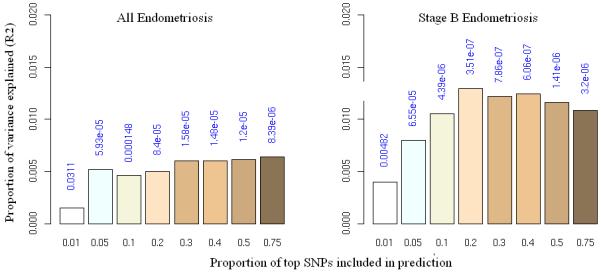
Allele specific score prediction for (a) “all” endometriosis and (b) stage B endometriosis, using Oxford as the “discovery” and QIMR as the “target” dataset. Variance explained in the target dataset on the basis of allele specific scores derived in the discovery dataset for eight significance thresholds (P<0.01, P<0.05, P<0.1, P<0.2, P<0.3, P<0.4, P<0.5, P<0.75, plotted left to right in each study). The y-axis indicates Nagelkerke’s pseudo R2 representing the proportion of variance explained. The number above each bar is the P value for the target dataset analysis. This figure shows that the results were not driven by a few highly associated regions, indicating a substantial number of common variants underlying disease.
We performed two GWA analyses stratified by dataset (QIMR and Oxford) using: 1) “all” 3,194 endometriosis cases, and 2) 1,364 stage B cases, given their substantially greater genetic loading (Online Methods). For ‘all’ endometriosis, the strongest signal was observed for rs12700667 in an inter-genic region on chromosome 7p15.2 (P = 2.6 × 10−7, OR = 1.22 (1.13-1.32), Table 2). As predicted from our quantitative genetic analyses, stronger signals of association across the genome were observed for stage B disease compared to ‘all’ endometriosis (Supplementary Figure 3). The 7p15.2 signal for stage B endometriosis was considerably stronger producing P = 1.5 × 10−9, OR = 1.38 (1.24-1.53) (Table 2) for rs12700667, and P = 6.0 × 10−8, OR = 1.34 (1.21-1.49) for nearby rs7798431 (r2 = 0.87). A second strong association was found for rs1250248 (2q35) within the FN1 gene (P = 3.2 × 10−8) (Supplementary Table 3). Results for SNPs rs12700667, rs7798431 and rs1250248 remained genome-wide significant after adjustment for multiple testing in the two non-independent GWA analyses via permutation (Online Methods). Only one of the permuted GWAs produced an independent P value less than that observed for rs12700667 (P = 0.001). The SNPs rs12700667 and rs7798431 lie in a narrow region of strong LD (r2 > 0.8) that extends for approximately 48 kb. Following imputation with 1000 Genomes and HapMap data (Fig 2; Supplementary Methods), conditioning on the effect of rs12700667 in logistic regression analysis showed no other independent associations with “all” or stage B endometriosis in the region.
Table 2.
GWA, replication and meta-analysis results for rs12700667
| Analysis | Number of Cases/Controls |
Risk allele (A) frequency in controls |
P value | OR (95% CIs) | Heterogeneity test P value |
|---|---|---|---|---|---|
| 1. GWA – All endometriosis | |||||
| QIMR | 2270/1870 | 0.73 | 1.5 × 10−5 | 1.25 (1.13-1.38) | - |
| Oxford | 924/5190 | 0.74 | 3.9 × 10−3 | 1.19 (1.06-1.34) | - |
| Combined | 3194/7060 | 0.74 | 2.6 × 10−7 | 1.22 (1.13-1.32) | 0.56 |
| 2. GWA – Stage B | |||||
| QIMR | 910/1870 | 0.73 | 8.3 × 10−7 | 1.40 (1.22-1.60) | - |
| Oxford | 454/5190 | 0.74 | 4.2 × 10−4 | 1.35 (1.14-1.60) | - |
| Combined | 1364/7060 | 0.74 | 1.5 × 10−9 | 1.38 (1.24-1.53) | 0.75 |
| 3. Replication NHSII – All endometriosis a |
2392/2271 | 0.73 | 1.2 × 10−3 | 1.17 (1.06-1.28) | - |
| 4. Meta-analysis – All Endometriosis (1+3) |
5586/9331 | 0.74 | 1.4 × 10−9 | 1.20 (1.13-1.27) | 0.64 |
Stage was unknown for cases in the NHSII replication cohort, though estimated to include ~40% stage B21.
Figure 2.
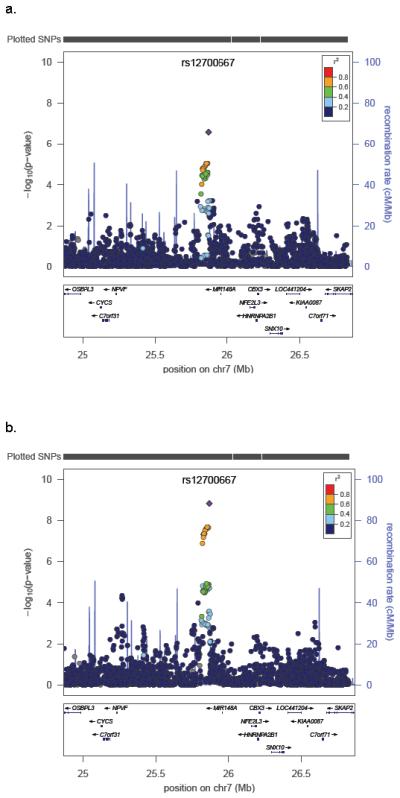
Evidence for association with (a) “all” endometriosis and (b) stage B endometriosis across the chromosome 7 region following imputation using HapMap 3 and 1000 Genomes project CEU and TSI reference panels. SNP rs12700667 is represented by a purple diamond. All other SNPs are colour coded according to the strength of LD (as measured by r2) with rs12700667.
In addition to the three genome-wide significant SNPs, we genotyped 70 SNPs producing nominal evidence of association with “all” (P < 1.0 × 10−4) or stage B endometriosis (P <1.0 × 10−4 in stage B and P <1.0 × 10−3 in “all” endometriosis analyses; Online Methods) in an independent IEC dataset encompassing 2,392 self-reported surgically confirmed cases from the Nurses’ Health Study II (NHSII) and 2,271 controls from GWAs of breast cancer20 and kidney function from the Nurses’ Health Study (NHS) I and II. Stage information was not available for NHSII cases, but the proportion likely to have stage B disease has been estimated at approximately 40%21, similar to that observed in the QIMR case set (Supplementary Table 1). Association with “all” endometriosis for the two SNPs on 7p15.2 was replicated in the US dataset, with P = 1.2 × 10−3, OR 1.17 (1.06-1.28) for rs12700667 and P = 1.6 × 10−3, OR = 1.17 (1.06-1.28) for rs7798431 (Supplementary Table 3). There was no evidence (nominal P ≤ 0.05) for replication of rs12540248 (FN1), or association with the remaining 70 SNPs (Supplementary Table 3).
Analysis of all 5,586 cases and 9,331 controls from the combined QIMR, Oxford and NHS cohorts further confirmed association between “all” endometriosis and 7p15.2, producing P values of 1.4 × 10−9 (OR = 1.20 (1.13-1.27)) for rs12700667 and 1.1 × 10−7 (OR = 1.18 (1.11-1.25)) for rs7798431 (Table 2). Although effect sizes from discovery datasets may be inflated22, the similarity of ORs for “all” endometriosis in our discovery (GWA) and replication datasets (Table 2), suggests this type of bias has not played a major role. Assuming the estimated OR of 1.20 and allele frequency of 0.74 for the rs12700667 A allele, a multiplicative risk model, and a population prevalence of 8%10,21,23, the estimated percentage of “all” endometriosis variance explained by rs12700667 is 0.36 (or 0.69% of the estimated 51% heritability of endometriosis9).
The associated SNPs are located in a ~924 kb inter-genic region containing at least one non-coding RNA (AK057379), predicted transcripts and regulatory elements and a miRNA (hsa-mir-148a) ~88 kb upstream of rs12700667. The closest gene NFE2L3, highly expressed in placenta, is located ~331 kb downstream of rs12700667. Two endometriosis candidate genes HOXA10 and HOXA1124,25, members of the homeobox A family of transcription factors that play a role in uterine development, lie ~1.35 Mb downstream.
Among reported candidate gene associations for endometriosis14 the only gene with a P value <10−3 for SNPs in the GWA data was PGR on chromosome 11 (Supplementary Table 3), but the result for this SNP was not significant in the replication stage. A recent GWA scan in Japanese women reported significant association of endometriosis with rs10965235 (P = 5.8 × 10−12, OR = 1.44) located on chromosome 9p21, and possible associations with rs13271465 on 8p22 and rs16826658 on 1p3626. The Japanese GWA study did not report our 7p15.2 signal among their 100 top SNPs followed up for replication, but with 1,423 cases and 1,318 controls they would have had only 13% power to detect the effect of rs12700667 with P ≤ 1.8 × 10−4 (Online Methods). We found no evidence for association with rs10965235 (which is monomorphic in individuals of European descent reflecting the different genetic (“ancestral”) backgrounds between the studies) or any other SNP in LD (r2 > 0.5 in HapMap JPT) in the QIMR/Oxford data (Supplementary Table 4). We also found no evidence of association to 8p22. We did find evidence for replication of SNP rs7521902 on 1p36 close to WNT4 for both “all” endometriosis (P = 9.0 × 10−5; OR = 1.16 (1.08-1.25)) and stage B (P = 7.5 × 10−6; OR = 1.25 (1.13-1.38)), the stronger signal in stage B providing additional empirical evidence for the benefit of examining stage B cases. Importantly, meta-analysis of the QIMR and Oxford “all” endometriosis OR with the reported Japanese OR of 1.25 (1.12-1.39) for rs7521902 produced a genome-wide significant P value of 4.2 × 10−8 (OR = 1.19 (1.12-1.27)). The frequency of the rs7521902 risk allele (A) was 0.57 and 0.51 in the Japanese GWAs cases and controls, and 0.26 and 0.24 in our combined GWAs cases and controls. WNT4 is important for development of the female reproductive tract27, ovarian follicle development and steroidogenesis28,29, and a plausible biological candidate.
We have identified a novel locus on chromosome 7p15.2 significantly associated with risk of endometriosis in women of European ancestry and confirm a previously reported suggestive association for SNPs close to the WNT4 locus. Our analyses also demonstrate a higher genetic loading for moderate-severe (stage B) endometriosis, and consistent with these results the strongest association signals were observed with stage B disease. Our predictive modelling demonstrates there are additional common variants contributing to risk for this disease and that future larger studies enriched for laparoscopically-confirmed moderate-severe cases will be better powered to identify risk loci and aberrant pathways contributing to the development of endometriosis.
Supplementary Material
Acknowledgements
We acknowledge with appreciation all women who participated in the QIMR, OXEGENE and NHS studies. We thank Endometriosis Associations for supporting study recruitment. We also thank the many hospital directors and staff, gynaecologists, general practitioners, and pathology services in Australia, the UK and the US who provided assistance with confirmation of diagnoses. We thank Sullivan Nicolaides and Queensland Medical Laboratory for pro bono collection and delivery of blood samples and other pathology services for assistance with blood collection.
The QIMR Study was supported by grants from the National Health and Medical Research Council (NHMRC) of Australia (241944, 339462, 389927,389875, 389891, 389892, 389938, 443036, 442915, 442981, 496610, 496739, 552485, 552498), the Cooperative Research Centre for Discovery of Genes for Common Human Diseases (CRC), Cerylid Biosciences (Melbourne), and donations from Neville and Shirley Hawkins. DRN is supported by the NHMRC Fellowship (339462 and 613674) and ARC Future Fellowship (FT0991022) schemes. SM is supported by NHMRC Career Development Awards (496674, 613705). PMV (442915) and GWM (339446, 619667) are supported by the NHMRC Fellowships Scheme. We thank B. Haddon, D. Smyth, H. Beeby, O. Zheng, B. Chapman and S. Medland for project and database management, sample processing, genotyping and imputation. We thank Brisbane gynaecologist Dr Daniel T O’Connor for his important role in initiating the early stages of the project and for confirmation of diagnosis and staging of disease from clinical records of many cases including 251 in these analyses. We are grateful to the many research assistants and interviewers for assistance with the studies contributing to the QIMR collection.
The work presented was supported by a grant from the Wellcome Trust (WT084766/Z/08/Z), and makes use of WTCCC2 control data generated by the Wellcome Trust Case-Control Consortium. A full list of the investigators who contributed to the generation of these data is available from www.wtccc.org.uk. Funding for the WTCCC project was provided by the Wellcome Trust under award 076113 and 085475. Imputation analyses were conducted using computational resources at the Oxford Supercomputing Centre (OCS). CAA is funded by the Wellcome Trust (WT91745/Z/10/Z). APM is supported by a Wellcome Trust Senior Research Fellowship. SK is supported by the Oxford Partnership Comprehensive Biomedical Research Centre with funding from the Department of Health NIHR Biomedical Research Centres funding scheme. KTZ is supported by a Wellcome Trust Research Career Development Fellowship (WT085235/Z/08/Z). We thank Louise Cotton, Lesley Pope, Gillian Chalk, Gail Farmer (University of Oxford). We also thank Philippe Koninckx, (Leuven, Belgium), Martin Sillem (Heidelberg, Germany), Colm O’Herlihy and Mary Wingfield (Dublin, Ireland), Mette Moen (Trondheim, Norway), Leila Adamyan (Moscow, Russia), Enda McVeigh (Oxford, UK), Christopher Sutton (Guildford, UK), David Adamson (Palo Alto, USA), and Ronald Batt (Buffalo, USA), for providing diagnostic confirmation.
The Nurses Health Studies I and II were supported by grants from the National Institutes of Health (NIH) of the United States (NHS1 cohort (PI: Dr. Susan Hankinson) - P01 CA087969, NHS1 blood cohort (PI: Dr. Susan Hankinson) – R01 CA049449, NHS1 Breast Cancer GWAS (PI: Dr. David Hunter) - UO1 CA098233, NHS1/NHS2 Kidney Stones GWAS (PI: Dr. Gary Curhan) - P01 DK070756, NHS2 cohort (PI: Dr. Walter Willett) - R01 CA050385, NHS2 blood cohort (PI: Dr. Susan Hankinson) - R01 CA067262, NHS2 endometriosis (PI: Dr. Stacey Missmer) -R01 HD052473 and R01 HD057210). We thank Drs. Lynn Marshall, David Hunter, and Robert Barbieri for their contributions to the endometriosis case validation study, and Ms. Barbara Egan and Lori Ward for surgical records procurement.
Appendix
ONLINE METHODS
GWA samples and phenotyping
For the current study, 2,351 surgically-confirmed endometriosis cases were drawn from individuals recruited by The Queensland Institute of Medical Research (QIMR)30 and a further 1,030 cases from individuals recruited by the Oxford Endometriosis Gene (OXEGENE) study. Controls consisted of 1,870 individuals recruited by QIMR31,32, and a further 6,000 individuals provided by the Wellcome Trust Case Control Consortium 2 (WTCCC2) (Supplementary Methods). Approval for the studies was obtained from the QIMR HREC and the Australian Twin Registry and the Oxford regional Multi-centre and Local Research Ethics Committees. Informed consent was obtained from all participants prior to testing.
GWA genotyping and quality control
QIMR and Oxford cases and QIMR controls were genotyped at deCODE Genetics on Illumina 670Quad (cases) and 610Quad (controls) BeadChips (Illumina Inc, San Diego, USA). The WTCCC2 controls were genotyped at the Wellcome Trust Sanger Institute using Illumina HumanHap1M Beadchips.
Genotypes for QIMR cases and controls were called with the Illumina BeadStudio software. Standard quality control procedures were applied as outlined previously (Supplementary Methods)33. Following exclusions 509,138 SNPs, 2,270 cases and 1,870 controls remained in the QIMR dataset. Oxford case and WTCCC2 control genotypes for all SNPs on the Illumina 670Quad BeadChip were called using Illuminus34. Following exclusions (Supplementary Note), 540,082 SNPs, 924 cases and 5,190 controls remained in the dataset. Post-QC genotype data from QIMR and Oxford were combined across 504,723 SNPs passing QC in both datasets.
Proportion of variation explained by all markers and predictive modelling
Using a novel method18 we estimated the proportion of variation explained by all markers. As genotyping artefacts can severely bias these estimates, the SNP data were subjected to more restrictive QC than utilised for the GWA analyses. SNPs with MAF <0.01, missing rates >0.001, P values for HWE <10−4, and non-autosomal SNPs were excluded. Individuals with missing rates >0.01, as well as one of any pair of individuals with estimated relationship >0.05, were also excluded. After QC 2,235 cases and 1,827 controls with 454,193 SNPs were used for analysis of the QIMR data, and 921 cases and 5,158 controls with 453,663 SNPs were used for analysis of the Oxford data (Supplementary Table 2). When combining the data, we first pruned SNPs to a common set and excluded closely linked SNPs having r2>0.5 in sliding 50 SNP windows using PLINK35. Again, one of any pair of individuals with estimated relationship >0.05 was excluded. The combined analysis included 3,154 cases, 6,981 controls and 203,826 SNPs (Table 1).
Estimation of variation explained by all SNPs on the observed scale
Pairwise realized relationships were estimated as described previously18,36,37. Phenotypic observations (affected or unaffected, coded as 0 or 1) were modelled as a linear function of the sum of the additive effects due to all SNPs and residuals. For the combined analysis (QIMR and Oxford) cohort information was modelled as a covariate, which adjusts for the mean difference in the proportion of cases between the two cohorts. Variance components were estimated by residual maximum likelihood (REML)38,39.
The proportion of variation in case-control status explained by all SNPs simultaneously is not a heritability in the conventional sense. Firstly, the observations are on the risk scale, whereas heritability estimates for disease from pedigree data are usually parameterized on an underlying unobserved liability scale. Secondly, the proportion of cases in the study is not the same as the proportion of cases in the population so the estimate we obtain is with respect to a case-control population and not the population at large. Thirdly, conventional heritability from pedigree data captures the additive genetic variation due to all causal variants, whereas we capture only the variation due to causal variants tagged by SNPs on the arrays. Despite these caveats, the comparison of proportion of variation estimates and resulting P values from stage A and stage B cases remains valid because the test statistics would not change after scale transformation and ascertainment correction.
Case status prediction
The aim of the prediction analysis was to evaluate the aggregate effects of many variants of small effect. We summarized variation across nominally associated loci into quantitative scores and related the scores to disease state in independent samples. Although variants of small effect (e.g. genotype relative risk of 1.05) are unlikely to achieve even nominal significance, increasing proportions of “true” effects will be detected at increasingly liberal P value thresholds e.g. P<0.1 (i.e. 10% of all SNPs), P<0.2, etc. Using such thresholds we defined large sets of “allele specific scores” in the “discovery” sample of the Oxford dataset to generate risk scores for individuals in the “target” sample of the QIMR dataset. The term risk score is used instead of risk, as it is impossible to differentiate the minority of true risk alleles from non-associated variants. In the discovery sample we selected sets of allele specific scores for SNPs with P<0.01, P<0.05, P<0.1, P<0.2, P<0.3, P<0.4, P<0.5 and P<0.75. For each individual in the target sample we calculated the number of score alleles they possessed, each weighted by the log10 odds ratio from the discovery sample. To assess whether the aggregate scores reflect endometriosis risk, we tested for a higher mean score in cases compared to controls. Logistic regression was used to assess the relationship between target sample disease status and aggregate risk score. Nagelkerke’s pseudo R-squared (henceforth R2) was used to assess the variance explained. Autosomal SNPs with MAF <0.01 and SNPs in high linkage disequilibrium were pruned, resulting in a set of 225,955 SNPs.
GWA analyses
Although endometriosis is a condition exclusive to women, male and female controls were used in analyses of autosomal markers to maximise power, a method adopted previously in GWAs of breast cancer by the WTCCC40,41. No significant allele frequency differences were detected between male and female controls. Moreover, the genome-wide significant SNPs rs12700667 (7p15) and rs7521902 (WNT4) showed no heterogeneity between male and female controls (P = 0.52 and P = 0.91, respectively). Cochran-Mantel-Haenszel (CMH) tests of association with “all” endometriosis or stage B alone were conducted in PLINK35, with QIMR and Oxford data as different strata (to account for between population differences in baseline effect). Breslow-Day (BD) tests were conducted to check that the assumptions of the CMH test (i.e. same effect size across strata) were true.
Permutation approach to correct for multiple testing
To address the non-independence between the “all” and stage B GWA analyses, we utilised a permutation approach where case/control status was randomly shuffled separately within the QIMR and Oxford datasets to break the relationship between phenotype and genotype, while retaining the relationship between “all” and stage B endometriosis. Of 1,000 permuted GWAs, recording the minimum P value for each SNP after analysis of both “all” and stage B cases, a P value ≤6.5 × 10−8 was obtained 50 times (genome-wide P ≤0.05). Hence rs12700667, rs7798431 and rs1250248 remain genome-wide significant after adjustment for multiple testing. Only one permuted GWAs produced an independent P value less than that observed for rs12700667 (P = 0.001, corrected for testing of both multiple markers and disease definitions).
Replication samples and genotyping
Endometriosis cases (2,392) for the replication samples were drawn from the US Nurses’ Health Study (NHS) II21,42. Replication controls were selected from two previous GWAs conducted in the NHSI and NHSII, including 1,142 postmenopausal, breast-cancer-free subjects from a breast cancer GWAs genotyped using the Illumina HumanHap 550 platform20 and 1,129 subjects from a GWAs for kidney function43 genotyped using Illumina 610 BeadChips. QC procedures have been described previously20.
SNPs selected for replication were genotyped in the 2,392 NHS cases. Multiplex assays were designed using the Sequenom MassARRAY Assay Design software (version 3.0: Sequenom Inc., San Diego, CA, USA) and samples genotyped using standard methods44,45. Post-laboratory QC was performed in PLINK35.
Replication and meta-analyses
GWA SNPs reaching genome-wide significance were tested for replication in the NHS cohort. Also, to help direct further studies we examined SNPs if they surpassed the following thresholds: (1) P <1.0 × 10−4 in the “all” endometriosis analysis (61 SNPs), or (2) any SNPs not already included with P <1.0 × 10−4 in the stage B analysis and P <1.0 × 10−3 in the “all” endometriosis analysis (14 SNPs). Genotype data were available for 2,271 NHS controls for 73 of these SNPs (Supplementary Table 3).
We estimated the association ORs and P values for the NHS cohort using PLINK35 (Supplementary Table 3), performing CMH and BD tests with the QIMR, Oxford and NHS datasets as distinct clusters. Meta-analysis for the QIMR, Oxford, and Japanese26 P values for 93 of the top 100 Japanese SNPs for which we had genotype data were conducted in GWAMA46.
Power of the Japanese GWA study26 including 1,423 endometriosis cases (unknown stage) and 1,318 controls to detect an OR of 1.20 for a risk allele frequency of 0.80 (HapMapII - JPT) of rs12700667, with a type I error of 1.8×10−4 (the threshold to select the top 100 SNPs for follow-up in their replication dataset), was calculated using the Genetic Power Calculator47.
URLs. ECR Browser http://ecrbrowser.dcode.org/; SNPTESTv2 http://www.stats.ox.ac.uk/~marchini/software/gwas/snptest.html; 1000 Genomes http://www.1000genomes.org/; HapMap http://hapmap.ncbi.nlm.nih.gov/
Additional references in Online Methods
- 30.Treloar SA, et al. Genomewide linkage study in 1,176 affected sister pair families identifies a significant susceptibility locus for endometriosis on chromosome 10q26. Am. J. Hum. Genet. 2005;77:365–376. doi: 10.1086/432960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McGregor B, et al. Genetic and environmental contributions to size, color, shape and other characteristics of melanocytic naevi in a sample of adolescent twins. Genet. Epidemiol. 1999;16:40–53. doi: 10.1002/(SICI)1098-2272(1999)16:1<40::AID-GEPI4>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 32.Zhu G, et al. A major quantitative-trait locus for mole density is linked to the familial melanoma gene CDKN2A: a maximum-likelihood combined linkage and association analysis in twins and their sibs. Am. J. Hum. Genet. 1999;65:483–492. doi: 10.1086/302494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Medland SE, et al. Common variants in the trichohyalin gene are associated with straight hair in Europeans. Am. J. Hum. Genet. 2009;85:750–755. doi: 10.1016/j.ajhg.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teo YY, et al. A genotype calling algorithm for the Illumina BeadArray platform. Bioinformatics. 2007;23:2741–2746. doi: 10.1093/bioinformatics/btm443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hayes BJ, Visscher PM, Goddard ME. Increased accuracy of artificial selection by using the realized relationship matrix. Genet. Res. 2009;91:47–60. doi: 10.1017/S0016672308009981. [DOI] [PubMed] [Google Scholar]
- 37.Oliehoek PA, Windig JJ, van Arendonk JA, Bijma P. Estimating relatedness between individuals in general populations with a focus on their use in conservation programs. Genetics. 2006;173:483–496. doi: 10.1534/genetics.105.049940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patterson HD, Thompson R. Recovery of interblock information when block sizes are unequal. Biometrika. 1971;58:545–554. [Google Scholar]
- 39.Gilmour AR, Gogel BJ, Cullis BR, Thompson R. ASReml User Guide Release 2.0. VSN International; Hemel Hempstead, UK: 2006. [Google Scholar]
- 40.Wellcome Trust Case Control Consortium et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat. Genet. 2007;39:1329–1337. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wellcome Trust Case Control Consortium et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464:713–720. doi: 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vitonis AF, Baer HJ, Hankinson SE, Laufer MR, Missmer SA. A prospective study of body size during childhood and early adulthood and the incidence of endometriosis. Hum. Reprod. 2010;25:1325–1334. doi: 10.1093/humrep/deq039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Curhan GC, Taylor EN. 24-h uric acid excretion and the risk of kidney stones. Kidney Int. 2008;73:489–496. doi: 10.1038/sj.ki.5002708. [DOI] [PubMed] [Google Scholar]
- 44.Zhao ZZ, et al. Genetic variation in tumour necrosis factor and lymphotoxin is not associated with endometriosis in an Australian sample. Hum. Reprod. 2007;22:2389–2397. doi: 10.1093/humrep/dem182. [DOI] [PubMed] [Google Scholar]
- 45.Zhao ZZ, et al. Common variation in the fibroblast growth factor receptor 2 gene is not associated with endometriosis risk. Hum. Reprod. 2008;23:1661–1668. doi: 10.1093/humrep/den035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mägi R, Morris AP. GWAMA: software for genome-wide association meta-analysis. BMC Bioinformatics. 2010;11:288. doi: 10.1186/1471-2105-11-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Purcell S, Cherny SS, Sham PC. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics. 2003;19:149–150. doi: 10.1093/bioinformatics/19.1.149. [DOI] [PubMed] [Google Scholar]
Footnotes
The authors declare no competing financial interests.
Conflict of interest statements
None
References
- 1.Giudice LC, Kao LC. Endometriosis. Lancet. 2004;364:1789–1799. doi: 10.1016/S0140-6736(04)17403-5. [DOI] [PubMed] [Google Scholar]
- 2.Simoens S, Hummelshoj L, D’Hooghe T. Endometriosis: cost estimates and methodological perspective. Hum. Reprod. Update. 2007;13:395–404. doi: 10.1093/humupd/dmm010. [DOI] [PubMed] [Google Scholar]
- 3.Jones GL, Kennedy SH, Jenkinson C. Health-related quality of life measurement in women with common benign gynecologic conditions: a systematic review. Am. J. Obstet. Gynecol. 2002;187:501–511. doi: 10.1067/mob.2002.124940. [DOI] [PubMed] [Google Scholar]
- 4.Kjerulff KH, Erickson BA, Langenberg PW. Chronic gynecological conditions reported by US women: findings from the National Health Interview Survey, 1984 to 1992. Am. J. Public Health. 1996;86:195–199. doi: 10.2105/ajph.86.2.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Revised American Fertility Society classification of endometriosis: 1985. Fertil. Steril. 1985;43:351–352. doi: 10.1016/s0015-0282(16)48430-x. [DOI] [PubMed] [Google Scholar]
- 6.Koninckx PR, Oosterlynck D, D’Hooghe T, Meuleman C. Deeply infiltrating tendometriosis is a disease whereas mild endometriosis could be considered a non-disease. Ann. N. Y. Acad. Sci. 1994;734:333–341. doi: 10.1111/j.1749-6632.1994.tb21763.x. [DOI] [PubMed] [Google Scholar]
- 7.Hadfield RM, Mardon HJ, Barlow DH, Kennedy SH. Endometriosis in monozygotic twins. Fertil. Steril. 1997;68:941–942. doi: 10.1016/s0015-0282(97)00359-2. [DOI] [PubMed] [Google Scholar]
- 8.Kennedy S. The genetics of endometriosis. J. Reprod. Med. 1998;43:263–268. [PubMed] [Google Scholar]
- 9.Treloar SA, O’Connor DT, O’Connor VM, Martin NG. Genetic influences on endometriosis in an Australian twin sample. Fertil. Steril. 1999;71:701–710. doi: 10.1016/s0015-0282(98)00540-8. [DOI] [PubMed] [Google Scholar]
- 10.Zondervan KT, Cardon LR, Kennedy SH. The genetic basis of endometriosis. Curr. Opin. Obstet. Gynecol. 2001;13:309–314. doi: 10.1097/00001703-200106000-00011. [DOI] [PubMed] [Google Scholar]
- 11.Simpson JL, Bischoff FZ. Heritability and molecular genetic studies of endometriosis. Ann. N. Y. Acad. Sci. 2002;955:239–251. doi: 10.1111/j.1749-6632.2002.tb02785.x. [DOI] [PubMed] [Google Scholar]
- 12.Stefansson H, et al. Genetic factors contribute to the risk of developing endometriosis. Hum. Reprod. 2002;17:555–559. doi: 10.1093/humrep/17.3.555. [DOI] [PubMed] [Google Scholar]
- 13.Zondervan KT, et al. Familial aggregation of endometriosis in a large pedigree of rhesus macaques. Hum. Reprod. 2004;19:448–455. doi: 10.1093/humrep/deh052. [DOI] [PubMed] [Google Scholar]
- 14.Montgomery GWM, et al. The search for genes contributing to endometriosis risk. Hum. Reprod. Update. 2008;14:447–457. doi: 10.1093/humupd/dmn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Treloar S, et al. The International Endogene Study: a collection of families for genetic research in endometriosis. Fertil. Steril. 2002;78:679–685. doi: 10.1016/s0015-0282(02)03341-1. [DOI] [PubMed] [Google Scholar]
- 16.Sturm RA, et al. A single SNP in an evolutionary conserved region within intron 86 of the HERC2 gene determines human blue-brown eye color. Am. J. Hum. Genet. 2008;82:424–431. doi: 10.1016/j.ajhg.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferreira MA, et al. Quantitative trait loci for CD4:CD8 lymphocyte ratio are associated with risk of type 1 diabetes and HIV-1 immune control. Am. J. Hum. Genet. 2010;86:88–92. doi: 10.1016/j.ajhg.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang J, et al. Common SNPs explain a large proportion of heritability for human height. Nat. Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.The International Schizophrenia Consortium Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hunter DJ, et al. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat. Genet. 2007;39:870–874. doi: 10.1038/ng2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Missmer SA, et al. Incidence of laparoscopically confirmed endometriosis by demographic, anthropometric, and lifestyle factors. Am. J. Epidemiol. 2004;160:784–796. doi: 10.1093/aje/kwh275. [DOI] [PubMed] [Google Scholar]
- 22.Kraft P. Curses--winner’s and otherwise--in genetic epidemiology. Epidemiology. 2008;19:649–651. doi: 10.1097/EDE.0b013e318181b865. [DOI] [PubMed] [Google Scholar]
- 23.Zondervan KT, Cardon LR, Kennedy SH. What makes a good case-control study? Design issues for complex traits such as endometriosis. Hum. Reprod. 2002;17:1415–1423. doi: 10.1093/humrep/17.6.1415. [DOI] [PubMed] [Google Scholar]
- 24.Taylor HS, Bagot C, Kardana A, Olive D, Arici A. HOX gene expression is altered in the endometrium of women with endometriosis. Hum. Reprod. 1999;14:1328–1331. doi: 10.1093/humrep/14.5.1328. [DOI] [PubMed] [Google Scholar]
- 25.Wu Y, et al. Aberrant methylation at HOXA10 may be responsible for its aberrant expression in the endometrium of patients with endometriosis. Am. J. Obstet. Gynecol. 2005;193:371–380. doi: 10.1016/j.ajog.2005.01.034. [DOI] [PubMed] [Google Scholar]
- 26.Uno S, et al. A genome-wide association study identifies genetic variants in the CDKN2BAS locus associated with endometriosis in Japanese. Nat. Genet. 2010;42:707–710. doi: 10.1038/ng.612. [DOI] [PubMed] [Google Scholar]
- 27.Vainio S, Heikkilä M, Kispert A, Chin N, McMahon AP. Female development in mammals is regulated by Wnt-4 signalling. Nature. 1999;397:405–409. doi: 10.1038/17068. [DOI] [PubMed] [Google Scholar]
- 28.Naillat F, et al. Wnt4/5a signalling coordinates cell adhesion and entry into meiosis during presumptive ovarian follicle development. Hum. Mol. Genet. 2010;19:1539–1550. doi: 10.1093/hmg/ddq027. [DOI] [PubMed] [Google Scholar]
- 29.Boyer A, et al. WNT4 is required for normal ovarian follicle development and female fertility. FASEB J. 2010;24:3010–3025. doi: 10.1096/fj.09-145789. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.