

SUMMARY
The SMK box riboswitch, which represents one of three known classes of S-adenosylmethionine (SAM)-responsive riboswitches, regulates gene expression in bacteria at the level of translation initiation. In contrast to most riboswitches, which contain separate domains responsible for ligand recognition and gene regulation, the ligand-binding and regulatory domains of the SMK box riboswitch are coincident. This property was exploited to allow the first atomic-level characterization of a functionally intact riboswitch in both the ligand-bound and ligand-free states. NMR spectroscopy revealed distinct mutually exclusive RNA conformations that are differentially populated in the presence or absence of the effector metabolite. Isothermal titration calorimetry and in vivo reporter assay results revealed the thermodynamic and functional consequences of this conformational equilibrium. We present a comprehensive model of the structural, thermodynamic, and functional properties of this compact RNA regulatory element.
Keywords: RNA, NMR spectroscopy, isothermal titration calorimetry, gene regulation, pre-existing equilibrium, riboswitch
INTRODUCTION
Riboswitches are regulatory RNA elements that are usually located in the 5’-untranslated regions (5’-UTRs) of certain mRNAs. These elements respond directly to environmental signals to regulate expression of cis-encoded genes without the requirement for additional regulatory factors or proteins1; 2; 3. Riboswitches are classified based on the environmental signal that they recognize, as well as conserved sequence and structural elements involved in signal recognition. Most riboswitches discovered to date regulate genes involved in metabolism and transport of vitamins, amino acids, nucleic acids, enzymatic cofactors, and metal ions2. In many instances, the ligand recognized by an individual riboswitch is a metabolic end-product or intermediate in the pathway regulated by that riboswitch. While most known riboswitches have been identified in bacteria, one class of riboswitch, the THI box, has been found in all three domains of life4; 5, demonstrating the prevalence of these regulatory RNAs.
Signal recognition by riboswitch RNAs typically results in a structural rearrangement that alters the efficiency of expression of an adjacent open reading frame (ORF). The two most common riboswitch mechanisms involve regulation at the level of premature termination of transcription or translation initiation. Riboswitches that affect transcription termination undergo a conformational switch that either stabilizes or destabilizes the helix of an intrinsic transcription terminator. Translational regulation utilizes a conformational change that affects accessibility of the Shine-Dalgarno (SD) sequence, which is the binding site for the 30S ribosomal subunit. In each of these mechanisms, the riboswitch element is predicted to favor one of two mutually exclusive alternate conformations depending on the presence or absence of the specific effector molecule or physiological signal to which it responds.
The SMK box riboswitch is one of three known riboswitch classes (S box/SAM-I, SAM-II, and SMK box/SAM-III) that bind the enzymatic cofactor S-adenosylmethionine (SAM)6; 7; 8. Each of the SAM-binding riboswitches forms a unique ligand binding pocket (characterized by conserved sequence elements and structural features) that specifically accommodates SAM9; 10; 11. All known SMK box elements regulate expression of the metK gene, which encodes SAM synthetase, the enzyme responsible for the production of SAM from adenosine and methionine7; 12. This riboswitch element regulates metK expression at the level of translation initiation through sequestration of a portion of the SD sequence by pairing with an upstream anti-Shine Dalgarno (ASD) sequence (Figure 1, Supplemental Figure 1a, b). The ASD-SD interaction is stabilized in the presence of SAM, resulting in SAM-dependent inhibition of binding of the ribosome12.
Figure 1.
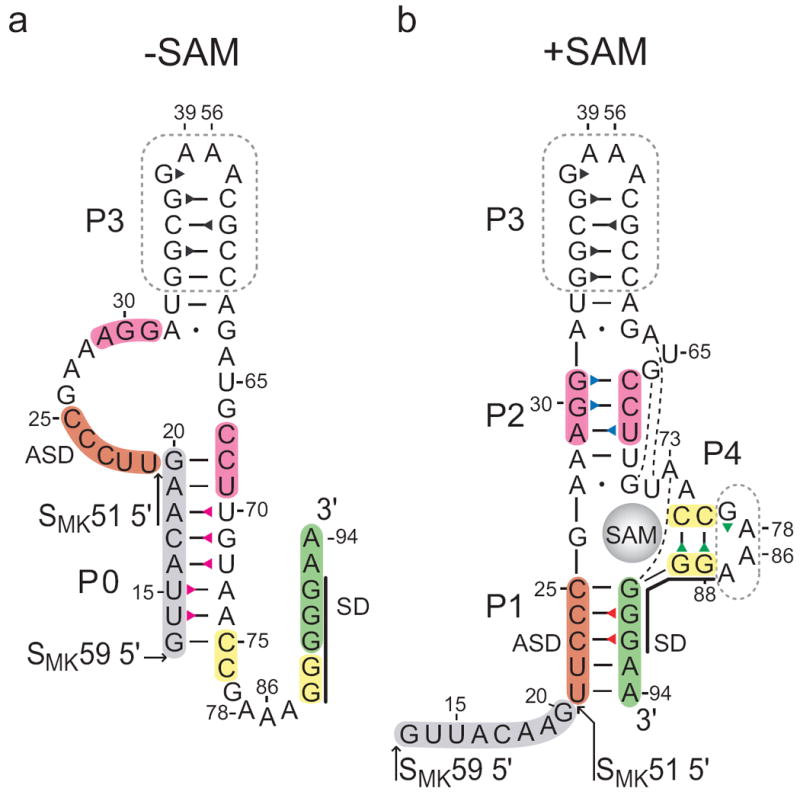
Secondary structure of the SMK box riboswitch. Previously proposed SAM-free (a) and SAM-bound (b) secondary structure models of the SMK box, represented as the SMK59 construct used in this study. The SMK51 construct lacks the eight 5’ nucleotides (grey). The boxed regions were truncated from hypervariable regions in the naturally-occurring E. faecalis metK sequence. Numbering is based on the E. faecalis metK sequence and is discontinuous at the engineered tetraloops. The Shine-Dalgarno (SD) sequence is marked with a black line. Base pairs observed in the crystal structure10 are marked: dash, Watson-Crick; dot, non-canonical; broken line, extrahelical. Triangles represent assigned imino protons and are colored corresponding to the labels in Figures 2 and 3.
While most riboswitch RNAs have a modular architecture composed of two separate domains (i.e., an aptamer domain for signal recognition and a regulatory domain), the SMK box riboswitch has a simpler architecture that utilizes a single domain for both ligand binding and gene regulation10; 12. This compact design facilitates high-resolution structural investigations using the complete riboswitch element. The crystal structure of a 53-nt minimal SMK box element (based on the Enterococcus faecalis metK sequence) in complex with SAM revealed that the RNA forms a Y-shaped arrangement with SAM located in the center of a three-way junction at the intersection of helices P1, P2, and P4 (Figure 1b, Supplemental Figure 2)10. Pairing interactions in each of these helical domains is required for SAM binding, and formation of helices P1 and P4 results in sequestration of residues 88-92 that constitute the metK SD sequence, providing a structural basis for SAM-dependent translational repression. These results verified the prediction that the SMK box riboswitch assumes a single-domain architecture and demonstrated that the SD sequence is directly involved in SAM binding through base-specific contacts.
Less evidence is available about the ligand-free conformation of the SMK box riboswitch. It was predicted based on phylogenetic analysis that helices P1, P2, and P4 are absent and that another helix (referred to here as P0) is formed in the absence of SAM (Figure 1a)7. Helix P0 is predicted to comprise residues 13-20 and residues 68-75 (relative to the predicted E. faecalis metK transcription start-site), which include nucleotides that overlap helices P2 and P4 in the ligand-bound conformation. Therefore, the two conformations (ligand-bound and unbound) are mutually exclusive and stabilization of helices P2 and P4 upon SAM binding is predicted to disrupt helix P0. Results from a variety of experiments (e.g., enzymatic probing, RNase H cleavage assays, and 30S ribosomal toeprinting) provide evidence that the RNA assumes an alternate conformation in the absence of ligand and exhibits secondary structural features consistent with the model7; 12.
In the current study we used NMR spectroscopy to investigate pairing interactions of the SMK box riboswitch in the presence and absence of SAM. We show that a 51-nt minimal construct (SMK51; Figure 1) lacking residues 13-20 at the 5’ end of the RNA favors a conformation resembling the ligand-bound state even in the absence of SAM. Inclusion of these eight additional 5’ nucleotides to generate a 59-nt construct (SMK59; Figure 1) resulted in an RNA capable of assuming two alternate conformations depending on the presence of SAM. These results validate the predicted secondary structural features of the SMK box riboswitch in each conformational state and demonstrate that residues at the 5’ end of the element (which are not required for ligand binding) are essential for stabilization of the ligand-free conformation. Moreover, we detail the thermodynamic and functional implications of the ability of the riboswitch to adopt two distinct and mutually exclusive structures by isothermal titration calorimetry (ITC) and in vivo reporter assays. Taken together, our results provide new insights into the interplay between structure and function in ligand-sensing regulatory RNA elements.
RESULTS
NMR spectroscopy of SAM-bound SMK51 and SMK59
SMK51, the initial RNA construct used for NMR studies (Figure 1), consists of 51 nucleotides equivalent to the minimal aptamer sequence used to determine the crystal structure of the SAM-bound SMK box (Supplemental Figure 2)10. Because of its known secondary and tertiary structure, the SAM-bound state of SMK51 served as a starting point for interpretation of NMR spectra. NMR structural characterization of the two SMK box riboswitch constructs is based primarily on the detection of exchangeable imino proton resonances of the 51- or 59-nucleotide RNAs. These resonances are direct reporters of base pairing interactions since the signals can be observed only if the protons are protected from solvent, while shifts in peak position reflect changes in the local chemical environment, including (but not limited to) conformational changes. The SMK51 RNA binds SAM more tightly than does the 106 nt wild-type sequence (corresponding to residues 15-118, see Supplemental Figure 1) in a size-exclusion filtration assay (Supplemental Figure 3), indicating that it is fully active in SAM binding.
The one-dimensional proton spectrum of SMK51 in the presence of a 10% stoichiometric excess of SAM reveals fourteen imino resonances (Figure 2a). The two overlapped signals upfield of 11 ppm can be assigned to the two guanines in tetraloops that cap helices P3 and P4 because these iminos have diagnostic chemical shifts13; 14. The remaining twelve signals arise from imino protons participating in helical base pairs. Analysis of the SAM-bound crystal structure suggests that 21 imino protons participate in base pairs (none of which are to the SAM ligand). Comparison to NMR data indicates that seven of the crystallographically-observed base pairs do not yield observable imino resonances, which is likely to be due to local “breathing” motions in the RNA that deprotect the protons from solvent or otherwise lead to resonance broadening.
Figure 2.
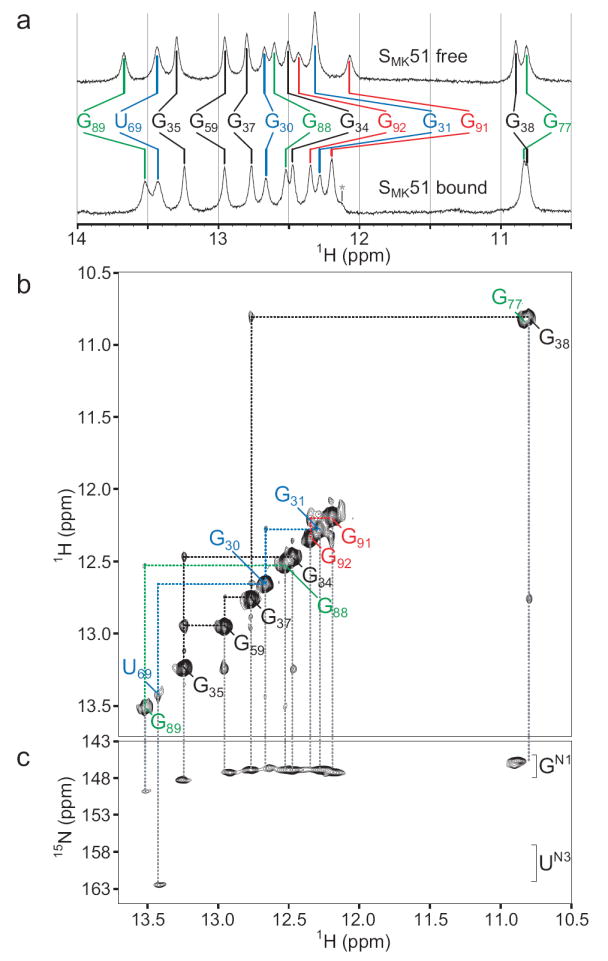
NMR spectra of SMK51 at 4°C reveal a folded aptamer domain. (a) The imino region of 1D 1H spectra of SAM-free (top) or SAM-bound (bottom) SMK51. The number of observed resonances indicates the number of imino protons protected from solvent exchange (e.g., via base pairing) while changes in peak position between states reflect a difference in the chemical environment of a given proton. An unassigned resonance is marked by an asterisk. (b) Imino region of the 1H-1H NOESY spectrum of SAM-bound SMK51. Signals along the diagonal are analogous to those observed in the 1D spectra. Off-diagonal cross peaks reflect through-space magnetization transfer between two protons within ~5 Å of each other. In the context of regular nucleic acid structure, protons whose signals are serially connected by cross peaks belong to a continuous region of secondary structure. NOE spin networks of helical regions are labeled: P1, red; P2, blue; P3, black; P4, green (represented as color-coded triangles in Figure 1b). (c) 1H-15N HSQC spectrum of SAM-bound SMK59, whose imino spectra are near-identical to those of SAM-bound SMK51. The 15N chemical shift allows assignment of nucleotide type (G or U).
SMK51 lacks eight nucleotides at the 5’ end (Figure 1) hypothesized to participate in formation of helix P0 in the absence of SAM; this is supported by the observation that a P0-containing conformation is not adopted by ligand-free SMK51 (see below). A second construct, SMK59, contains these additional eight nucleotides and was therefore predicted to be able to form the P0 helix in the absence of SAM. No additional signals were expected in the imino region of SAM-bound SMK59 since the additional nucleotides unique to this RNA are predicted to be single-stranded and deprotected from solvent in this state. Accordingly, spectra of SAM-bound SMK59 are nearly identical to those of SAM-bound SMK51 (Figure 3a, top and middle spectra). Four spin networks in SMK51 and SMK59 could be confidently assigned to bases in helices P1, P2, P3, and P4 using NOE spectroscopy (Figure 2, see Supplemental Results). These networks correspond to the four helical regions observed in the crystal structure.
Figure 3.
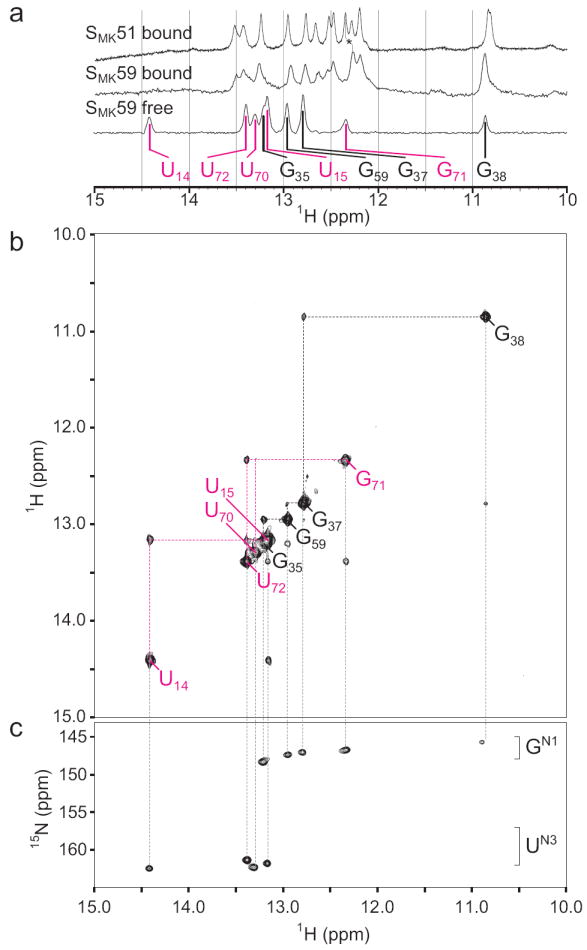
NMR spectra of SMK59 at 4°C reveal a novel fold in the absence of ligand. (a) The imino region of 1D 1H spectra of SAM-bound (top) SMK51, SAM-bound SMK59 (middle), or SAM-free SMK59 (bottom). Similarity of chemical shifts indicates near-identical structures for the SAM-bound constructs. A resonance shifted between the two bound states was used for assignment of G92 and is marked with an asterisk. The helix P3 spin network is labeled in black as in Figure 2; signals from the helix P0 are labeled in magenta (represented as color-coded triangles in Figure 1a). (b) Imino region of the 1H-1H NOESY spectrum of SAM-free SMK59. The helix P3 spin network is largely unperturbed compared to the SAM-bound state, but a novel spin network corresponding to helix P0 is detected. (c) 1H-15N HSQC spectrum of SAM-free SMK59 allows assignment of nucleotide type.
NMR spectroscopy of SAM-free SMK51
NMR spectra of ligand-free SMK51 were recorded to investigate the structure of this RNA in the absence of SAM. At 4°C, NMR spectra of SAM-free SMK51 are similar to those of the SAM-bound RNA (Figure 2, Supplemental Figure 4). This similarity suggests that the global structure is unperturbed, while minor chemical shift perturbations reflect the altered chemical environment near the binding pocket due to the absence of ligand. In contrast, the NOESY spectrum of SAM-free SMK51 recorded at 25°C shows extensive loss of imino proton signals compared to that of SAM-bound SMK51 at the same temperature (Supplemental Figure 5). Under these conditions, spin networks from helices P1 and P4 in SAM-free SMK51 were undetected while helices P2 and P3 are largely intact. This observation reflects the reduced stability in the absence of SAM of the ligand-binding region of an otherwise unperturbed SMK51.
NMR spectroscopy of SAM-free SMK59
In contrast to SMK51, the 1H-1H NOESY spectrum of SAM-free SMK59 reports the absence of all structural elements detected in the SAM-bound state with the exception of helix P3, and the appearance of a new spin network containing five base pairs (Figure 3b). Although the P3 helix resonances are subject to slight chemical shift perturbations, the data indicate that this helix remains intact with the exception of the imino signal from G34, which is not observed. This suggests increased breathing in SAM-free SMK59 brought about by unfolding of the P2 helix, whose resonances are not observed. The new NOE spin network was revealed to contain a U·U·U·G·U pattern via a 1H-15N correlated spectrum (Figure 3c), and thus this network was assigned to helix P0: residues U14·U15·U72·G71·U70.
A two-dimensional 1H-15N correlated spectrum was used to confirm that the assigned P0 iminos participate in canonical Watson-Crick base pairs (Supplemental Figure 6). In light of weak diagnostic G71·U70 cross peaks, 1H-1H NOESY spectra of model helices were used to verify assignment of nucleotides in helix P0 (Supplemental Figure 7). Taken together, these data reveal that the SMK59 RNA adopts a unique fold in the absence of SAM; three of the helices (P1, P2, P4) found in the SAM-bound structure are absent and a new helix (P0) is formed.
Isothermal titration calorimetry
ITC was used to measure the binding affinity of the two SMK box constructs for SAM at 15°C. The ligand was titrated into either SMK51 or SMK59 RNA (Figure 4a, b). Data were fit to a single-site binding model, yielding the following values: SMK51, ΔH = -18.7 ± 0.2 kcal/mol, KD = 63 ± 11 nM, ΔS = -32.0 ± 0.8 cal K-1 mol-1 (Figure 4a); SMK59, ΔH = -10.7 ± 0.3 kcal/mol, KD = 400 nM ± 78 nM, ΔS = -7.9 ± 1.1 cal K-1 mol-1 (Figure 4b). Relative to SMK59, SMK51 exhibits a ~6-fold higher affinity for SAM, consistent with the observation that the SMK51 RNA binds SAM more efficiently than does the full-length E. faecalis metK sequence in filtration experiments (Supplemental Figure 3).
Figure 4.
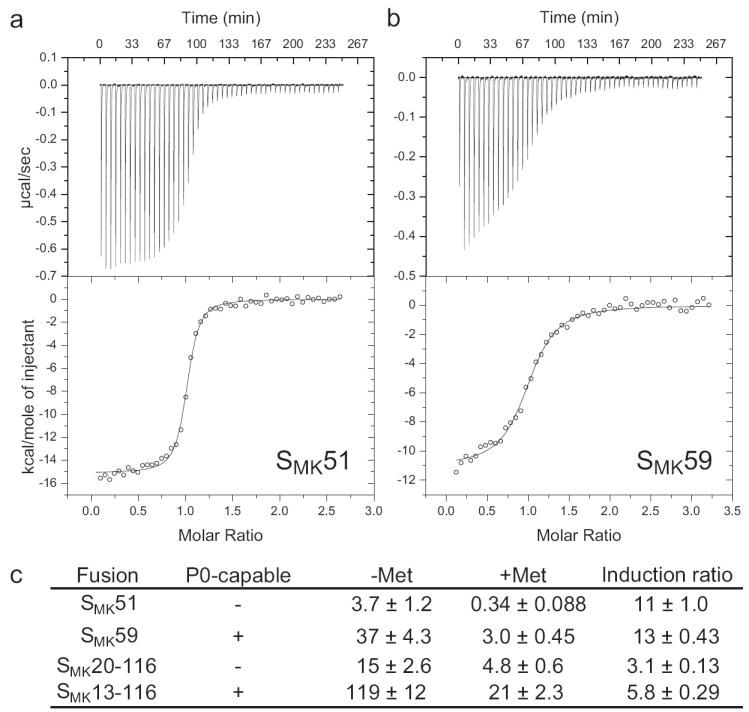
Isothermal titration calorimetry and in vivo assays with the SMK box. (a) Thermogram generated upon titration of SAM into SMK51. Fitting to a one-site model reveals a ΔH of -18.7 ± 0.2 kcal/mol and a KD of 63 ± 11 nM. (b) Analogous data for SMK59, with a ΔH of -10.7 ± 0.3 kcal/mol and a KD of 400 ± 78 nM. (c) Expression of Pgly-metK-lacZ fusions in B. subtilis during growth under high SAM (+Met) or low SAM (Met) conditions. β-galactosidase activities are expressed in Miller units. Induction ratio is the β-galactosidase activity in cells grown in the absence of methionine divided by the activity in cells grown in the presence of methionine.
Because the ligand-bound states of the SMK51 and SMK59 constructs contain similar, spectroscopically indistinguishable binding sites (Figure 3a), we attribute the observed differences in SAM-binding affinity of SMK51 and SMK59 primarily to differences in the relative stabilities of their unliganded states. The NMR data report that in the absence of ligand, SMK51 remains in a state similar to that adopted in the presence of SAM; this state, designated PRIMEDSMK, may represent a structural intermediate in which the RNA is primed for SAM binding. In contrast, the alternative structure adopted by SMK59 in the absence of SAM, designated ISOSMK, must undergo conformational isomerization in order to bind the ligand.
Because the secondary structure of the ISOSMK state is incompatible with RNA-SAM contacts in the BOUNDSMK state, we judged the isomerization between ISOSMK and BOUNDSMK states of SMK59 as most likely to occur through conformational sampling via a pre-existing equilibrium15. If SAM can bind only to one of the two equilibrating SMK59 states (i.e., mandatory coupling)16, the weaker effective affinity, Keff, of SMK59 can be quantitatively linked to the thermodynamic equilibrium, KISO, between the isomerizing states:
| (1) |
| (2) |
where ISOSMK is the P0-containing state observed in SAM-free SMK59 (Figure 6a, left), PRIMEDSMK is the binding-ready unliganded state observed in SAM-free SMK51 (Figure 6a, center), and BOUNDSMK is the SAM-bound state observed in both constructs (Figure 6a, right). KA in equations (1, 2) is the affinity of the primed structure for SAM, and is likely to be approximated well by the affinity of SMK51 for SAM. Keff in equation (2) is the effective affinity when the free state is subject to conformational isomerization (i.e., KISO > 0); this is approximated by the affinity of SMK59 for SAM. Via this analysis, we find that KISO is ~5.3, which implies that under the conditions sampled, ~80% of SAM-free SMK59 exists in the ISOSMK conformation while the remaining ~20% adopts an ensemble of conformations approximating the primed-like species. Simulated ITC data are consistent with this interpretation (Supplemental Figure 8, see also Supplemental Results). Thus the conformational equilibrium present in the switching-competent SMK59 construct can be seen to have the following thermodynamic consequences compared to the switching-incompetent SMK51 construct: (1) Populating the competing structure in ligand-free state necessarily decreases the net affinity of the RNA for its ligand. (2) The binding enthalpy gain is reduced at least in part by the cost of breaking hydrogen bonds in helix P0. (3) The entropic penalty for binding is reduced by the fewer degrees of freedom lost in rearranging the structured RNA (SMK59) compared to ordering the disordered one (SMK51).
Figure 6.
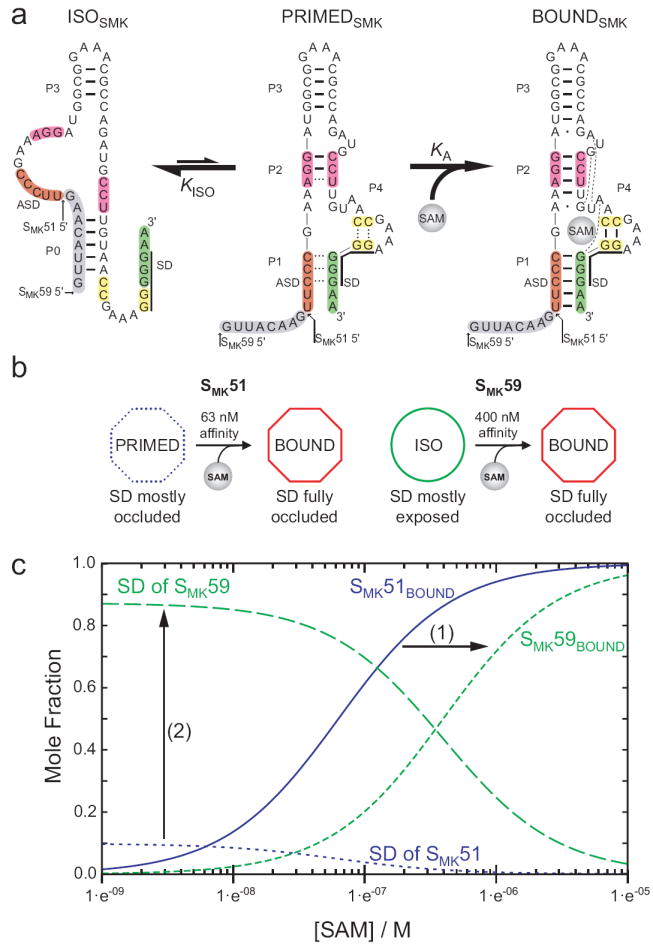
Model of SMK box structural transitions in response to SAM. (a) Observed conformations of SMK box. Left, the free state observed in SAM-free SMK59 via NMR, with accessible SD sequence. Center, the poorly-folded state observed via NMR in SAM-free SMK51 (which is missing the eight 5’ nucleotides shown here in grey), with a partially-accessible SD sequence. Right, the well-folded SAM-bound state with occluded SD sequence, observed in ligand-bound SMK51 and SMK59 and the previously-determined crystal structure. Hydrogen bonds are represented as lines as observed via NMR for ISOSMK and PRIMEDSMK, while hydrogen bonds expected based on the crystal structure10 are shown for BOUNDSMK. Dashed lines in the PRIMEDSMK state represent transiently formed base pairs as detected via NMR. (b) A schematic representing the transitions observed in ITC for SMK51 or SMK59 and how the analogous constructs behave while undergoing similar transitions inside the cell, as measured by β-galactosidase reporter assays. (c) Effect of the alternatively folded conformation on the populations of riboswitch conformations. SAM dependence of molecular species are simulated from equations (2, 3) describing the fractional populations of the SAM-bound species (SMK51BOUND, and SMK59BOUND) and of the effective concentration of exposed SD sequence for each construct. Curves were generated with the experimentally determined values of KA and KISO, and using a relative accessibility of the SD in the PRIMEDSMK form, α, estimated to be 0.1. The equilibrium between the ISOSMK and PRIMEDSMK conformations (1) shifts the affinity of the riboswitch into the biologically-relevant micromolar range, and (2) amplifies the response of the switch to SAM by promoting greater SD accessibility at low [SAM].
In vivo riboswitch activity assays
To investigate the effect of deletion of the 5’ residues on SAM-dependent repression in vivo, four metK-lacZ translational fusion constructs incorporating varying SMK box sequences were compared. Two fusion constructs contained the SMK51 and SMK59 sequences and are named accordingly. Another pair of fusion constructs was based on the naturally-occurring E. faecalis SMK box; these constructs are named SMK13-116 and SMK20-116, respectively (Supplemental Figure 1). Fusion constructs SMK59 and SMK13-116 contain the residues required for formation of helix P0 (Figure 1a) while SMK51 and SMK20-116 do not. Fusions were integrated into the chromosome of B. subtilis strain BR151, a methionine auxotroph in which the intracellular SAM pools can be modulated via the presence of methionine in the growth media17. In this strain, a metK-lacZ fusion corresponding to residues 15-118 exhibits high expression when SAM pools are low, and ~5-fold repression when SAM levels are high7. Comparison of fusions corresponding to the wild-type E. faecalis metK sequence either containing (SMK13-116) or lacking (SMK20-116) the 5’ side of the P0 helix showed that while both were capable of SAM-dependent repression, the ability to form the P0 helix resulted in higher expression during growth in both high SAM and low SAM conditions (Figure 4c). A similar pattern was observed for the model SMK51 and SMK59 RNAs, which exhibited lower expression than the full-length E. faecalis metK constructs under both high and low SAM growth conditions, consistent with higher affinity for SAM. These results are consistent with the model that the P0 helix stabilizes the SAM-free form, and that absence of the helix facilitates formation of the SAM-bound conformation, resulting in decreased expression (Figure 4c, Figure 6).
Small-angle X-ray scattering
To investigate the overall shape of the RNA molecules in various states, SAXS data were recorded for SMK51 and SMK59 in the absence and presence of SAM. SMK51 was also examined in the absence of Mg2+ and SAM. Guinier analysis shows potential moderate aggregation of the SMK51 sample in the absence of SAM, while the other samples appear well-behaved (Supplemental Figure 9). Kratky plots were used to assess the relative foldedness of the various states. Increased folding is observed upon addition of 5 mM Mg2+ to SMK51, with continued folding in the presence of SAM (Figure 5a). Intramolecular distance pair distribution histograms (Figure 5c) reveal a similar but less compact structure for SMK51 in the absence of SAM compared to when the ligand is bound. These results are consistent with partial unfolding between the proposed BOUNDSMK and PRIMEDSMK states (Figure 6). Free and bound solution states of SMK51 have a greater radius of gyration than that estimated based on the crystal structure of the slightly larger 53-nucleotide SMK box RNA used for crystallization10, suggesting a less compact structure in solution. This is consistent with the observation that not all crystallographically-inferred hydrogen bonds give rise to observable imino proton resonances in NMR spectra.
Figure 5.
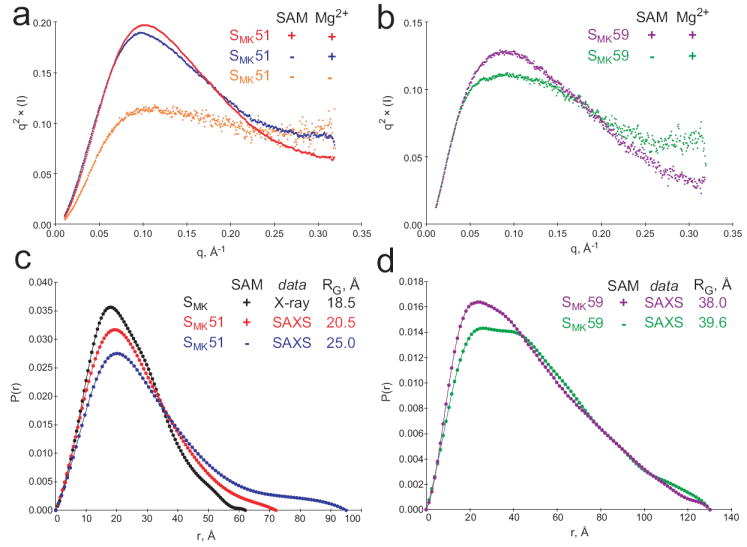
SAXS analysis of the SMK box in the presence and absence of ligand. (a) Kratky plots reveal increased folding of SMK51. Unfolded biopolymer is typified by a q2 × (I) plateau at high scattering angles (q > 0.2 Å-1), and this character decreases upon the addition of Mg2+ (blue) and SAM (red). (b) SAM-associated folding can also be observed in Kratky plots of SMK59 in the presence (purple) or absence (green) of SAM. (c) Intramolecular pair distribution functions for the SMK box. A simulated curve was generated for the previously-determined crystal structure of 53-nucleotide SMK box bound to SAM (black) and is compared to curves resulting from SAXS analysis of the 51-nucleotide SMK51 in solution (red, with SAM; blue, without SAM). Comparison of the radius of gyration (RG) suggests that the solution conformation is less compact than that adopted in the crystal. The binding of SAM is associated with a compaction of the solution structure of SMK51 based on the similar curve shapes and markedly different RG for free (25.01 Å, blue) versus bound (20.52 Å, red). (d) Comparison of SAXS-derived pair distribution curves for SMK59 with (purple) and without (green) SAM reveals a noteworthy change in the overall shape of the molecule. While the SAM-bound state generates a curve resembling those from the SAM-bound SMK51 species (c), the bimodal distribution in the absence of SAM is indicative of a two-lobed structure as might be expected for the ISOSMK state which is composed of two helical segments connected by a single-stranded region.
Kratky plots reveal that SMK59 also becomes more well-folded in the presence of SAM (Figure 5b). In contrast to SMK51, the intramolecular distance pair distribution histograms for SMK59 reveal a marked change in the structure of the RNA in response to SAM binding (Figure 5d). In the presence of SAM, the SMK59 histogram is roughly Gaussian with a maximum at ~20 Å, consistent with the roughly prolate spheroidal BOUNDSMK state (Figure 6). In the absence of SAM, the SMK59 pair distribution is bimodal with maxima at ~25 Å and ~40 Å; this is consistent with rearrangement of the RNA into the ISOSMK conformation, whose two helical regions (separated by poorly structured single-stranded RNA) would give rise to a bimodal distribution. Although we expect that the minor population of PRIMEDSMK molecules present in solution will also contribute to the net scattering profile, the data clearly indicate adoption of a significantly more extended conformation upon removal of SAM.
Using SAXS data, an ab initio three-dimensional reconstruction of SAM-bound SMK51 was generated. This model exhibits the same size and general features of the previously determined SMK box crystal structure and verifies that the global fold of SAM-bound SMK51 in solution resembles that observed in the crystal10 (Supplemental Figure 11).
DISCUSSION
The previously determined crystal structure of the SMK box bound to SAM10 (Supplemental Figure 2) reveals the structural details of ligand recognition, but does not address the conformational changes required for the RNA to fulfill its role in gene regulation. No high-resolution structural study to date has characterized a riboswitch containing the aptamer and regulatory domains in both the ligand-free and ligand-bound conformations, which have opposing influences on gene regulation and are mutually exclusive. Several NMR and crystallographic studies have compared the ligand-free and ligand-bound states of isolated aptamer domains that lack residues to promote competing structures, and in turn detected changes ranging from a minor rearrangement of the active site18; 19 to larger impacts on tertiary structure and dynamics20; 21; 22; 23; 24. Here we utilize two constructs to probe the role of competing conformations in riboswitch gene control: SMK51, which favors a bound-like conformation even in the absence of SAM and SMK59, which is capable of switching between regulatory states.
The non-switching construct SMK51 corresponds to the RNA whose crystal structure was previously solved in a ligand-bound state, and is shown via NMR spectroscopy to adopt a secondary structure in solution resembling that observed in the crystal10. The SAM-bound solution structure of SMK51 is consistent with the crystal structure based on imino proton assignments obtained using 2D 1H-1H NOESY spectroscopy (Figure 2b). Furthermore, an ab initio SAXS reconstruction verifies that the overall structure of SAM-bound SMK51 is extremely similar to that of the SMK box crystal structure (Supplemental Figure 11). However, fewer imino proton resonances were observed than might be expected from the crystallographically observed base pairing, implying dynamics that result in exchange with solvent and/or resonance broadening. Indeed, comparison of small-angle X-ray scattering (SAXS) data for SMK51 to scattering data simulated from coordinates of the SMK box crystal structure are consistent with a less compact structure in solution than in the lattice (Figure 5c). In the absence of ligand, the structure of this switching-incompetent RNA is largely unchanged at 4°C based on detection of the same NOE spin networks (Supplemental Figure 4a) and observation of only small chemical shift perturbations with respect to the bound state (Figure 2a), characteristic of local changes expected upon ligand removal. At higher temperatures (25°C), the RNA undergoes a loss of secondary structure in helices near the binding pocket (P1 and P4), with only helix P3 retaining bound-like structure (Supplemental Figure 5). The NMR data are supported by the SAXS-based observation of a less well-folded and less compact SMK51 in the absence of ligand (Figure 5a, c). These results are generally consistent with the range of findings from previous studies of riboswitch aptamer domain structure, from minor changes localized to the ligand-binding site18; 19 (observed at 4°C in SMK51) to a larger-scale loss of structure in the absence of ligand24; 25 (observed at 25°C in SMK51).
To investigate the conformational rearrangement at the crux of riboswitch function, we utilized the SMK59 construct, which contains eight 5’ nucleotides necessary for formation of helix P0, the paired region predicted from phylogenetic analysis to be formed in the SAM-free state (Figure 1a)7. Near-identical NMR spectra of ligand-bound states confirm that the conformation of the SMK59-SAM complex is equivalent to that of SMK51 (Figure 3a), as would be expected since the single-stranded nucleotides exclusive to SMK59 should not produce detectable imino proton signals. In contrast, spectra of ligand-free SMK59 reveal an extensive change in the RNA structure relative to the SAM-bound state (Figure 3b). Although helix P3 remains largely unperturbed, the remaining three imino spin networks near the SAM-binding pocket are replaced with a novel five-nucleotide spin network corresponding to helix P0 (Figure 3, Supplemental Figure 6). As with the SAM-bound state, the SAM-free state is less well-folded than would be predicted based on examination of the sequence; P0 could potentially comprise eight base pairs yet only five are detected via NMR. This reconfiguration upon ligand binding is corroborated by SAXS data revealing globally distinct conformations of SMK59 in the presence or absence of SAM (Figure 5d). SMK59 therefore is revealed to be a minimal riboswitch capable of adopting two mutually exclusive, biologically-relevant folds in response to its ligand.
Three states of the SMK riboswitch were observed via NMR: 1) a SAM-free “on” state where the SD sequence is exposed (ISOSMK, Figure 6a, left); 2) a potential intermediate state consisting of a poorly-folded structure resembling the SAM-bound state (PRIMEDSMK, Figure 6a, center); and 3) a SAM-bound “off” state that is well-folded and resembles the crystal structure (BOUNDSMK, Figure 6a, right). Consideration of these states is useful in interpreting thermodynamic data obtained via ITC and functional data from in vivo reporter assays.
The functional implications of the First, we note that the eight nucleotides that distinguish the switchable SMK59 from the switching-incompetent SMK51 shift the affinity of the aptamer for SAM from ~60 nM (SMK51) to ~400 nM (SMK59), closer to the physiologically-relevant micromolar concentrations of the metabolite under repressing conditions17; 26; 27 (Figure 4a). Second, the response of the riboswitch is determined by its affinity for SAM, and by the exposure of the SD sequence in the accessible conformations of the RNA. The NMR data presented here indicate that the SAM-free states of the SMK box riboswitch, ISOSMK and PRIMEDSMK, are not equivalent in terms of accessibility of their SD sequences (Figure 6a).
In order to better understand the role of ISOSMK in translation regulation, we considered the effect of the ISOSMK ⇄ PRIMEDSMK equilibrium on both SAM binding affinity and the concentration of exposed ribosome binding sites, [SD]free. If the SD sequence is fully exposed in ISOSMK and α represents the relative accessibility of the SD sequence in PRIMEDSMK, the SAM-dependence of the fractional exposure, f, of the SD sequence can be written as follows (see Supplemental Results):
| (3) |
In this model, the equilibrium between two SAM-free RNA conformations has two effects (Figure 6c): (1) the competing equilibrium defined by KISO decreases the effective affinity of the SAM aptamer; and (2) the increased exposure of the SD sequence in the ISO form results in a greater amplitude change in the translational regulatory signal than that achieved by simply destabilizing helix P1 and leaving the SD largely occluded. These expectations are consistent with in vivo reporter assays (Figure 4c) which show that despite its less robust ability to repress gene expression when SAM pools are high (SMK59, 3.0 Miller units; SMK51, 0.34 Miller units), the SMK59 construct is more responsive to reduction in SAM pools as its induction ratio (13-fold) is greater than that of the SMK51-like construct (11-fold) (Figure 6b). The differential between induction ratios is more pronounced when comparing the naturally-occurring SMK box sequence with its P0-incompetent partner (induction ratios of 5.8 and 3.1 for fusion constructs SMK13-116 and SMK20-116, respectively), underscoring the functional importance of an SD-exposing conformational isomer. Similar analyses have been performed with respect to the kinetics of ligand-binding aptamer RNAs28; 29, although those systems lack the context of a corresponding regulatory element and therefore differ from the present characterization of the SMK box.
Prior studies of ligand-free aptamer domains of other riboswitches have revealed a variety of structural responses, the most extreme of which parallel the structural changes we observed with the truncated SMK51, i.e., the absence of ligand results in destabilization of secondary structural elements. Our thermodynamic analysis and in vivo results demonstrate that such an RNA can still operate as a switch; however, the response of this system is inferior to a switch that populates two mutually exclusive folds (e.g., ISO and BOUND), which enables both greater gene expression in the absence of SAM and a larger range of SAM-dependent response that can be titrated to the physiologically relevant flux in SAM pools.
Taken together, these results provide insight into the fine-tuning implicit in the evolution of an effective, metabolite-responsive RNA element. For transcriptional riboswitches, the fate of the mRNA is kinetically determined during transcription and thus thermodynamics are of less importance1. In contrast, the SMK box is a translationally-controlled RNA element, which acts via a thermodynamic (i.e., reversible) mechanism. The half-life of the metK mRNA in vivo (~3 min) exceeds the half-life of the RNA-SAM complex in vitro (~8 sec), and switching between the bound and free forms can be observed in vitro on a physiologically relevant timescale27. This suggests that a single transcript may have multiple opportunities for binding and release of SAM, and therefore multiple opportunities to regulate gene expression, underscoring the significance of the two interconverting folds observed in the present study.
The SMK box is particularly amenable to structural study due to its compact nature with overlapping aptamer and regulatory domains. This property has enabled the first detailed insights into the interconverting structures unique to this switchable RNA molecule in both its ligand-bound and ligand-free states. Its interdependent structural and regulatory transitions nevertheless obey the same principles at work in larger, extended riboswitches, and the present mechanistic framework for interpreting the transitions inherent to riboswitch function (Figure 6c) should aid in understanding and engineering this class of regulatory RNAs.
METHODS
Sample preparation
Full details of RNA transcription and purification are available in the Supplemental Methods. Briefly, the SMK leader sequence was positioned downstream of a T7 RNA polymerase promoter between two ribozyme sequences which allowed preparation of RNA constructs with homogeneous termini corresponding to sequences depicted in Figure 1. The RNA was transcribed in vitro, followed by ribozyme cleavage and denaturing PAGE purification to remove ribozyme fragments. Anion exchange chromatography was performed to remove contaminating acrylamide and the RNA was precipitated with 75% ethanol and 0.3 M NaOAc, pH 5.2. The RNA was resuspended in NMR buffer (25 mM potassium phosphate, pH 6.2, 50 mM NaCl, 5 mM MgCl2), dialyzed against the same buffer, and stored at -20°C. Before use, samples were folded by incubating at 65°C for 5 min followed by slow cooling to 4°C.
Model RNAs (5’-GUUACAGAAAUGUAAC-3’ and 5’-GCUACAGAAAUGUAGC-3’) were purchased from Dharmacon (Fisher Scientific) and used for confirmation of helix P0 resonance assignment in SAM-free SMK59. To aid assignment of resonances in SAM-bound SMK51, paired point sequence alterations were introduced into the model RNAs at helix P1 (C22G and G92C) and helix P2 (G30C and C68G).
NMR spectroscopy
Samples (0.5-1.0 mM) were dialyzed into NMR buffer containing 10% D2O. SAM-bound samples were prepared by mixing a 10% stoichiometric excess of the ligand from a 5 mM stock solution prepared in NMR buffer. Spectra were recorded at 4, 15, or 25°C on 600 and 800 MHz Bruker Avance DRX spectrometers (Billerica, MA) equipped with triple resonance pulsed-field z-axis gradient cryogenic probes. Pulse sequences (1D: 1H, 2D: NOESY, 1H-15N HSQC, HNN-COSY30) were employed with WATERGATE or flip-back water suppression31. Homonuclear NOESY spectra used to determine imino proton connectivities were recorded with a mixing time of 300 ms, with 256 complex points in the indirect dimension. 1H-15N HSQC spectra were recorded with 64 complex points in the indirect dimension. HNN-COSY spectra used 30 ms 15N-15N COSY transfer period, with 32 complex points in the indirect dimension. Spectra were processed using sine bell or Gaussian apodization, with zero-filling once in each dimension. NMRPipe32 and NMRView33 were used for processing and analysis, respectively.
Isothermal titration calorimetry
SMK51 and SMK59 RNA samples (typically ≤10 ml) were dialyzed for at least 24 h total against at least three 0.5-1.0 liter volumes of NMR buffer. The final dialysate was used to dissolve crystalline SAM (Sigma-Aldrich #A7007), creating a 5 mM stock solution (based on FW) that was subsequently diluted to a concentration of 250 μM using the same dialysate. RNA concentrations were estimated by absorbance at 260 nm to be 18.2 μM and 13.6 μM for SMK51 and SMK59, respectively. ITC experiments were performed using a VP-ITC instrument (Microcal) at 15°C with a reference power of 15 μcal s-1. For each experiment, fifty 5 μl injections of SAM were performed into the 1.4 ml cell containing the RNA, with a duration of 8.5 s per injection and 300 s spacing. Experiments were performed in triplicate for both SMK51 and SMK59. The first injection was discarded and the data from each experiment were fit to a single-site binding model using Origin (v7.0383, Microcal). Upon fitting, obtained RNA:SAM stoichiometries slightly less than 1:1 revealed that the active SAM concentration in the syringe was less than expected, and decreased between individual runs. This is consistent with the ~80% purity of commercially available SAM and its routinely-observed degradation when dissolved in water34; 35. To correct for this and ensure a proper fit of the other parameters, the effective concentration of SAM was allowed to vary during fitting from 180-250 μM, and a stoichiometry of 1 was assumed, which is expected based on the SMK box crystal structure10 and is consistent with the NMR-based observations (data not shown). SMK59 thermograms exhibit a small, reproducible feature within the first few injections; this may be due to a minor conformation of the RNA otherwise undetected via NMR experiments or via native and denaturing PAGE analysis.
Translational fusions and β-galactosidase assays
Full-length and truncated E. faecalis metK leader sequences (see Supplemental Figure 1), including the first 15 nucleotides of the coding region, were positioned downstream of a B. subtilis glyQS promoter (Pgly) and fused to codon 18 of a lacZ reporter in plasmid pFG32836. Constructs were integrated in single copy into the chromosome of B. subtilis strain BR151 (metB10 lys-3 trpC2) using specialized transducing phage SPβ as previously described36; 37. Strains containing lacZ translational fusions were selected using chloramphenicol (5 μg ml-1). Cells were grown shaking at 37°C in Spizizen minimal media38 containing methionine (50 μg ml-1) until early exponential phase. Cells were harvested by centrifugation at 6,000 × g and resuspended in Spizizen minimal media with or without methionine (50 μg ml-1). Aliquots were collected after 4 h of growth at 37°C and used to measure β-galactosidase activity after permeabilization of the cells with toluene39. β-Galactosidase activity assays were carried out at least in triplicate, and standard deviations are reported.
Small-angle X-ray Scattering (SAXS)
SAXS data for SMK51 and SMK59 samples were acquired at beamline 12.3.1 of the Advanced Light Source at Lawrence Berkeley National Labs. The samples were passed through a 0.22 μm filter before data acquisition. RNA concentrations were between 1-20 mg/ml, and NMR buffer was used (with omission of MgCl2 as a variable). Data collection was performed at 22°C. Buffer-only datasets were collected and used for buffer subtraction from the RNA datasets. Exposures of 10 s were collected for interpretation, while 1 s exposures were collected before and after to test for possible radiation damage and report on intensities in the low-q range (< 0.1 Å-1). Radiation-damaged samples were not included in analysis. For each sample, data were collected for RNA concentrations covering an order of magnitude. Datasets were excluded from analysis if the minimum- and maximum-concentration scattering profiles were not superimposable.
Scattering intensity (I) was obtained as a function of momentum transfer (q) in the range 0.02<q<0.32 Å-1. PRIMUS was used to generate Guinier plots which were used to check for aggregation and obtain the radius of gyration (Supplemental Figure 9)40; 41. Kratky plots were generated in Excel (Figure 5a, b). The program GNOM was used to generate histograms of intramolecular distances (P(r)) by regularized transformation of the scattering intensity42. The maximum intramolecular distance (Dmax) is provided as an input parameter to GNOM, and was initially estimated by setting the value to several hundred Å in order to reveal the point at which upper-limit edge the histogram nears zero. This initial estimate was then varied until the best fit of the data was obtained. The program DAMMIN was used to generate three-dimensional bead models based on the SAXS data of SMK51 bound to SAM43. The program was run in “slow” mode, with the model built within a virtual prolate cylinder of 45 × 130 Å. Ten independent DAMMIN runs were performed; the resulting models were aligned using SUPCOMB and averaged using DAMAVER 44; 45. The resulting model was aligned to the previously determined X-ray structure of the SMK box10 using the program MASSHA46 and visualized using PyMol47.
Research highlights.
- The SMK box riboswitch changes conformation in response to its ligand SAM.
- This conformational equilibrium takes place in the absence of ligand.
- Exchange between folds diminishes the affinity of the RNA for SAM.
- Diminished affinity is one way the riboswitch is tuned for biological function.
Supplementary Material
Acknowledgments
The authors thank members of the Foster and Henkin labs for invaluable discussions, Michal Hammel (LBNL/SIBYLS Beamline), Elihu Ihms and the OSU CCIC staff for assistance with data collection, and Harald Schwalbe (Frankfurt University) for helpful discussions on experimental design. This work was funded by NIH grant GM063615 (to TMH) and ARRA supplement GM063615S1 (TMH and MPF). RCW was supported in part by a predoctoral fellowship from the American Heart Association.
Abbreviations
- SAM
S-adenosylmethionine
- SAXS
small-angle X-ray scattering
- ITC
isothermal titration calorimetry
Footnotes
CONTRIBUTIONS RCW, AMS, and RTF prepared samples and performed the experiments. IRK contributed to data collection and analysis. RCW, AMS, TMH, and MPF contributed to experimental design, data analysis, and writing the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Garst AD, Batey RT. A switch in time: Detailing the life of a riboswitch. Biochim Biophys Acta. 2009 doi: 10.1016/j.bbagrm.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henkin TM. Riboswitch RNAs: using RNA to sense cellular metabolism. Genes Dev. 2008;22:3383–90. doi: 10.1101/gad.1747308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith AM, Fuchs RT, Grundy FJ, Henkin TM. Riboswitch RNAs: regulation of gene expression by direct monitoring of a physiological signal. RNA Biol. 2010;7:104–10. doi: 10.4161/rna.7.1.10757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miranda-Rios J. The THI-box riboswitch, or how RNA binds thiamin pyrophosphate. Structure. 2007;15:259–65. doi: 10.1016/j.str.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 5.Sudarsan N, Barrick JE, Breaker RR. Metabolite-binding RNA domains are present in the genes of eukaryotes. RNA. 2003;9:644–7. doi: 10.1261/rna.5090103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Corbino KA, Barrick JE, Lim J, Welz R, Tucker BJ, Puskarz I, Mandal M, Rudnick ND, Breaker RR. Evidence for a second class of S-adenosylmethionine riboswitches and other regulatory RNA motifs in alpha-proteobacteria. Genome Biol. 2005;6:R70. doi: 10.1186/gb-2005-6-8-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fuchs RT, Grundy FJ, Henkin TM. The SMK box is a new SAM-binding RNA for translational regulation of SAM synthetase. Nat Struct Mol Biol. 2006;13:226–33. doi: 10.1038/nsmb1059. [DOI] [PubMed] [Google Scholar]
- 8.McDaniel BA, Grundy FJ, Artsimovitch I, Henkin TM. Transcription termination control of the S box system: direct measurement of S-adenosylmethionine by the leader RNA. Proc Natl Acad Sci U S A. 2003;100:3083–8. doi: 10.1073/pnas.0630422100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilbert SD, Rambo RP, Van Tyne D, Batey RT. Structure of the SAM-II riboswitch bound to S-adenosylmethionine. Nat Struct Mol Biol. 2008;15:177–82. doi: 10.1038/nsmb.1371. [DOI] [PubMed] [Google Scholar]
- 10.Lu C, Smith AM, Fuchs RT, Ding F, Rajashankar K, Henkin TM, Ke A. Crystal structures of the SAM-III/SMK riboswitch reveal the SAM-dependent translation inhibition mechanism. Nat Struct Mol Biol. 2008;15:1076–83. doi: 10.1038/nsmb.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Montange RK, Batey RT. Structure of the S-adenosylmethionine riboswitch regulatory mRNA element. Nature. 2006;441:1172–5. doi: 10.1038/nature04819. [DOI] [PubMed] [Google Scholar]
- 12.Fuchs RT, Grundy FJ, Henkin TM. S-adenosylmethionine directly inhibits binding of 30S ribosomal subunits to the SMK box translational riboswitch RNA. Proc Natl Acad Sci U S A. 2007;104:4876–80. doi: 10.1073/pnas.0609956104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jucker FM, Heus HA, Yip PF, Moors EH, Pardi A. A network of heterogeneous hydrogen bonds in GNRA tetraloops. J Mol Biol. 1996;264:968–80. doi: 10.1006/jmbi.1996.0690. [DOI] [PubMed] [Google Scholar]
- 14.Rudisser S, Tinoco I., Jr Solution structure of Cobalt(III)hexammine complexed to the GAAA tetraloop, and metal-ion binding to G•A mismatches. J Mol Biol. 2000;295:1211–23. doi: 10.1006/jmbi.1999.3421. [DOI] [PubMed] [Google Scholar]
- 15.Monod J, Wyman J, Changeux JP. On the Nature of Allosteric Transitions: A Plausible Model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 16.Eftink MR, Anusiem AC, Biltonen RL. Enthalpy-entropy compensation and heat capacity changes for protein-ligand interactions: general thermodynamic models and data for the binding of nucleotides to ribonuclease A. Biochemistry. 1983;22:3884–96. doi: 10.1021/bi00285a025. [DOI] [PubMed] [Google Scholar]
- 17.Tomsic J, McDaniel BA, Grundy FJ, Henkin TM. Natural variability in S-adenosylmethionine (SAM)-dependent riboswitches: S-box elements in Bacillus subtilis exhibit differential sensitivity to SAM in vivo and in vitro. J Bacteriol. 2008;190:823–33. doi: 10.1128/JB.01034-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Serganov A, Huang L, Patel DJ. Structural insights into amino acid binding and gene control by a lysine riboswitch. Nature. 2008;455:1263–7. doi: 10.1038/nature07326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garst AD, Heroux A, Rambo RP, Batey RT. Crystal structure of the lysine riboswitch regulatory mRNA element. J Biol Chem. 2008;283:22347–51. doi: 10.1074/jbc.C800120200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ottink OM, Rampersad SM, Tessari M, Zaman GJ, Heus HA, Wijmenga SS. Ligand-induced folding of the guanine-sensing riboswitch is controlled by a combined predetermined induced fit mechanism. RNA. 2007;13:2202–12. doi: 10.1261/rna.635307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Noeske J, Buck J, Furtig B, Nasiri HR, Schwalbe H, Wohnert J. Interplay of ’induced fit’ and preorganization in the ligand induced folding of the aptamer domain of the guanine binding riboswitch. Nucleic Acids Res. 2007;35:572–83. doi: 10.1093/nar/gkl1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buck J, Furtig B, Noeske J, Wohnert J, Schwalbe H. Time-resolved NMR methods resolving ligand-induced RNA folding at atomic resolution. Proc Natl Acad Sci U S A. 2007;104:15699–704. doi: 10.1073/pnas.0703182104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Serganov A, Polonskaia A, Phan AT, Breaker RR, Patel DJ. Structural basis for gene regulation by a thiamine pyrophosphate-sensing riboswitch. Nature. 2006;441:1167–71. doi: 10.1038/nature04740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lang K, Rieder R, Micura R. Ligand-induced folding of the thiM TPP riboswitch investigated by a structure-based fluorescence spectroscopic approach. Nucleic Acids Res. 2007;35:5370–8. doi: 10.1093/nar/gkm580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stoddard CD, Montange RK, Hennelly SP, Rambo RP, Sanbonmatsu KY, Batey RT. Free State Conformational Sampling of the SAM-I Riboswitch Aptamer Domain. Structure. 18:787–797. doi: 10.1016/j.str.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wabiko H, Ochi K, Nguyen DM, Allen ER, Freese E. Genetic mapping and physiological consequences of metE mutations of Bacillus subtilis. J Bacteriol. 1988;170:2705–10. doi: 10.1128/jb.170.6.2705-2710.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith AM, Fuchs RT, Grundy FJ, Henkin TM. The SAM-responsive SMK box is a reversible riboswitch. Mol Micro. 2010 doi: 10.1111/j.1365-2958.2010.07410.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jucker FM, Phillips RM, McCallum SA, Pardi A. Role of a heterogeneous free state in the formation of a specific RNA-theophylline complex. Biochemistry. 2003;42:2560–7. doi: 10.1021/bi027103+. [DOI] [PubMed] [Google Scholar]
- 29.Gilbert SD, Stoddard CD, Wise SJ, Batey RT. Thermodynamic and kinetic characterization of ligand binding to the purine riboswitch aptamer domain. J Mol Biol. 2006;359:754–68. doi: 10.1016/j.jmb.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 30.Dingley AJ, Nisius L, Cordier F, Grzesiek S. Direct detection of N-H[…]N hydrogen bonds in biomolecules by NMR spectroscopy. Nat Protoc. 2008;3:242–8. doi: 10.1038/nprot.2007.497. [DOI] [PubMed] [Google Scholar]
- 31.Liu M, Xi-an M, Ye Chaohui, Huang He, Nicholson Jeremy K, Lindon John C. Improved WATERGATE Pulse Sequences for Solvent Suppression in NMR Spectroscopy. Journal of Magnetic Resonance. 1998;132:125–129. [Google Scholar]
- 32.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–93. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 33.Johnson BA. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol Biol. 2004;278:313–52. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- 34.Parks LW, Schlenk F. The stability and hydrolysis of S-adenosylmethionine; isolation of S-ribosylmethionine. J Biol Chem. 1958;230:295–305. [PubMed] [Google Scholar]
- 35.Desiderio C, Cavallaro RA, De Rossi A, D’Anselmi F, Fuso A, Scarpa S. Evaluation of chemical and diastereoisomeric stability of S-adenosylmethionine in aqueous solution by capillary electrophoresis. J Pharm Biomed Anal. 2005;38:449–56. doi: 10.1016/j.jpba.2005.01.041. [DOI] [PubMed] [Google Scholar]
- 36.Grundy FJ, Henkin TM. tRNA as a positive regulator of transcription antitermination in B. subtilis. Cell. 1993;74:475–82. doi: 10.1016/0092-8674(93)80049-k. [DOI] [PubMed] [Google Scholar]
- 37.Grundy FJ, Henkin TM. The S box regulon: a new global transcription termination control system for methionine and cysteine biosynthesis genes in Gram-positive bacteria. Mol Microbiol. 1998;30:737–49. doi: 10.1046/j.1365-2958.1998.01105.x. [DOI] [PubMed] [Google Scholar]
- 38.Anagnostopoulos C, Spizizen J. Requirements for transformation in Bacillus subtilis. J Bacteriol. 1961;81:741–6. doi: 10.1128/jb.81.5.741-746.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller J. Experiments in molecular genetics. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press; 1972. [Google Scholar]
- 40.Guinier A, Fournet G. Small-Angle Scattering of X-Rays. Wiley; New York: 1955. [Google Scholar]
- 41.Konarev PV, Volkov VV, Sokolova AV, Kochb MHJ, Svergun DI. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr. 2003;36:1277–1282. [Google Scholar]
- 42.Konig S, Svergun DI, Koch MH, Hubner G, Schellenberger A. Synchrotron radiation solution X-ray scattering study of the pH dependence of the quaternary structure of yeast pyruvate decarboxylase. Biochemistry. 1992;31:8726–31. doi: 10.1021/bi00152a007. [DOI] [PubMed] [Google Scholar]
- 43.Svergun DI. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J. 1999;76:2879–86. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kozin M, Svergun DI. Automated matching of high and low resolution structural models. J Appl Cryst. 2001;34:33–41. [Google Scholar]
- 45.Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Cryst. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Konarev PV, Petoukhov MV, Svergun DI. MASSHA - a graphics system for rigid-body modelling of macromolecular complexes against solution scattering data. J Appl Cryst. 2001;34:527–532. [Google Scholar]
- 47.DeLano WL. The PyMOL Molecular Graphics System. 2002;2009 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.