

Abstract
We describe the facile generation of a stable recombinant antibody with intrinsic red fluorescent properties for qualitative and potentially quantitative immunofluorescence analysis. The REDantibody based on the X-ray crystallographic structures of the anti-sialyl-Tn antibody B72.3 and 3D model of the monomeric red fluorescent protein was designed to retain optimal spatial geometry between the C- and N- termini of the VH and VL chains respectively to mimic the domains interface pairing in antibody Fab fragments and to incorporate the red fluorescent protein as a bridging scaffold. The model was further validated by assembling a REDantibody based on CA19.9 the anti-sialylated Lewis (Le)a blood group antigen and 4D5-8 the anti-p185HER2 antibodies. The chimeric heavy and light chains containing red fluorescent protein as a bridge were correctly processed and secreted into E. coli periplasm for assembly and disulphide bond formation, further analysis revealed the molecules to be exclusively monomers. Purified anti-glycan proteins were used for an immunofluorescent analysis of Trypanosoma cruzi epimastigotes, and the anti-p185HER2 used to determine the binding properties. The REDantibody platform facilitates rapid generation of scFv chimeras that could be used for screening antibodies against cell surface markers. Furthermore, such modular assembly should permit the interchange of binding sites and of fluorophores to create robust panels of coloured antibodies.
Keywords: antibody engineering, fluorescent proteins, scFv, synthetic antibody
1. Introduction
The development and application of optical techniques to visualise the genetically encoded fluorescent proteins (FPs) in living systems has been recognised as an important tool for studies of cell biology and physiology. To allow multiplexing in these studies a palette of FPs have been created that span the visible spectrum from deep blue to near infra-red (Tsien, 2009). In parallel the advances in antibody engineering technology have generated a plethora of recombinant antibodies against a diverse range of targets (Winter and Milstein, 1991; Winter, 1998; Lerner, 2006). Although these two genetically encoded molecules would appear to readily permit further modular combinatorial expansion as fusions to produce bifunctional molecules with combined binding and fluorescent properties, such as single chain fragment variable (scFv) regions fused directly to a fluorescent protein either at the C- or N-termini (Griep et al., 1999; Bazl et al., 2007; Olichon and Surrey, 2007; Serebrovskaya et al., 2009), the uptake and application of scFv-fluorophor fusions has been limited. The FP’s have in common a very rigid β-barrel structure that can withstand fusions to either the N- or C- termini (Hink et al., 2000) and extensive permutations to two of the exposed loops without impacting on the optical fluorescence properties (Pavoor et al., 2009). On the other hand the scFv’s stability is not as robust and varies on a case by case basis, in some instances may be prone to aggregation, thus present stability isuues. To date many antibody structures have been determined by X-ray crystallography and the distance between the C-terminal on the variable heavy chain and the N-terminal on the variable light chain has been determined to be approximately 34-35 Å (Arndt et al., 1998). To generate conventional scFv antibodies a 20-30 amino acid flexible linker is inserted between these two termini which can span more than 35 Å (Bird et al., 1988; Huston et al., 1988). This is important because the non-covalent interactions between variable heavy and light (VH / VL) interfaces are critical for antigen recognition. Nevertheless, in scFv’s with such long flexible linkers the VH / VL pairing exist in equilibrium with the unpaired state, often resulting in aggregation, reduced efficacy and decreased stability relative to the Fab fragment or whole IgG, where the dissociation is restricted. This may be addressed by engineering the VH and VL interface residues to enhance association and thus stability (Worn and Pluckthun, 2001), but this would need to be done on a cases by case for each scFv. However, alternatively linking the VH and VL via a correctly folded rigid β-barrel structure such as a monomeric FP could provide a generic solution to facilitate optimal interface pairing. Using such a rigid linker would favour VH/VL orientation, association and restrict dissociation, ensure monomeric assembly, thus potentially confer Fab like stability at the same time introducing the fluorophor properties. In essence replacing the two stabilising CH1 and CL domains with a single FP domain.
Here we report the design, assembly, production and characterisation of a VH–RFP–VL–His-tag (REDantibody) molecule, where monomeric red fluorescent protein (mRFP) from Discosoma (Campbell et al., 2002) is inserted as a rigid linker between the VH and VL domains of three recombinant distinct antibodies, anti-carbohydrate antibodies B72.3 (Brady et al., 1991), CA19.9 (Koprowski et al., 1979) and 4D5-8 anti-p185HER2 (Eigenbrot et al., 1993). The resulting recombinant molecules are characterised by SDS-PAGE, size exclusion chromatography, spectrophotometry, surface plasmon resonance and by utility in immunofluorescence detection of Trypanosoma cruzi epimastigotes by confocal microscopy to demonstrate that the two functionalities are retained i.e., binding affinity and optical properties.
2. Materials and methods
2.1Molecular design and visualisation
Structure of B72.3 and 4D5-8 antibodies were downloaded from PDB database (PBD: 1BBJ and 1FVC respectively). RFP structure was predicted using Swiss-Model Workspace server. Further modelling was performed using MIFit+ software version 2009.09-1 (Rigaku) and protein models were viewed using PyMol software version 1.1 (DeLano Scientific).
2.2 Plasmids, primers and synthetic DNA
Plasmid pBAK1, previously constructed in our laboratory is based on pET-26b vector (Novagen). All primers were purchased from Invitrogen. Synthetic DNA sequences of B72.3 and CA19.9 antibody variable domains in VH-VL orientation were codon optimised for Escherichia coli (E. coli)expression and purchased from Epoch Biolabs as plasmids pBSK-B72.3 and pBSK-CA19.9 respectively, with NcoI, BamHI and NotI restriction sites to facilitate construction of the expression vectors. Plasmid pAK19 encoding the 4D5-8 VH and VL was provided by Dan Yansura (Genentech, USA). Plasmid pMT-RFP with mRFP1 gene was a gift from Professor Ray St.Ledger (University of Maryland, USA).
2.2 Bacterial strains, growth media and recombinant DNA technique
XL1-Blue E. coli strain (Stratagene) was used for plasmid construction steps. To express recombinant antibodies BL21 (DE3) strain of E. coli (Novagen) were used. E. coli cells were grown in Lysogeny Broth (LB) (Bertani, 2004) or LB agar plates. Kanamycin sulfate and carbenicillin were used at 30 μg/mL and 100 μg/mL final concentrations respectively. Plasmid DNA was isolated using QIAprep Spin Miniprep Kit (Qiagen) and DNA from the gel was purified using QIAquick Gel Extraction Kit (Qiagen). The E. coli cells were transformed using standard heat shock methods. Restriction and modification enzymes were purchased from New England Biolabs (NEB). Final plasmid constructs were confirmed by DNA sequence analysis.
2.3 Construction of the expression plasmid
Antibody scFv encoding fragments were either digested directly from pBSK-B72.3, pBSK-CA19.9 or assembled from VH and VL domains encoded by pASK19 plasmids respectively and inserted into NcoI, and NotI restriction sites of previously digested plasmid pBAK1 to make pBAK1B72.3, pBAK1CA19.9 and pBAK14D5-8 respectively. The competent E.coli XL1 Blue cells were transformed using ligation mixtures and the clones were selected on the LB plates containing kanamycin. Positive clones were confirmed by DNA sequencing. To make RFP chimeras in VH-RFP-VL orientation, plasmids pBAK1B72.3, pBAK1CA19.9 and pBAK14D5-8 were digested with BamHI restriction enzyme and PCR product of mRFP1 gene obtained using pMT-RFP plasmid template and oligonucleotide primers RFPBamHIF and RFPBamHIR (Table 1), inserted to produce pBAK1B72.3RFP, pBAK1CA19.9RFP and pBAK14D5-8RFP respectively. Colonies were initially screened by colony PCR using primers T7F and RFPBamHIR (Table 1) and selected clones confirmed by plasmid DNA sequencing.
Table 1.
PCR primers used in REDantibody Assembly
| Name | Oligonucleotide sequence | Cloning site |
|---|---|---|
| RFPBamHIF | CAGTGGATCCGAGGACGTCATCAAGGAGTTC | BamHI |
| RFPBamHIR | CAGTGGATCCGCCTCCGCCTGTGCGGCCCTCGGC GCGCTCGTAC |
BamHI |
| T7F | GCAGCTAATACGACTCACTATAGG |
2.4 Antibody expression in E. coli
To express scFv and REDantibody chimeras, E. coli BL21 (DE3) (Novagen) cells were transformed with the appropriate plasmid and plated onto LB agar supplemented with kanamycin sulfate (30 μg/mL final concentration). The cells were allowed to grow at 37°C for 18 h and the following day, five fresh colonies were inoculated into 10 mL of LB media (with antibiotics) and grown at 37°C (with shaking at 250 rpm) for 16 h. Next day, 200 mL of pre-warmed LB media, prepared in 1 L conical flasks (with antibiotics) were inoculated with 10 mL of the overnight culture and grown at 37°C (with shaking at 250 rpm) until the optical density at 600 nm had reached 0.5, then the cells were placed on ice for 30 min and Isopropyl β-D-1-thiogalactopyranoside (IPTG) added (final concentration 0.3 mM) to the cultures and the cells were grown at 20°C for an additional 20 h with shaking at 250 rpm. Bacterial cells were pelleted by centrifugation for 20 min, 5,000 rpm at 4°C (using Sorvall SuperT 21 bench top centrifuge, with SL-250T rotor) the supernatant discarded and the pellets retained for periplasmic protein extraction.
2.5 Antibody purification from E. coli periplasm
Bacterial cell pellets from 200 mL culture were resuspended in 10 mL of periplasmic buffer (30 mM Tris-base, pH 8.0, 20% sucrose and 1 mM EDTA) supplemented to a final concentration of 0.1 mM phenylmethylsulfonyl fluoride (PMSF). The cells were incubated on ice for 10 min and centrifuged at 9000 rpm for 10 min at 4 °C (using Sorvall SuperT 21 bench top centrifuge, with SL-50T rotor). The supernatants were collected and stored on ice, whilst cell pellets were resuspended in 7 mL of 5 mM MgCl2 (4 °C). After incubation for 5 min on ice, bacterial cells were pelleted by centrifugation as described before, and the supernatants combined to give the periplasmic fraction.
2.6 Ni-NTA purification of recombinant proteins
Recombinant proteins were purified using a 1 mL HisTrap HP column (GE Healthcare) fitted to an ÄKTAprime™ plus (GE Healthcare) liquid chromatography system. First, the 1 mL column was equilibrated with 5 column volumes of equilibration buffer (20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 10 mM imidazole). Following this, 17 mL E. coli periplasmic fraction (diluted two times with 20 mM Tris-HCl, pH 8.0, 500 mM NaCl buffer) was loaded onto the column and the column was washed with 5 column volumes of wash buffer (20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 20 mM imidazole). The protein was eluted with 5 column volumes of elution buffer (20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 500 mM imidazole) and collected in 1 mL aliquots. Protein elution was monitored at 280 nm. Eluted fractions 1-5 were analysed by SDS-PAGE.
2.7 Desalting and Anion Exchange chromatography
Recombinant antibody obtained by Ni-NTA purification were desalted using 40 mL Sephadex G25 column (GE Healthcare) fitted to an ÄKTAprime™ plus (GE Healthcare) liquid chromatography system in 20 mM Tris-HCl, pH 8.0 buffer and further purified using 1 mL HiTrap Q Sepharose FF column (GE Healthcare) and eluted by increasing buffer salt concentration. First, a 1 mL column was equilibrated with 5 column volumes of equilibration buffer (20 mM Tris-HCl, pH 8.0). Following this, 5 mL of desalted antibody fraction was loaded onto the column and protein was eluted by increasing concentration of NaCl in 20 mM Tris-HCl, pH 8.0 buffer and collected into 1 mL fractions. Eluted antibodies were concentrated and the buffer exhanged to 20 mM Tris-HCl, pH 8.0 and 150 mM NaCl using Ultracel YM-10 Amicon centrifugal devices from Millipore. Protein concentration was determined using Bradford assay kit (Bio-Rad)(Bradford, 1976).
2.8 Size Exclusion Chromatography
SEC was performed on an ÄKTAprime™ plus using a HiLoad 16/60 Superdex 200 size-exclusion column (GE Healthcare, Amersham, UK) equilibrated with degassed phosphate buffered saline (PBS). The purified proteins were separated by loading 1 mL of the sample at a concentration of 100-200 μg/mL. The flow rate was 1 mL/min, and the absorbance of the eluted protein was monitored at 280 nm. The column was calibrated with the following protein standards: β-amylase (200 kDa), bovine serum albumin (66 kDa), carbonic anhydrase (29 kDa) and cytochrome c (12.4 kDa).
2.9 SDS-PAGE
The fractions eluted from the HisTrap column (1-5) were analysed by SDS-PAGE using 12% Tris-Glycine gels. Proteins were stained with Coomassie Blue R250 as follows: after electrophoresis gel was submerged in plastic container into 50 mL of 0.025% Coomassie Blue in 10% acetic acid solution and heated in the microwave until boiling (approximately 1 min), cooled down for 2 min on bench and destained in 50 mL of 10% acetic acid by repeating previous procedure of heating in microwave and cooling down. Finally, the gel was kept in 10% acetic acid before scanning.
2.10 Optical properties determination
The fluorescent measurements were carried out using SpectraMAX Gemini EM fluorescence microplate reader with Gemini EM software (Molecular Devices, UK) using Optiplex 96F microtitre plates (Perkin Elmer, UK) with REDantibody 4D5-8, 50 μg in 0.1mL/well in phosphate buffered saline (137 mM NaCl, 2.5 mM KCl, 10 mM Na2HPO4 and 1.8 mM KH2PO4) pH7.4. Excitation and emission spectra between 500-650nm were determined at 2 nm intervals and the arbitrary fluorescence units (AFU) recorded. The concentration dependant fluorescence with excitation 584nm and emission 607nm was determined for tenfold serial dilutions of REDantibody 4D5-8 from a stock solution of 95 nmoles.
2.11 Surface plasmon resonance (SPR) measurements
The SPR measurements were carried out on the BIAcore 3000 following the procedure previously described to determine the binding of 4D5-8 scFv with p185HER2-ECD (Worn and Pluckthun, 1998).The p185HER2-ECD antigen (Sino Biological Inc, Beijing, China) 100μg/mL in 20 mM citrate buffer pH4.0 was coupled to the CM-5 research grade sensor chip using an amine coupling kit (GE Healthcare). The 4D5-8 REDantibody were applied at 50, 100, 175, 250 and 500 μg/mL to the chip at a flow rate of 20 μl/min at 20°C. The surface was regenerated by injection of 45 μl of 0.1M glycine-HCL, pH2.2, 0.5M NaCl. Data were analysed using the global fit in the BIAevaluation program version 4.1
2.12 Immunofluorescent confocal microscopy
Wild-type T. cruzi epimastigote 4 days old cultures were fixed with 2% paraformaldehyde in phosphate buffer and allowed to adhere onto glass slides. Adhered cells were washed with PBS and non-specific antigen-binding sites were blocked with 2% BSA in PBS, pH 8.0, for 1 h at room temperature. Samples were incubated for 60 min at room temperature with recombinant REDantibodies. REDantibody 4D5-8 was used as a negative control. Slides were mounted in N-propylgalate (Sigma P130) to reduce photobleaching and observed on a Leica SP2 Confocal Laser Scanning Microscope, using 543-nm He-Ne laser. All images were analysed using IMARIS software version 6.3 (Bitplane). Formaldehyde fixed Trypanosoma cruzi epimastigotes TC1 strain were a gift from Professor Michael Miles and Michael Lewis.
3. Results
3.1 Molecular design
The X-ray crystallographic structure of Fab B72.3 (PDB: 1BBJ) reveals that the distance between the C-terminal Ser114 on the variable heavy chain and the N-terminal Asp1 on the variable light chain is approximately 34 Å. A search of the Protein Data Base (PDB) was carried out to identify structures with N- and C- termini protruding from the same plane with spacing of 20-30 Å apart without intersecting the rest of the protein. Fortuitously, mRFP1 met these criteria with the distance between Val7 and His221 of the termini in fast maturing red fluorescent protein DsRed variant (PDB 2VAE) being approximately 26 Å. Direct fusion of the VH to the N-terminus and VL to the C-terminus would have resulted in a linkage 8 Å shorter than the optimal spacing revealed by X-ray crystallographic structure of B72.3. This was overcome by modelling the N- and C- termini to include Gly4Ser linkers on each end of RFP (Figure 1). The resulting model could accommodate the variable regions of the antibody with a molecular geometry approaching that determined from the X-ray studies.
Figure 1. Ribbon representation of a REDantibody molecule 3D model.
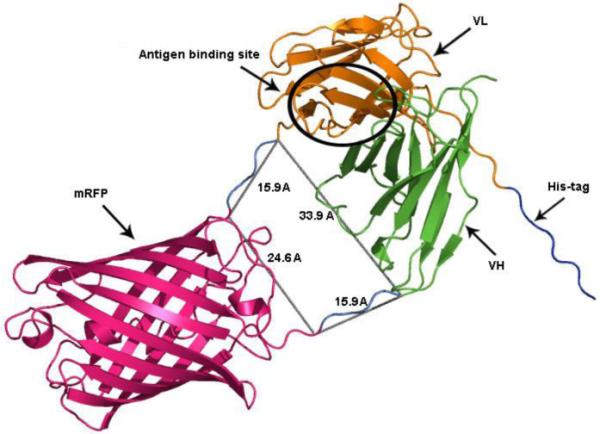
VH chain is shown in green, RFP domain in red, VL chain in yellow. His-tag is shown in blue and linkers between VH-RFP and RFP-VL are in cyan with the distances indicated. Antigen binding site is indicated as a black circle on antibody structure. All distances are shown as a grey lines and are indicated in the angstroms (Å).
3.2 DNA manipulation
The REDantibody constructs were assembled in a modified pET-26b vector (Novagen) with a pelB leader sequence to direct secretion to the periplasm of E. coli (Figure 2A). The synthetic genes of the VH-VL scFv antibodies were designed to encode an in-frame BamHI site between the VH and VL regions and the whole flanked by NcoI and NotI sites for in-frame directional cloning. The mRFP gene was inserted into each of these plasmids to make expression plasmids pBAK1B72.3RFP and pBAK1CA19.9RFP as shown in Figure 2A. The control pBAK14D5-8RFP was assembled in a similar manner after removing an internal BamHI site. The expression cassettes starts from pelB leader sequence followed by VH chain, RFP, VL chain and an octa-histidine-tag at the carboxyl terminus of the resulting protein sequence as shown in Figure 2B.
Figure 2. Generation of expression plasmid (A) and E. coli expression cassette (B) of REDantibody.

(A) The single chain Fv encoding fragment flanked by NcoI and NotI sites with an inframe BamHI site between the VH and VL was inserted into pBAK1 vector to generate pBAK1B72.3, pBAK1CA19.9 and pBAK14D5. The mRFP encoding fragment flanked by BamHI sites was inserted into the pBAK1B72.3, CA19.9 and 4D5 to generate pBAK172.3RFP, pBAK1CA19.9RFP and pBAK14D5RFP.
(B) The expression cassette has a T7 promoter to initiate transcription using T7 RNA polymerase, lac operator is for the control of transcription using IPTG. RBS is ribosome binding site where translation begins, pelB sequence to guide newly synthesised molecule into the E. coli periplasm where this sequence is deleted and disulphide bonds are formed. VH is variable heavy chain of antibody, Link is an additional 5 amino acids to the mRFP1 gene, mRFP is a gene of red fluorescent protein and VL is variable light domain of antibody. This is followed by His-tag sequence for immobilised metal affinity purification and stop codon. T7 terminator is to terminate transcription by T7 RNA polymerase.
3.3 Protein expression & purification
The recombinant scFv and the corresponding REDantibody proteins were expressed in BL 21 (DE3) E. coli strain and recovered from the periplasmic extract and purified via Ni-NTA affinity chromatography. Protein expression and purification processes were monitored by SDS-PAGE. The protein concentration of the recovered functional protein was determined for each construct; mRFP (20mg/L), REDantibody 4D5-8 (5mg/L), REDantibody CA19.9 (0.88mg/L) and REDantibody (72.3 0.9mg/L). The predicted molecular weights of the scFv and REDantibody recombinant proteins were approximately 25 and 51 kDa respectively as shown in Figure 3. The SDS-PAGE analysis of the Ni-NTA affinity enriched recombinant antibodies also had other protein bands corresponding to co-enriched E.coli host proteins. The size exclusion column was calibrated using a standard range 200-12kDa (Sigma, UK) prior to use (Figure 4A). The mRFP protein elution corresponded to monomer of 25kDa (Figure 4B). A single REDantibody peak eluted at fractions/time corresponding to ~51 kDa, (Figure 4C). Moreover the colour of REDantibody E. coli culture, periplasmic and elution fractions were pink (data not shown).
Figure 3. Expression and purification (A) CA19.9 scFv and (B) CA19.9 REDantibody.

The purified scFvCA19.9 (A) and REDantibodyCA19.9 (B) were resolved by 12% SDS–PAGE and stained with Coomassie Brilliant Blue R250. Lane 1: molecular weight markers, 70, 40, 30 and 20kDa; lane 2: Ni-NTA column elution 1; lane 3: Ni-NTA column elution 2; lane 4: Ni-NTA column elution 3; lane 5: Ni-NTA column elution 4.
Figure 4. Gel filtration on Sephadex G200 column.
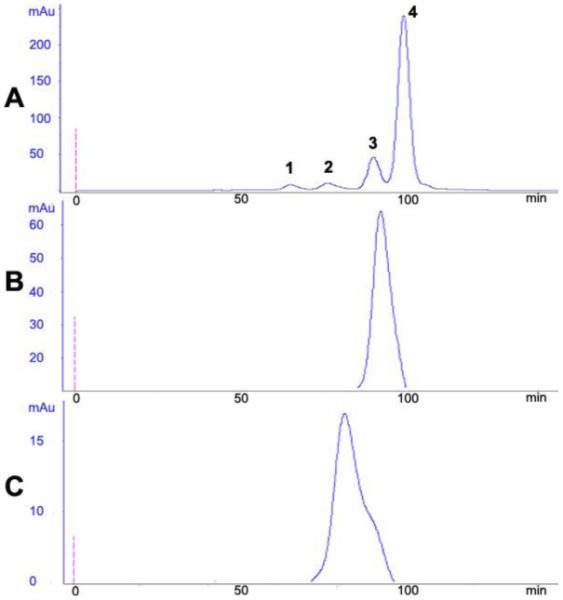
Panel A - standard proteins (peak1- 200 kDa, peak 2 - 66 kDa, peak 3 - 29 kDa, peak 4 - 12.4 kDa); Panel B - mRFP1 and panel C - REDantibodyCA19.9.
3.4 Fluorophor measurements
The excitation and emission peaks of REDantibody 4D5-8 were determined to be 584 and 607nm as shown in figure 5A. The concentration dependent emission at 607nm permitted accurate detection of 9.5 pmole of this REDantibody as shown in figure 5B.
Figure 5. Spectrophotometric analysis of REDantibody.

Scanning fluorescent spectrophoptometry of REDantibody 4D5-8 (A) and concentration dependent fluorescent intensity of REDantibody 4D5-8 (B).
3.5 BIAcore measurements
The association and disassociation rate for REDantibody 4D5-8 binding to p185HER2-ECD was determined to be 2.19 ×10−4 ±0.13 ×10−4 M−1s−1 and 4.43 × 10−5 ±1.04 × 10−5 s−1 respectively and the KD calculated to be 2.2 ± 0.8 nM (n=3, data not shown).
3.6 Immunofluorescent staining and confocal microscopy
Functional analysis of the REDantibody was based on well-characterised properties of B72.3 and CA19.9 antibodies to recognise sialyl-Tn and sialylated Lewis (Le)a blood group antigen respectively (Reddish et al., 1997; Pant et al., 2008) which are part of a panel of markers used in cancer diagnostics (Betta et al., 1991). Interestingly the same antigen has been detected on the surface of the human pathogen Trypanosoma cruzi (Freire et al., 2003). The 4D5-8 REDantibody was constructed for use as a negative control since it recognises a peptide epitope on p185HER2 and not sialyl glycan. This was confirmed by the fluorescent staining of T. cruzi epimastigotes using purified recombinant anti-glycan REDantibodies shown in Figure 6 A&B. The control REDantibody 4D5-8 did not label the parasites (Figure 6C). The maximum intensity fluorescence staining image with REDantibody CA19.9 (Figure 6D) revealed the appearance of a cleft area on the parasite surface with reduced staining. Confocal three-dimensional imaging (Figure 6E) confirmed the presence of a depression corresponding to the cleft observed earlier.
Figure 6. Immunofluorescent microscopic analysis of T.cruzi epimastigotes.

T.cruzi epimastigotes stained with REDantibody. CA19.9 (A), REDantibody B72.3 (B) and negative control REDantibody 4D5-8 (C). T.cruzi epimastigote is stained with REDantibody CA19.9 and viewed by microscopy reveals a maximum intensity fluorescent image (D). The surface staining combined with confocal microscopy reveals a slight invagination on the parasite surface (E).
4. Discussion
Molecules tagged with fluorophores in particular antibodies are highly sensitive tools used in imaging and for cell sorting. Conventional organic fluorophores exemplified by FITC, TRITC, CY-3, and CY-5 are chemically conjugated to the antibodies in vitro. Chemical conjugation is dependent on the availability of particular amino acid side chains. The inappropriate conjugation in or in close proximity of the binding site can reduce the binding efficacy of the parental antibody. The number of labels incorporated into each antibody molecule in the preparation also varies and limits the utility of these reagents in quantitative analysis. The actual chemical link itself may be prone to cleavage over time resulting in leaching of fluorophor. The chemical fluorophores also fade due to photobleaching upon excitation. Also in the case with whole immunoglobulin the binding to Fc receptors presents problems in immunohistochemical analysis. Significant advances have been made in conjugation chemistry to attempt to overcome some of these difficulties with whole antibodies or Fab fragments. With the application of recombinant DNA technology, scFv’s have been engineered to incorporate a free cysteine at the C-terminus for specific conjugation chemistry or have been fused directly to fluorescent proteins for prokaryotic (Griep et al., 1999; Casey et al., 2000; Hink et al., 2000; Schwalbach et al., 2000; Morino et al., 2001) or eukaryotic expression (Peipp et al., 2004). However conventional scFv, are not ideally suited for these applications. The scFv’s are not as stable as whole antibody or the Fab fragment (Worn and Pluckthun, 2001). In the scFv if the flexible linker that joins the VH and VL domains is long (>20 amino acids) it is prone to aggregation since such a long linker permits the dissociation of the VH and VL domains. If the linker length is reduced scFv is prone to form a mixture of multimers (dimers, trimers) (Holliger et al., 1993). However the multimeric assemblies may have altered binding kinetics.
Fusing a scFv assembled with a long linker directly to GFP would not necessarily reduce the aggregation properties of the scFv. Likewise using a scFv with shorter linkers would favour multimer formation which may or may not retain optimal affinity for the target. Despite these issues the concept of having a genetically encoded affinity label is very appealing since it provides a simple platform for creating uniform diagnostic reagents. An alternative approach to this problem has been to modify an FP to incorporate two extended loops that can form recognition motifs, whilst retaining the fluorophor properties, although not antibodies as such, they have potential (Pavoor et al., 2009). However the antibody based binding site composed of a VH and VL with a total of six loops with an extremely high combinatorially diverse CDRH3 and CDRL3 from natural and synthetic repertoires provide a vast range of high affinity binding sites (Winter, 1998; Lerner, 2006). We sought to combine the best features of both the antibody binding sites and the properties of encoded fluorophores (Tsien, 2009) in a single molecule, by inserting the FP between the VH and VL domains to spatially orientate the VH/VL interfaces to mimic the pairing observed in Fab structures. Our earlier attempts to construct similar bridged molecules using a related β-barrel structure of green fluorescent protein EGFP (PDB 1GFL) (Yang et al., 1996) resulted in molecules that did not bind to the target antigen. We speculate that since the EGFP could exist as a dimer at higher protein concentrations its assembly could sterically hinder VH / VL domain pairing, with or without the introduction of Gly4Ser linkers (data not shown). This obstacle was overcome by using a monomeric fluorescent protein with a β-barrel structure exemplified by mRFP1 (Campbell et al., 2002) as shown in Figure 1.
The vectors constructed encoding the REDantibodies are based on the pET series (Studier, 1991) as shown in Figure 2. The use of the T7 promoter and the BL21(DE3) host results in high level of expression of the recombinant protein, the majority of which is retained within the cell cytoplasm despite having a pelB leader sequence to direct the polypeptide into the bacterial periplasm. The amount of REDantibody recovered varied for each antibody construct, 4D5-8 had the highest recovery (5mg/L), whereas the recovery of the corresponding anti-sialyl glycan antibodies were both just below 1mg/L, both levels are comparable to the recovery of scFv from E.coli. It may be the recovery of anti-sialyl glycan REDantibodies was reduced since the secreted, correctly folded and functional REDantibody in both cases bound to sialic acid, which is present on the host bacterial surface (Barry and Goebel, 1957). Notwithstanding, sufficient REDantibody was recovered from the periplasmic extract to permit further affinity purification and functional analysis. The SDS-PAGE of the affinity isolated CA19.9 scFv and the corresponding REDantibody had polypeptides of 25 and 51kDa respectively as shown in Figure 3 A and B respectively. Size exclusion chromatography on the affinity purified proteins was used to determine whether the molecules were monomeric or multimeric. The mRFP and REDantibody each eluted with a single peak corresponding to 25 kDa and 51 kDa as shown in Figure 4 panel B and C respectively, indicating that both proteins are monomers.
A key feature of this REDantibody platform, is the intrinsic colour of the bacteria producing the active molecule and of the purified proteins having a visible pink-red colour, thus not necessarily requiring sophisticated detection equipment to follow the purification steps. The excitation and emission wavelengths of REDantibody were determined to be 584nm and 607nm respectively (as shown in Figure 5A), are identical to that of mRFP (Campbell et al., 2002; Khrameeva et al., 2008), confirming that the fusing immunoglobulin domains to both ends of a FP did not compromise the fluorescent properties. Moreover, it may be possible to detect as little as 9.5 pmoles of REDantibody using a standard fluorometer as shown in figure 5B. This may permit quantitative analysis of target antigen. The kinetics of binding for the anti-glycan REDantibodies was difficult to perform since a defined antigen preparation was not readily available. Thus we carried out the binding studies on the REDantibody 4D5-8. The calculated KD value for REDantibody 4D5-8 of 2.19 nM was within the values determined independently for the 4D5-8 scFv with a VH-(Gly4 Ser)3 linker VL orientation of 194 pM (Worn and Pluckthun, 1998), and 9.4 nM for Herceptin™ (Troise et al., 2008), further confirming that the binding characteristics are not compromised in this format.
The use of this FP would also permit combined use with other conventional dyes for FACs analysis, immunochemistry and confocal microscopy that utilize a range up to 543nm. In this study we used the REDantibody B72.3 and CA19.9 to visualise the carbohydrate antigens sialyl-Tn and sialylated Lewis (Le)a on the surface of T.cruzi epimastigotes respectively, using immunofluorescence (Figure 6 A,B & D) and confocal microscopy (Figure 6E). The negative control REDantibody 4D5-8 did not label the parasites when examined using immunofluorescence microscopy implying the specificity was introduced by the antibody binding domains (Figure 6C). It was observed that the slides prepared for this study could be viewed for at least a week, without noticeable loss of signal implying robust stability of the REDantibody. The mRFP1 derived from DsRed is stable within a wide range of pH 5.0-12.0 and in 6M urea (Stepanenko et al., 2005), thus the antibody antigen interaction may be disrupted by adjusting the pH and or the addition of urea and the released fluorophore quantitatively determined in solution. The resulting REDantibody molecule is also stable to photobleaching, thus potentially permitting a higher sensitivity by use of time-resolved pulsed analysis. Another feature of REDantibody is the generation of reactive oxygen species H2O2 upon exposure to white light: this may potentially have a wide range of applications in vitro and possibly in vivo, i.e., photo-ablation of target cells (Serebrovskaya et al., 2009). Additionally with a far red emission spectra, these types of targeted molecules may have utility in in vivo whole body imaging (Shcherbo et al., 2007). Furthermore, this platform may also be applied to other existing mAbs to create the next generation of diagnostic and cell sorting molecules. The platform may also be used to create libraries of VH and VL domains that may be readily accessed using high throughput non-isotopic screening as an alternative phage or other types of display selection technologies. In general the use of other pre-folded protein domains with N- and C- termini in the same spatial plane with spacing close to 20-30 Å may be used in this modular approach to create bifunctional antibodies. The monomeric β-barrel fluorophor architecture used in this study is essentially the same in all the coloured variant (Tsien, 2009), thus enabling the assembly of a palette of genetically encoded antibody based reagents for simultaneous multi-analyte detection.
5. Conclusions
Assembling the REDantibody using the fluorophore as a bridge introduces the following properties: firstly, with an integrated fluorophore, a single reagent has been created, which reduces time, cost and increases reproducibility of the binding assay. Secondly the antibody binding site and fluorophore are stoichiometric, and thus the signal generated is directly proportional to the amount of antibody bound (i.e., potentially quantitative). Finally, expression and purification steps can be monitored without the need for sophisticated detection equipment since the bacteria and the recombinant protein are visible to the naked eye with a distinct red colour.
Acknowledgements
We thank Professor Michael Miles for constructive comments, Michael Lewis for providing trypanosomes, Professor Ray St.Ledger for providing the mRFP1 DNA and Dr. Mark JP Kerrigan for assistance with the confocal microscopy. BA is a recipient of a University of Westminster Cavendish Studentship award.
Funding
This work was supported by the National Institutes of Health [R01 AI066045].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arndt KM, Muller KM, Pluckthun A. Factors influencing the dimer to monomer transition of an antibody single-chain Fv fragment. Biochemistry. 1998;37:12918–26. doi: 10.1021/bi9810407. [DOI] [PubMed] [Google Scholar]
- Barry GT, Goebel WF. Colominic acid, a substance of bacterial origin related to sialic acid. Nature. 1957;179:206. doi: 10.1038/179206a0. [DOI] [PubMed] [Google Scholar]
- Bazl MR, Rasaee MJ, Foruzandeh M, Rahimpour A, Kiani J, Rahbarizadeh F, Alirezapour B, Mohammadi M. Production of chimeric recombinant single domain antibody-green fluorescent fusion protein in Chinese hamster ovary cells. Hybridoma (Larchmt) 2007;26:1–9. doi: 10.1089/hyb.2006.037. [DOI] [PubMed] [Google Scholar]
- Bertani G. Lysogeny at mid-twentieth century: P1, P2, and other experimental systems. J Bacteriol. 2004;186:595–600. doi: 10.1128/JB.186.3.595-600.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betta PG, Pavesi M, Pastormerlo M, Tallarida F, Bellingeri D, Bocca R. Use of monoclonal antibody B72.3 as a marker of metastatic carcinoma cells in neoplastic effusions. Pathologica. 1991;83:99–104. [PubMed] [Google Scholar]
- Bird RE, Hardman KD, Jacobson JW, Johnson S, Kaufman BM, Lee SM, Lee T, Pope SH, Riordan GS, Whitlow M. Single-chain antigen-binding proteins. Science. 1988;242:423–6. doi: 10.1126/science.3140379. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Brady RL, Hubbard RE, King DJ, Low DC, Roberts SM, Todd RJ. Crystallization and preliminary X-ray diffraction study of a chimaeric Fab’ fragment of antibody binding tumour cells. J Mol Biol. 1991;219:603–4. doi: 10.1016/0022-2836(91)90656-q. [DOI] [PubMed] [Google Scholar]
- Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. A monomeric red fluorescent protein. Proc Natl Acad Sci U S A. 2002;99:7877–82. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey JL, Coley AM, Tilley LM, Foley M. Green fluorescent antibodies: novel in vitro tools. Protein Eng. 2000;13:445–52. doi: 10.1093/protein/13.6.445. [DOI] [PubMed] [Google Scholar]
- Eigenbrot C, Randal M, Presta L, Carter P, Kossiakoff AA. X-ray structures of the antigen-binding domains from three variants of humanized anti-p185HER2 antibody 4D5 and comparison with molecular modeling. J Mol Biol. 1993;229:969–95. doi: 10.1006/jmbi.1993.1099. [DOI] [PubMed] [Google Scholar]
- Freire T, Robello C, Soule S, Ferreira F, Osinaga E. Sialyl-Tn antigen expression and O-linked GalNAc-Thr synthesis by Trypanosoma cruzi. Biochem Biophys Res Commun. 2003;312:1309–16. doi: 10.1016/j.bbrc.2003.11.060. [DOI] [PubMed] [Google Scholar]
- Griep RA, van Twisk C, van der Wolf JM, Schots A. Fluobodies: green fluorescent single-chain Fv fusion proteins. J Immunol Methods. 1999;230:121–30. doi: 10.1016/s0022-1759(99)00131-3. [DOI] [PubMed] [Google Scholar]
- Hink MA, Griep RA, Borst JW, van Hoek A, Eppink MH, Schots A, Visser AJ. Structural dynamics of green fluorescent protein alone and fused with a single chain Fv protein. J Biol Chem. 2000;275:17556–60. doi: 10.1074/jbc.M001348200. [DOI] [PubMed] [Google Scholar]
- Holliger P, Prospero T, Winter G. “Diabodies”: small bivalent and bispecific antibody fragments. Proc Natl Acad Sci U S A. 1993;90:6444–8. doi: 10.1073/pnas.90.14.6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huston JS, Levinson D, Mudgett-Hunter M, Tai MS, Novotny J, Margolies MN, Ridge RJ, Bruccoleri RE, Haber E, Crea R, et al. Protein engineering of antibody binding sites: recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc Natl Acad Sci U S A. 1988;85:5879–83. doi: 10.1073/pnas.85.16.5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khrameeva EE, Drutsa VL, Vrzheshch EP, Dmitrienko DV, Vrzheshch PV. Mutants of monomeric red fluorescent protein mRFP1 at residue 66: structure modeling by molecular dynamics and search for correlations with spectral properties. Biochemistry (Mosc) 2008;73:1085–95. doi: 10.1134/s0006297908100040. [DOI] [PubMed] [Google Scholar]
- Koprowski H, Steplewski Z, Mitchell K, Herlyn M, Herlyn D, Fuhrer P. Colorectal carcinoma antigens detected by hybridoma antibodies. Somatic Cell Genet. 1979;5:957–71. doi: 10.1007/BF01542654. [DOI] [PubMed] [Google Scholar]
- Lerner RA. Manufacturing immunity to disease in a test tube: the magic bullet realized. Angew Chem Int Ed Engl. 2006;45:8106–25. doi: 10.1002/anie.200603381. [DOI] [PubMed] [Google Scholar]
- Morino K, Katsumi H, Akahori Y, Iba Y, Shinohara M, Ukai Y, Kohara Y, Kurosawa Y. Antibody fusions with fluorescent proteins: a versatile reagent for profiling protein expression. J Immunol Methods. 2001;257:175–84. doi: 10.1016/s0022-1759(01)00462-8. [DOI] [PubMed] [Google Scholar]
- Olichon A, Surrey T. Selection of genetically encoded fluorescent single domain antibodies engineered for efficient expression in Escherichia coli. J Biol Chem. 2007;282:36314–20. doi: 10.1074/jbc.M704908200. [DOI] [PubMed] [Google Scholar]
- Pant KD, Jain A, McCracken JD, Thompson K. Immunohistochemical examination of anti-STn monoclonal antibodies LLU9B4, B72.3, and B35.2 for their potential use as tumor markers. Dig Dis Sci. 2008;53:2189–94. doi: 10.1007/s10620-007-0137-2. [DOI] [PubMed] [Google Scholar]
- Pavoor TV, Cho YK, Shusta EV. Development of GFP-based biosensors possessing the binding properties of antibodies. Proc Natl Acad Sci U S A. 2009;106:11895–900. doi: 10.1073/pnas.0902828106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peipp M, Saul D, Barbin K, Bruenke J, Zunino SJ, Niederweis M, Fey GH. Efficient eukaryotic expression of fluorescent scFv fusion proteins directed against CD antigens for FACS applications. J Immunol Methods. 2004;285:265–80. doi: 10.1016/j.jim.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Reddish MA, Jackson L, Koganty RR, Qiu D, Hong W, Longenecker BM. Specificities of anti-sialyl-Tn and anti-Tn monoclonal antibodies generated using novel clustered synthetic glycopeptide epitopes. Glycoconj J. 1997;14:549–60. doi: 10.1023/a:1018576224062. [DOI] [PubMed] [Google Scholar]
- Schwalbach G, Sibler AP, Choulier L, Deryckere F, Weiss E. Production of fluorescent single-chain antibody fragments in Escherichia coli. Protein Expr Purif. 2000;18:121–32. doi: 10.1006/prep.1999.1185. [DOI] [PubMed] [Google Scholar]
- Serebrovskaya EO, Edelweiss EF, Stremovskiy OA, Lukyanov KA, Chudakov DM, Deyev SM. Targeting cancer cells by using an antireceptor antibody-photosensitizer fusion protein. Proc Natl Acad Sci U S A. 2009;106:9221–5. doi: 10.1073/pnas.0904140106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbo D, Merzlyak EM, Chepurnykh TV, Fradkov AF, Ermakova GV, Solovieva EA, Lukyanov KA, Bogdanova EA, Zaraisky AG, Lukyanov S, Chudakov DM. Bright far-red fluorescent protein for whole-body imaging. Nat Methods. 2007;4:741–6. doi: 10.1038/nmeth1083. [DOI] [PubMed] [Google Scholar]
- Stepanenko OV, Verkhusha VV, Shavlovskii MM, Aleinikova TD, Uverskii VN, Kuznetsova IM, Turoverov KK. [The role of quarternary structure in fluorescent protein stability] Tsitologiia. 2005;47:1017–27. [PubMed] [Google Scholar]
- Studier FW. Use of bacteriophage T7 lysozyme to improve an inducible T7 expression system. J Mol Biol. 1991;219:37–44. doi: 10.1016/0022-2836(91)90855-z. [DOI] [PubMed] [Google Scholar]
- Troise F, Cafaro V, Giancola C, D’Alessio G, De Lorenzo C. Differential binding of human immunoagents and Herceptin to the ErbB2 receptor. FEBS J. 2008;275:4967–79. doi: 10.1111/j.1742-4658.2008.06625.x. [DOI] [PubMed] [Google Scholar]
- Tsien RY. Constructing and exploiting the fluorescent protein paintbox (Nobel Lecture) Angew Chem Int Ed Engl. 2009;48:5612–26. doi: 10.1002/anie.200901916. [DOI] [PubMed] [Google Scholar]
- Winter G. Synthetic human antibodies and a strategy for protein engineering. FEBS Lett. 1998;430:92–4. doi: 10.1016/s0014-5793(98)00628-0. [DOI] [PubMed] [Google Scholar]
- Winter G, Milstein C. Man-made antibodies. Nature. 1991;349:293–9. doi: 10.1038/349293a0. [DOI] [PubMed] [Google Scholar]
- Worn A, Pluckthun A. An intrinsically stable antibody scFv fragment can tolerate the loss of both disulfide bonds and fold correctly. FEBS Lett. 1998;427:357–61. doi: 10.1016/s0014-5793(98)00463-3. [DOI] [PubMed] [Google Scholar]
- Worn A, Pluckthun A. Stability engineering of antibody single-chain Fv fragments. J Mol Biol. 2001;305:989–1010. doi: 10.1006/jmbi.2000.4265. [DOI] [PubMed] [Google Scholar]
- Yang F, Moss LG, Phillips GN., Jr. The molecular structure of green fluorescent protein. Nat Biotechnol. 1996;14:1246–51. doi: 10.1038/nbt1096-1246. [DOI] [PubMed] [Google Scholar]