

Abstract
Oxidative damage by reactive oxygen species generated in mitochondria is a potential cause of stem-cell dysregulation. Little is known about how hematopoietic stem cells mitigate/lessen this risk in the face of upregulated mitochondrial biogenesis/function necessary for the energy needs of differentiation and progenitor expansion. Here we report that upregulation of mitochondrial mass in mouse hematopoietic stem cells is closely linked to the appearance of CD34 on their surface, a marker indicating loss of long-term repopulating ability. These mitochondria have low membrane potential initially, but become active before exiting the primitive LSK compartment. Steady-state hematopoiesis perturbed by global expression of SDF-1/CXCL12 transgene causes a shift in ratios of these mitochondrialy-distinct LSK populations. Based on known effects of SDF-1 and signaling by it's receptor, CXCR4, along with finding primitive progenitors with high mitochondrial mass but low activity, we suggest a model of asymmetric self-renewing stem cell division that could lessen stem cell exposure to oxidative damage.
Keywords: Mitochondria, Stem cells, Hematopoiesis, Asymmetric cell division, Oxidative stress, Mitochondrial membrane potential, Mitochondrial mass, CD34, LSK bone marrow cells, Asymmetric mitochondrial distribution
Introduction
Animal cells' use of oxygen as the terminal electron acceptor during mitochondrial oxidative-phoshporylation (OX-PHOS) makes it possible to derive vastly more energy from glucose than from glycolysis alone. However, this advantage comes at an increased risk of oxidative damage to proteins and lipids, primarily in the form of reactive oxygen species (ROS). Although some ROS appears necessary for balanced metabolism and homeostasis1-3, an overabundance can induce macromolecular damage leading to cell death.4 Hematopoietic stem cells (HSC), like many types of stem cells, are particularly vulnerable to ROS damage because once it arises, the damage can be transmitted to progenitors, and other differentiated cells, potentially resulting various pathological conditions.5-7 The principle source of ROS in cells is the mitochondrion, where it can be produced in increased quantities if the electron-transport chain is inhibited or damaged.4
Stem cells have several strategies to lessen the impact of ROS, which we have been investigating in embryonic stem cells (ESC).8 It is believed that many stem cells, like HSC, reside in hypoxic environments as one of these strategies.5 In addition to hypoxic stem cell niches, HSCs, as well as ESC, are known to have very low numbers of mitochondria, which also appear to have low OX-PHOS activity.9,10 Stem cell mitochondria have recently come under increased scrutiny because new information has revealed their role in numerous cellular processes, beyond ATP generation and apoptotic regulation11-14, and they recently have even been suggested to serve as a cell-fate or lineage determinant.6 We recently discussed the potential role of asymmetric segregation of mitochondria during mitosis, either numerically, or “old versus new” and in other capacities contributing to the aging process or pathology.15 Asymmetric cellular distribution/localization of mitochondria is a dynamic process and mechanisms/regulation of this in stem cells are of interest, especially as relates to hypoxia inducing factor (HIF-1) function.16 Asymmetric segregation of mitochondria during mitosis has been observed in some species and may play a role in cell-fate determination and differentiation, and also in self-renewal and pluripotency.6,17-22 The potential role of ROS and influence of oxidative damage in asymmetric mitochondrial distribution and segregation are unknown.
A challenge that HSC and other stem cells face is the necessity to upregulate mitochondrial biogenesis and OX-PHOS, which is required for energy needs of cellular expansion and differentiation22, but without subjecting stem cells to increased oxidative damage. Very little is know about this process in stem cells, especially HSC, owing primarily to their rarity which makes difficult the use of most traditional bioenergetic methodologies to study them. Using flow cytometry with improved flow cytometric probes of mitochondrial mass and function, we have begun to address how HSCs deal with oxidative damage or stress challenges during differentiation and how this could be linked to HSC self-renewal and pluripotency. Using transgenic mice engineered to express the HSC-homing, survival-enhancing and glycolysis-suppressing chemokine, SDF-1/CXCL1223, and normal mice as a control, we identified new, mitochondrialy distinct, sub-populations of primitive hematopoietic stem and progenitor cells, which suggest possible insight into how HSCs might deal with challenges of potential oxidative damage during upregulation of mitochondrial biogenesis and function.
Methods
Antibodies and mitochondrial probes
Anti-c-kit, -sca-1, -CD34, and –LIN cocktail and respective isotype control antibodies, in various fluorescent formats were either from Becton-Dickenson (Franklin Lakes, NJ) or BD/Pharmingen (San Jose, CA), and were used for phenotypic identification of lineage negative (LIN-), sca-1 positive (sca-1+), c-kit positive (c-kit+),(LSK) cells24. Mitochondrial probes, JC-1 and MitoTracker (MT) Green FM, were from Molecular Probes/Invitrogen (Carlsbad, CA), and were dissolved in DMSO immediately before use, and were used according to manufacturer's instructions with minor modification (final concentrations were half those suggested because excess probe can precipitate into a very fine, nearly invisible particulate that can foul the flow-cytometer intake port.) Bone marrow (BM) cells were stained with surface-marker antibodies, then promptly stained with mitochondrial probes. MT staining to quantitate mitochondrial was done at room temperature for 30 minutes in RPMI 1640 (without FCS) and washed twice with RPMI, then resuspended in cold RPMI before flow analysis. JC-1 staining to quantitate mitochondrial membrane potential was done at 37° C for 30 min. in RPMI, washed twice with cold RPMI and resuspended in cold RPMI before flow analysis. Flow analysis was done as soon as possible (usually within 10 min) after staining/incubation. We found this to be particularly important because these probes are not effectively amenable to formaldehyde fixation, as indicated by the manufacturer, and they are subject to slow leakage, even in cold RPMI and J-aggregate formation is reversible. We also found that another critical consideration for accurate measurement of mitochondrial activities is to incubate and wash cells in RPMI 1640 (with glucose and pyruvate (Gibco/Invitrogen; Carlsbad, CA) instead of PBS, because RPMI provides a source of substrates for glycolysis and mitochondrial metabolism. Pre-incubation with the uncoupler, CCCP, caused collapse of ΔΨm with concomitant inhibition of JC-1red fluorescence (data not shown).
Animals
Wild-type male and female C3hHej/Feb mice were from Jackson Laboratories (Bar Harbor, ME) and were maintained as previously described.23 In two separate experiments we observed no difference in results between male and female mice (data not shown). Female SDF-1 transgene-expressing C3hHej/Feb mice were developed and maintained as previously described by us.23 Only female pups were born that expressed the transgene after mating heterozygous mating male and female pairs.23 The reason for this sex-linked transgene expression is unknown.
Flow-Cytometry
Flow-cytometric analysis of stained whole BM cells was done with a Becton-Dickenson LSR II cytometer. Typically, 1 to 2 million events were recorded for each sample after instrument set-up using appropriate isotype control stained cells. This ensured abundant numbers of LSK cells for later analysis. Initial experiments also included single antibody stained cells to verify appropriate compensation settings. Instrument set-up and data acquisition was accomplished with FACSDiva software from Becton-Dickenson and was digitally stored for later analysis.
Data analysis and statistics
Experiments were conducted as pairs of samples from wild type and mutant animals and each pair was harvested, stained, and analyzed as independent experiments at different times. This approach reduced the time between the end of probe staining and data-acquisition, which was important because of reasons already stated above regarding probe stability. This also simplified statistical analysis of significance, which was done using a paired t-statistic test (two-tailed) with 95% confidence interval. Calculated p-values between data sets were considered significantly different if p was less than 0.05. Stored data (as FC5 3.0 files) was analyzed with WinList Software (Verity Software House; Topsham, MD) or with Cyflogic software (CyFlo Ltd; Turku, Finland). WinList uses “log-bias” and “hyperlog” mathematical transforms to more easily visualize events that are near or below the axis and more easily recognize discrete cell populations. It also uses a very robust post-acquisition re-compensation algorhythm. Cyflogic has an intuitive user interface that features a robust histogram modeler with “Silhouette statistics”. LSK cell identification and gating was done as previously described.24
Results
Understanding mitochondrial behavior during HSC differentiation and self-renewal could lead to a better understanding of how HSC deal with or respond to increased oxidative risk, information that could be of broad use in studies of stem cells in general.
We utilized a transgenic mouse model we had previously developed that globally expresses the chemokine SDF-1/CXCL12 transgene.23 Because SDF-1 is essential for proper HSC mobilization/homing to/from bone marrow (BM) and is important in stress-induced HSC survival25 we considered this a good choice to use as a model of perturbed steady-state hematopoiesis. The SDF-1 receptor, CXCR4, expressed on HSC25, is involved in inhibition of the glycolytic enzyme, PGK-1, suppression of glycolysis26,27, and is also linked to PPARs, which regulate mitochondrial biogenesis and are coupled to oxidative stress responses and cancer.28
Upregulation of mitochondrial biogenesis is directly associated with loss of HSC pluripotency
Mouse bone marrow long-term self-renewing HSC are highly enriched in a population of LSK cells.29 LSK cells are composed of both long-term and short-term repopulating stem cells and progenitor cells. Expression of surface-determinant CD34 can be used to distinguish between these short and long-term repopulating HSC.29 Appearance of CD34 on the surface of LSK cells is closely linked to loss of long-term serial repopulating ability and pluripotency, and is an early marker to assess pluripotency/differentiation status of LSK cells. We noted two discrete populations of LSK cells, either CD34lo or CD34hi, in mouse BM which have a direct relationship to mitochondrial mass (Mt-mass; Figure 1B). After rigorous analysis, both at the instrument-compensation level and with various combinations of software-applied post-acquisition re-compensation analysis, we concluded that there is a direct relationship between CD34 surface expression level and Mt-mass that was not due to compensation artifact. LSK cells also routinely had, varyingly, two populations differing in c-kit expression levels (Fig. 1B, C). However, gate analysis of the two populations showed no relationship to CD34 expression level and the nature of these subpopulations of LSK cells is, at present, unknown. The large population of Lin- c-kit+ sca-1- cells, believed to be more differentiated progenitors, also had as high or higher Mt-mass as the CD34hi LSK cells (data not shown). There was no apparent relationship of sca-1 expression density and Mt-mass in CD34hi or CD34lo LSK cells.
Figure 1. Mitochondrial mass is linked to CD34 expression in mouse HSC.
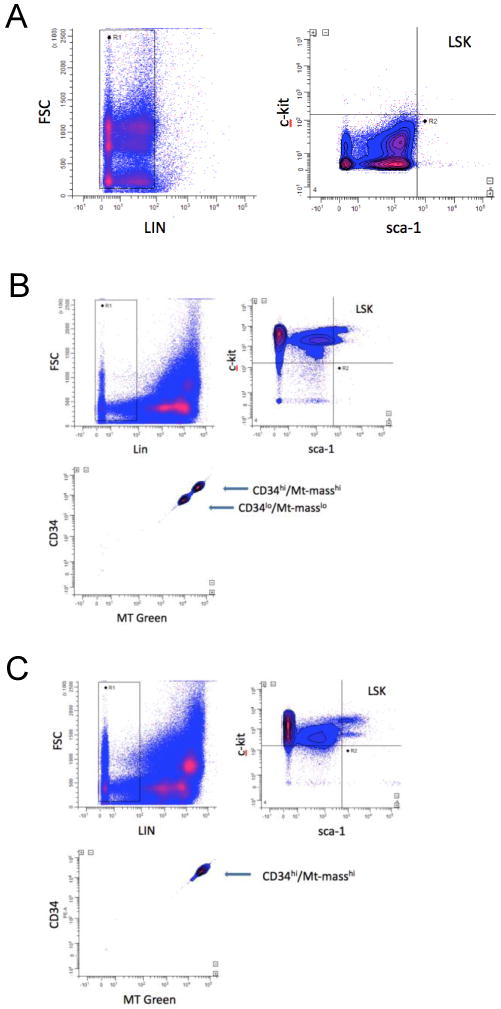
Flow cytometric analysis of mitochondrial mass in BM LSK cells using MitoTracker Green FM is shown. Dot-plots of isotype antibody control data (A), Wild Type (WT) mouse BM (B) and SDF-1 transgene (TG) expressing mouse BM cells (C). The top left panels show lineage gating (LIN), the top right panels show LSK gating, and the bottom left panels show CD34 expression and MT Green FM fluorescence in gated LSK cells. WinList software was used for analysis and re-compensation using hyperlog and log-bias transforms (see METHODS). Data are representative of at least six independent experiments.
Expression of the SDF-transgene in BM had no effect on Mt-mass levels per cell in the different cellular subpopulations (Fig.1C), but it did have a significant effect on the proportions of cells that were CD34lo/Mt-mass low, compared to CD34hi/Mt-mass high cells (Fig. 1C and Fig.2B). Also, overall LSK proportions were significantly increased in SDF-1 transgenic BM compared to wild-type (WT) BM (Fig. 2A), as was the proportion of CD34hi LSK cells compared to CD34lo LSK cells (Fig. 3B). These data suggest that Mt-mass increases in HSC very early, as they begin to differentiate and lose pluripotency (long-term repopulating ability), in close synchrony with expression of CD34. This suggests that SDF-1 transgenic expression-induced perturbation of this steady-state system results in increased total BM LSK content that is dominated by a proportional increase in CD34hi/Mt-mass high LSK cells.
Figure 2. Total LSK and CD34hi/lo LSK populations in mouse BM.

Quantitative analysis of % of total BM cells from WT and SDF-1 TG LSK cells is shown (A). LSK cells were gated as in Figure 1. The mean+/- S.E. from six independent experiments are shown. The p value indicates a significant increase of LSK cells in SDF-1 TG compared to WT BM (a: p=0.019). Comparative analysis of the proportions of CD34lo/hi LSK cells from WT and SDF-1 TG BM are shown in B. CD34 and LSK gating were done as in Figure 1. Data are expressed as mean % +/- S.E. of LSK cells. Data from five independent experiments are shown and values of p indicate a significant difference in both population types from WT compared to SDF-1 TG LSK cells (b: p=0.010;c: p=0.007).
Figure 3. Three distinct populations of LSK cells are seen in BM based on size and mitochondrial mass.
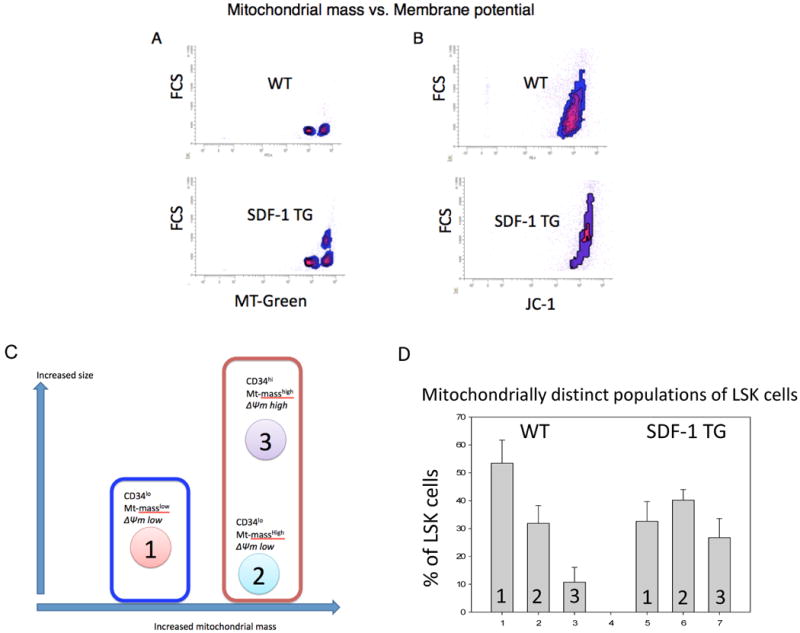
Density plots of flow cytometric data comparing cell size and MT Green FM fluorescence (A and C) or size and JC-1 polymeric (red) fluorescence intensity (B and D) are shown. Polymeric JC-1 fluorescence is proportional to mitochondrial membrane potential (see METHODS). Data from WT LSK cells (A and B) or from SDF-1 TG LSK cells (C and D) are shown. Three distinct populations are discernable (A and C) and are indicated numerically. LSK gating was done as in Figure 1. Data represent three independent experiments.
Three “mitochondrialy-distinct” LSK subpopulations are found in mouse BM
During our Mt-mass studies, we noticed a relationship between LSK cell size (FSC) and Mt-mass (Fig. 3A,C). There were three distinct populations of LSK cells that were consistently discernable in mouse BM. Population 1 was CD34lo and population 2 and 3 were CD34hi. To further distinguish these three populations, we assessed mitochondrial activity, as indicated by mitochondrial membrane potential (ΔΨm), of BM LSK cells using JC-1 probe (Fig. 3B,D). The larger cells had increased ΔΨm. Interpretation of these data was clarified when comparing WT versus SDF-1 transgenic mice BM LSK cells (Fig. 3A-D). Population 2 could not be seen in the ΔΨm analysis. The patterns observed in the SDF-1 transgenic BM LSK cells are consistent with the presence of a smaller, Mt-mass high population of LSK cells having lower ΔΨm than population 3, but these have ΔΨm similar to or slightly higher than that of population 1. Combined, our interpretation of these data is illustrated in Figure 4A, with analysis of the relative proportions of these populations in WT and SDF-1 transgenic mouse BM shown in Figure 4B. The influence of SDF-1 transgenic overexpression in this steady-state system appears to induce quantitative proportional shift in the three populations rather than have a direct influence on Mt-mass or activity. This population shift is primarily toward LSK cells with increased Mt-mass and activity and is consistent with proportionally increased differentiation of CD34lo LSK cells to CD34hi LSK cells (population 2 and 3). It is particularly noteworthy that there exists a population of CD34hi LSK cells with increased Mt-mass, but with low ΔΨm (population 2) in the BM. Our interpretation of the effects of SDF-1 transgene expression on steady-state hematopoiesis is illustrated in Figure 4C.
Figure 4. Illustration of three mitochondrialy distinct types of LSK cells and their properties.
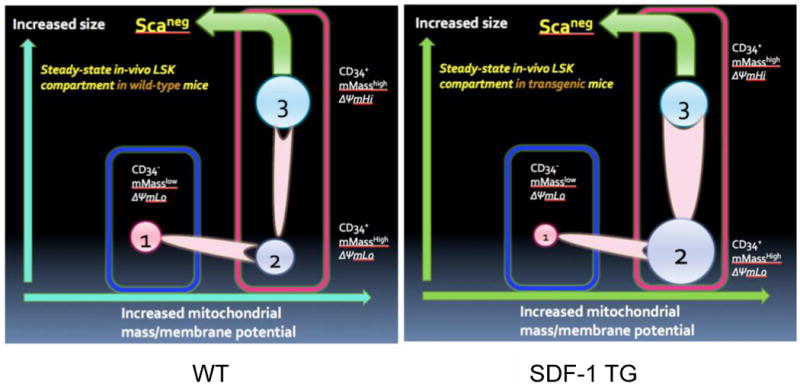
Based on data from Figures 1-3, a model of proposed types of LSK cells in mouse BM is shown (A). The relationships of cell size, mitochondrial mass, and CD34 expression are indicated. The relative proportions of the three types of LSK cells, expressed as mean % of LSK cells +/- S.E. are shown in B. Data from three independent experiments like Figure 3 and 4B, and using the proposed model shown in A, the influence of global SDF-1 transgene expression compared to wild type (WT) on the LSK compartment is modeled (C).
A more robust method of measurement of ΔΨm with the JC-1 probe is to compare the ratio of monomeric to polymeric intracellular JC-1, which have differing fluorescent emission spectra. Monomeric JC-1 can be found in cellular cytoplasm (cell size dependent) and even in mitochondria with very low activity.30 Polymeric JC-1 can only be found in active mitochondria because robust transport of the probe into the mitochondrial matrix is dependent on a polarized inner-membrane potential.30 As JC-1 accumulates in the matrix, it aggregates and its emission spectrum shifts from green to red. Thus, the ratio of monomer to polymer is a more accurate measure of ΔΨm, which takes into consideration Mt-mass and cell size. Figure 5B and C, respectively show JC-1 analysis of BM LSK cells from WT and mutant mice. LSK cells can be categorized into two types; those with high ΔΨm and those with low ΔΨm. The influence of SDF-1 transgene expression on LSK ΔΨm (Fig.5D) is seen to reduce overall LSK ΔΨm (Fig.5E), but this was due to a significant proportional decrease in ΔΨm-high cells with a concomitant increase in ΔΨm-low cells (Fig. 5E). There was also an increase in JC-1 monomer levels in LSK cells from SDF-1 transgene expressing mouse LSK cells (Fig. 5C). These data are consistent with the data in Figure 3, where SDF-1 transgene expression causes a shift to larger LSK cells with greater Mt-mass, but also shows that on a proportional basis, there is an accumulation of LSK cells with increased Mt-mass and low ΔΨm in the BM of mutant mice.
Figure 5. Two types of LSK cells with different mitochondrial membrane potential are in mouse BM.
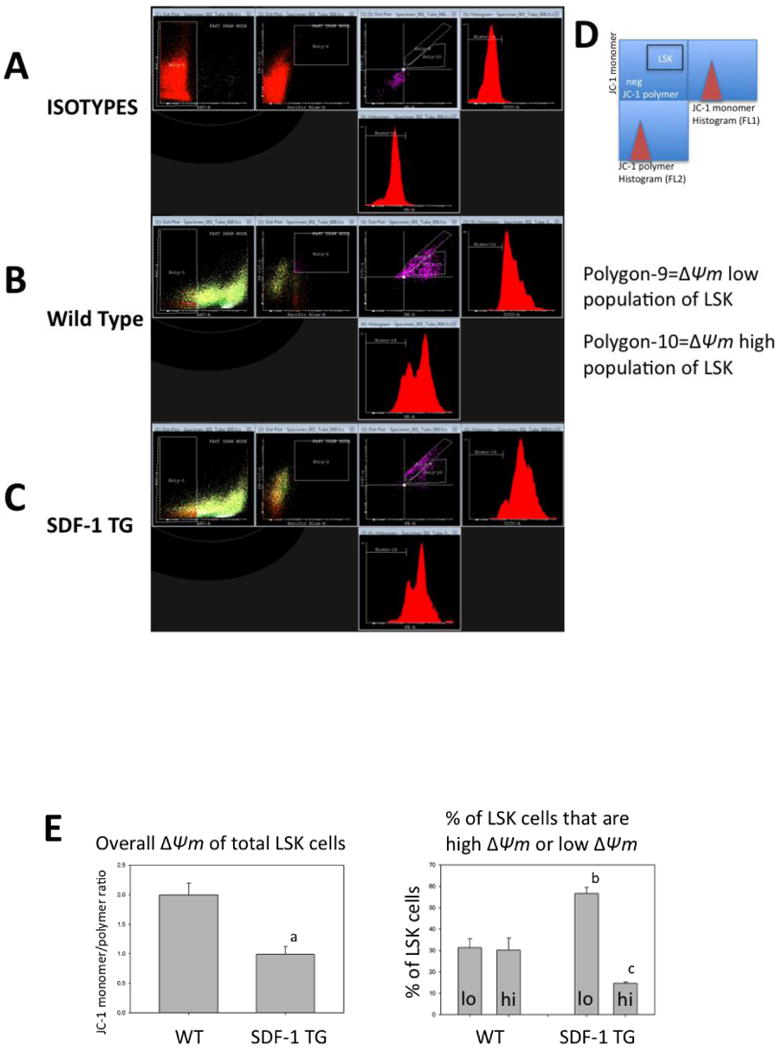
Dot-plots of flow cytometric data that measures mitochondrial membrane potential (ΔΨm) as indicated by relative fluorescence of monomeric (green, FL1) and polymeric (red, FL2) in LSK cells from WT (B) or SDF-1 TG (C) mouse BM are shown. Antibody isotype controls are shown in A. LSK gating was done as in Figure 1 and monomer versus polymer dot-plots are shown in the middle panels. Adjacent to the monomer and polymer axes is shown relative frequency histograms of the dot-plot data. The geometric means of these histograms were used to calculate the JC-1 monomer/polymer ratios, which are a measure of ΔΨm (see METHODS). The key for the right three panels is shown in D. The gated area for LSK cells (LSK) is indicated. Two populations of LSK cells are discernable based on JC-1 monomer/polymer ratios (polygon-9 and -10). Data are representative of five independent experiments. Cyflogic software was used for this analysis. The overall ΔΨm of total LSK cells, based on JC-1 monomer/polymer ratios for WT or SDF-1 TG BM LSK cells are shown in E (left panel). Mean JC-1 ratios +/- S.E. for five independent experiments are shown. The p value indicates that ΔΨm was significantly lower in SDF-1 TG LSK cells compared to WT LSK cells (a: p=0.005). Also shown in E are the relative proportions of the two populations with either low or high ΔΨm that were independently gated (polygon-9 and -10) and expressed as mean % of total LSK cells +/- S.E. for WT or SDF-1 TG LSK cells (right panel) (b: p=0.009; c: p=0.017).
Discussion
The quintessential property of HSCs is their ability to undergo an asymmetric, self-renewing cell division.31 There are two types of self-renewing cell divisions. One is asymmetric, when one daughter cell remains an HSC, and the other inherits various cell-fate determinants that cause it to initiate a program of differentiation. The other type is an “expansion” division when both daughter cells are pluripotent HSCs. Little is known about the regulation and mechanisms of self-renewing HSC divisions, which are believed to be tightly regulated by intrinsic determinants and extrinsic signals from the niche and cytokines. Cell polarization and asymmetric localization of cell-fate determinants prior to division is believed to be the fundamental driving force of HSC self-renewal and differentiation.32 Candidate protein cell-fate determinants have been identified in stem cells from various organisms.32,33 Recently, mitochondria have also been proposed to have a role as a cell-fate determinant.6 Mitochondria have a very complex “life-cycle” within cells which helps maintain undamaged mitochondria and degradation of damaged mitochondria (usually ROS damage) by autophagy.34 A potential role as cell-fate determinants for mitochondria has risen because new evidence suggests they have functions beyond ATP generation and apoptosis. They may act as scaffolds for integrating various signal transduction pathways. Proteins known to regulate cell cycle (Rb, p53) or cytokine signaling (STAT3) are known to bind to mitochondria and shuttle between the nucleus and mitochondria.11-14 Some of these proteins are known to directly influence mitochondrial OX-PHOS activity.13,14 Mitochondria asymmetrically localize in cells such as neurons17 or at the pseudopodial front of migrating cells16, but to our knowledge they have never been observed to asymmetrically segregate during mitosis in HSCs, although this has been observed in some lower species.21 Mitochondria are also linked to the mitotic spindle in some cells.18,19
HSCs and other stem cells have very low numbers/activity of mitochondria, a property used to advantage in purifying HSCs.35 ESC upregulate mitochondrial biogenesis as they differentiate, and suppression of this can influence differentiation programming.6,22 Upregulation of mitochondrial biogenesis is linked to the switch from glycolytic metabolism in ESC to OX-PHOS metabolism in embryoid body cells and differentiated cells, and is believed to be necessary for increased energy demands of differentiation and expansion.10 Our data presents evidence that mitochondria are upregulated during the early stages of HSC differentiation (i.e. CD34 expression and the known loss of long-term repopulation29), consistent with observations in ESC.
Because increased mitochondrial OX-PHOS subjects cells to increased risk of oxidative damage, primarily in the form of ROS, cells have several means to lessen these risks.5,8 One strategy for HSCs, may be to reside in a hypoxic niche, which appears to be located at/near the endosteum in trabecular bones in mammals.5 Another strategy is to maintain low numbers of mitochondria.35 However, the required upregulation of active mitochondria during differentiation presents an additional challenge to HSC to maintain themselves without subjecting the self-renewing stem cell to oxidative damage. Considering that mitochondria may be a cell-fate determinant, we hypothesize, but is yet to be proven, that asymmetric segregation of mitochondria could also serve the purpose of reducing risk of oxidative stress to HSCs during self-renewal and differentiation. Because mitochondria cannot be generated de-novo, at least one mitochondrion must be inherited in each daughter cell after mitosis. Therefore, we considered two models of asymmetric stem cell division (illustrated in Figure 6) where the mother stem cell is minimally exposed to oxidative stress during upregulation of mitochondrial biogenesis required for differentiation. Model 1 depicts how asymmetric segregation of mitochondria during an asymmetric self-renewing HSC division limits exposure of the resulting pluripotent HSC daughters to oxidative risk. Model 2 suggests how symmetric mitochondrial segregation during an asymmetric self-renewing stem cell division could protect the HSC daughter from increased oxidative risk.
Figure 6. Two potential models of HSC mitochondrial upregulation without exposure to increased oxidative risk.
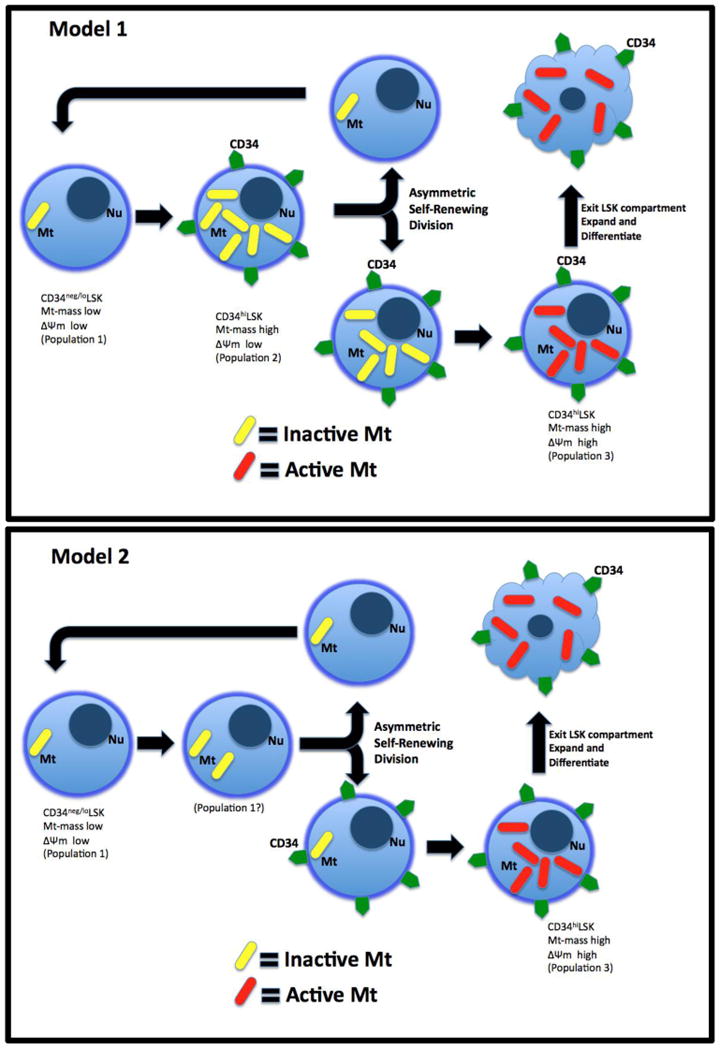
Shown are two different models of mitochondrial upregulation during HSC self-renewing cell divisions. Model 1 represents asymmetric co-segregation of CD34 and mitochondria and model 2 represents symmetric mitochondrial segregation and asymmetric CD34 segregation. Both models produce self-renewed HSC daughter cells that have had no exposure to active mitochondria.
Our data show a highly associated link between upregulation of mitochondrial biogenesis and CD34 expression, which is more consistent with model 1. We found no evidence of a CD34+ LSK cell population with low Mt-mass, which is a key part of model 2. Our study presents evidence supporting the existence of a CD34+ LSK cell population with high Mt-mass, but with low ΔΨm (population 2; Fig 4A). Therefore, we believe Model 2 is a more likely candidate strategy for HSC to undergo an asymmetric self-renewing cell division that limits exposure of the new (replacement) HSC daughter cell to oxidative risk while still allowing mitochondrial upregulation necessary for further differentiation of progenitor cells. How HSC could increase Mt mass without simultaneous activation is unknown. Our finding of an adult stem cell with large Mt-mass and low activity is similar to recent observations in the MRL mouse, which was shown to have adult tissues with increased numbers of inactive mitochondria and a large metabolic reserve.36 It was suggested that MRL mouse cells have a metabolic programming more similar to embryonic cells than normal somatic cells. However, the relationship between our observations and those in the MRL mouse are unclear at this time.
Numbers and proportions of the newly described mitochondrialy-distinct LSK cells in steady-state BM can be influenced by global perturbations in the hematopoietic system as demonstrated in our SDF-1 transgene expression model. These effects of SDF-1 transgene expression appear to be quantitative in nature rather than by induction of the appearance of “new/abnormal” subpopulations, or disappearance of the ones we have described. Our results suggest that constitutive hyperactivation of the SDF-1 receptor, CXCR4, results in a generalized “pressure” for HSC to move out of the CD34low LSK compartment and into the CD34high LSK compartment, a trait consistent with increased numbers of actively cycling progenitors.23 The known effects of SDF-1 on stem cell homing/mobilization25 could account for this shift, but there also appears to be an unusually high accumulation of CD34hi LSK cells with increased Mt-mass and low ΔΨm in the BM of these mice (not unlike the MRL mouse). CXCR4 activation inhibits PGK-1, a fundamentally important enzyme for glycolysis, and CXCR4 also suppresses glycolysis.27 In addition, mitochondrial biogenesis regulators, PPARs, downregulate CXCR4 expression.28 PPARs also inhibit the PDK enzyme complex, which in turn inhibits PDHs, the enzymes required for converting pyruvate, the end product of glycolysis, into acetyl-CoA, the substrate for the Krebs cycle and mitochondrial OX-PHOS. We speculate that the CXCR4/PPAR/PGK-1/PDH pathways may ultimately converge to suppress conversion of pyruvate to Acetyl-CoA in constitutively active CXCR4 mutant mouse HSCs, thus depriving their mitochondria of their substrate, keeping their activity low even when mitochondrial mass is elevated. This notion may also explain why CD34hi LSK cells accumulate and do not efficiently continue differentiation because without active mitochondria, energy needs of differentiating cells are not met.
Acknowledgments
This work was supported by grants to HEB from the National Institutes of Health; R01 HL56416 and HL67384, and a Project in PO1 HL53586.
References
- 1.Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 2.Duranteau J, Chandel NS, Kulisz A, Shao Z, Schumacker PT. Intracellular signaling by reactive oxygen species during hypoxia in cardiomyocytes. J Biol Chem. 1998;273:11619–24. doi: 10.1074/jbc.273.19.11619. [DOI] [PubMed] [Google Scholar]
- 3.Nemoto S, Takeda K, Yu ZX, Ferrans VJ, Finkel T. Role for mitochondrial oxidants as regulators of cellular metabolism. Molecular and Cellular Biology. 2000;20:7311–8. doi: 10.1128/mcb.20.19.7311-7318.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–95. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 5.Eliasson P, Jönsson JI. The hematopoietic stem cell niche: low in oxygen but a nice place to be. J Cell Physiol. 2010;222:17–22. doi: 10.1002/jcp.21908. [DOI] [PubMed] [Google Scholar]
- 6.Parker GC, Acsadi G, Brenner CA. Mitochondria: determinants of stem cell fate? Stem Cells and Development. 2009;18:803–6. doi: 10.1089/scd.2009.1806.edi. [DOI] [PubMed] [Google Scholar]
- 7.Schieke SM, Ma M, Cao L, Mccoy JP, Liu C, Hensel NF, et al. Mitochondrial Metabolism Modulates Differentiation and Teratoma Formation Capacity in Mouse Embryonic Stem Cells. Journal of Biological Chemistry. 2008;283:28506–12. doi: 10.1074/jbc.M802763200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Han MK, Song EK, Guo Y, Ou X, Mantel C, Broxmeyer HE. SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell. 2008;2:241–51. doi: 10.1016/j.stem.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jang YY, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood. 2007;110:3056–63. doi: 10.1182/blood-2007-05-087759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Facucho-Oliveira JM, Alderson J, Spikings EC, Egginton S, St John JC. Mitochondrial DNA replication during differentiation of murine embryonic stem cells. Journal of Cell Science. 2007;120:4025–34. doi: 10.1242/jcs.016972. [DOI] [PubMed] [Google Scholar]
- 11.Hsieh MCF, Das D, Sambandam N, Zhang MQ, Nahlé Z. Regulation of the PDK4 isozyme by the Rb-E2F1 complex. J Biol Chem. 2008;283:27410–7. doi: 10.1074/jbc.M802418200. [DOI] [PubMed] [Google Scholar]
- 12.Ferecatu I, Le Floch N, Bergeaud M, Rodriguez-Enfedaque A, Rincheval V, Oliver L, et al. Evidence for a mitochondrial localization of the retinoblastoma protein. BMC Cell Biol. 2009;10:50. doi: 10.1186/1471-2121-10-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–6. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reich NC. STAT3 revs up the powerhouse. Science signaling. 2009;2:pe61. doi: 10.1126/scisignal.290pe61. [DOI] [PubMed] [Google Scholar]
- 15.Mantel C, Broxmeyer HE. Sirtuin 1, stem cells, aging, and stem cell aging. Current Opinion in Hematology. 2008;15:326–31. doi: 10.1097/MOH.0b013e3283043819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hales KG. The machinery of mitochondrial fusion, division, and distribution, and emerging connections to apoptosis. Mitochondrion. 2004;4:285–308. doi: 10.1016/j.mito.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 17.Macaskill AF, Kittler JT. Control of mitochondrial transport and localization in neurons. Trends Cell Biol. 2009 doi: 10.1016/j.tcb.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Krüger N, Tolić-Nørrelykke IM. Association of mitochondria with spindle poles facilitates spindle alignment. Curr Biol. 2008;18:R646–R7. doi: 10.1016/j.cub.2008.06.069. [DOI] [PubMed] [Google Scholar]
- 19.Dinkelmann MV, Zhang H, Skop AR, White JG. SPD-3 is required for spindle alignment in Caenorhabditis elegans embryos and localizes to mitochondria. Genetics. 2007;177:1609–20. doi: 10.1534/genetics.107.078386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boldogh IR, Pon LA. Mitochondria on the move. Trends Cell Biol. 2007;17:502–10. doi: 10.1016/j.tcb.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 21.Staiber W. Asymmetric distribution of mitochondria and of spindle microtubules in opposite directions in differential mitosis of germ line cells in Acricotopus. Cell Tissue Res. 2007;329:197–203. doi: 10.1007/s00441-007-0400-z. [DOI] [PubMed] [Google Scholar]
- 22.Facucho-Oliveira JM, St John JC. The relationship between pluripotency and mitochondrial DNA proliferation during early embryo development and embryonic stem cell differentiation. Stem Cell Rev Rep. 2009;5:140–58. doi: 10.1007/s12015-009-9058-0. [DOI] [PubMed] [Google Scholar]
- 23.Broxmeyer HE, Cooper S, Kohli L, Hangoc G, Lee Y, Mantel C, et al. Transgenic expression of stromal cell-derived factor-1/CXC chemokine ligand 12 enhances myeloid progenitor cell survival/antiapoptosis in vitro in response to growth factor withdrawal and enhances myelopoiesis in vivo. J Immunol. 2003;170:421–9. doi: 10.4049/jimmunol.170.1.421. [DOI] [PubMed] [Google Scholar]
- 24.Christopherson KW, Hangoc G, Mantel CR, Broxmeyer HE. Modulation of hematopoietic stem cell homing and engraftment by CD26. Science. 2004;305:1000–3. doi: 10.1126/science.1097071. [DOI] [PubMed] [Google Scholar]
- 25.Broxmeyer HE. Chemokines in hematopoiesis. Current Opinion in Hematology. 2008;15:49–58. doi: 10.1097/MOH.0b013e3282f29012. [DOI] [PubMed] [Google Scholar]
- 26.Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A, et al. Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp Med. 2003;198:1391–402. doi: 10.1084/jem.20030267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, Wang J, Dai J, Jung Y, Wei CL, Wang Y, et al. A glycolytic mechanism regulating an angiogenic switch in prostate cancer. Cancer Research. 2007;67:149–59. doi: 10.1158/0008-5472.CAN-06-2971. [DOI] [PubMed] [Google Scholar]
- 28.Richard CL, Blay J. CXCR4 in Cancer and Its Regulation by PPARgamma. PPAR research. 2008;2008:769413. doi: 10.1155/2008/769413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blank U, Karlsson G, Karlsson S. Signaling pathways governing stem-cell fate. Blood. 2008;111:492–503. doi: 10.1182/blood-2007-07-075168. [DOI] [PubMed] [Google Scholar]
- 30.Reers M, Smith TW, Chen LB. J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry. 1991;30:4480–6. doi: 10.1021/bi00232a015. [DOI] [PubMed] [Google Scholar]
- 31.Mantel C, Broxmeyer HE. A new connection between the spindle checkpoint, asymmetric cell division and cytokine signaling. Cell Cycle. 2007;6:144–6. doi: 10.4161/cc.6.2.3791. [DOI] [PubMed] [Google Scholar]
- 32.Siller KH, Doe CQ. Spindle orientation during asymmetric cell division. Nat Cell Biol. 2009;11:365–74. doi: 10.1038/ncb0409-365. [DOI] [PubMed] [Google Scholar]
- 33.Shaheen M, Broxmeyer HE. The humoral regulation of hematopoiesis. In: Hoffman R, Benz EJ, Shatil SJ, Furie B, Silberstein LE, McGlave P, Helsop H, Anastasi J, editors. Hematology: Basic Principles and Practices. 5th. Philadelphia, PA: Elsevier Churchhill Livingston; 2009. pp. 253–275. [Google Scholar]
- 34.Scherz-Shouval R, Elazar Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007;17:422–7. doi: 10.1016/j.tcb.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 35.Mckenzie JL, Takenaka K, Gan OI, Doedens M, Dick JE. Low rhodamine 123 retention identifies long-term human hematopoietic stem cells within the Lin-CD34+CD38- population. Blood. 2007;109:543–5. doi: 10.1182/blood-2006-06-030270. [DOI] [PubMed] [Google Scholar]
- 36.Naviaux RK, Le TP, Bedelbaeva K, Leferovich J, Gourevitch D, Sachadyn P, et al. Retained features of embryonic metabolism in the adult MRL mouse. Mol Genet Metab. 2009;96:133–44. doi: 10.1016/j.ymgme.2008.11.164. [DOI] [PMC free article] [PubMed] [Google Scholar]