

Abstract
Bacteriophage phi29 DNA packaging motor is geared by a six-pRNA ring. pRNA is able to form a multimeric complex and patterned superstructures via the interaction of two reengineered interlocking loops. This unique feature makes it an ideal polyvalent vehicle for nanomachine fabrication, pathogen detection, and the delivery of therapeutics. This report describes novel approaches for the fabrication of polyvalent therapeutic pRNA nanoparticles, especially tetramers for specific siRNA delivery to cancer cells and for the silencing of targeted genes. RNA 3-D design, circular permutation, folding energy alteration, and nucleotide modification were applied to generate stable RNA nanoparticles with low toxicity. Animal trials demonstrated the high efficiency of the polyvalent RNA nanoparticles in the prevention and treatment of cancer. Using such protein-free nanoparticles as therapeutic reagents would allow for long-term administration to avoid the induction of antibody due to repeated treatment for chronic diseases.
I. INTRODUCTION
Biological macromolecules, including DNA, RNA and proteins, intrinsically have defined features at the nanometer scale. They could serve as unique and powerful building blocks for the bottom-up fabrication of nanostructures and nanodevices [1–9]. What makes RNA particularly useful is the amazing diversity evident in its functionality, which can be directly attributed to the versatility present in its structure. RNA molecules can easily be designed and manipulated, similar to DNA, and offer flexibility of structure and function similar to that of proteins. These features make RNA a favorable candidate for nanotechnological applications. Typically, RNA molecules contain a large variety of single-stranded stem-loops for inter- and/or inter-molecular interactions. These loops can serve as mounting dovetails, and thus, external linking dowels might not be needed in fabrication and assembly [7–9]. RNA molecules are polymers made up of four nucleotides, A, C, G, and U. A 100-nucleotide RNA polymer can generate as many as 1060 (4100) different RNA molecules. Since the size of RNA ranges from the angstrom to the nanometer, bottom-up approaches would be reasonable for use with RNA in nanotechnological applications.
The pRNA (DNA packaging RNA) of the DNA packaging motor of bacteriophage phi29 can serve as a building block for nanomaterials via bottom-up assembly. Through the interaction of two interlocking loops, phi29 pRNA forms dimers, trimers, hexamers, and patterned superstructures [7]. This unique property makes pRNA an ideal building block for bottom-up assembly in nanotechnology.
Recent development of molecular therapy is one of the most promising applications of modern biological science. siRNA [10–13], ribozyme [14], and antisense RNA [15] have all shown great potential as novel molecular approaches to down-regulate specific gene expression. The successful application of siRNA for the treatment of cancer and infectious diseases requires the development of a safe, efficient, specific, and nonpathogenic delivery system. There are several barriers which make it difficult to develop a successful delivery system. Firstly, therapeutic particles greater than 100 nm are difficult for cell entry via receptor-mediated endocytosis. Secondly, molecules smaller than 20 nm can move out of blood vessels or the kidney quickly during circulation and have a shorter retention time in the body. pRNA multimers may provide the answer because they range in size of 20 ~ 40nm. The unique structural and molecular features of pRNA allow easy manipulation, making it possible to redesign for gene targeting and delivery vehicles.
II. SYNTHESIS OF AMP AND GMP DERIVATIVES FOR RNA LABELING
Different kinds of AMP and GMP derivatives can be synthesized by conjugating different chemical groups with AMP or GMP through linker molecules by established chemistry [16–20]. GMPS or Biotin-AMP (Fig. 1) was introduced to the 5′-end of pRNA chimeras since GMP and AMP derivatives are efficient initiators in RNA transcription by T7 RNA polymerase but cannot be used during chain elongation due to the one phosphate. It was confirmed that a pRNA with the first nucleotide replaced by GMPS [18] or Biotin-AMP [17] did not interfere with pRNA multimerization. The SH-group of the biotin at the 5′-end of the pRNA was used as active groups for conjugation of ligands and detection markers for the production of deliverable polyvalent therapeutic particles. SH-groups can be used to link either a NH2-group via BMPS or any common chemicals that contain maleimide. Using similar approaches, it was possible to efficiently synthesize RNA directly and incorporate certain kinds of groups, such as amino-NH2, -COOH, Maleimide, or NHS for constructing polyvalent RNA nanoparticles. The NH2-group was used to link any particles with a COOH-group with the help of EDC. NH2/NH2 interaction was achieved via heterobifunctional crosslinkers.
Fig. 1.
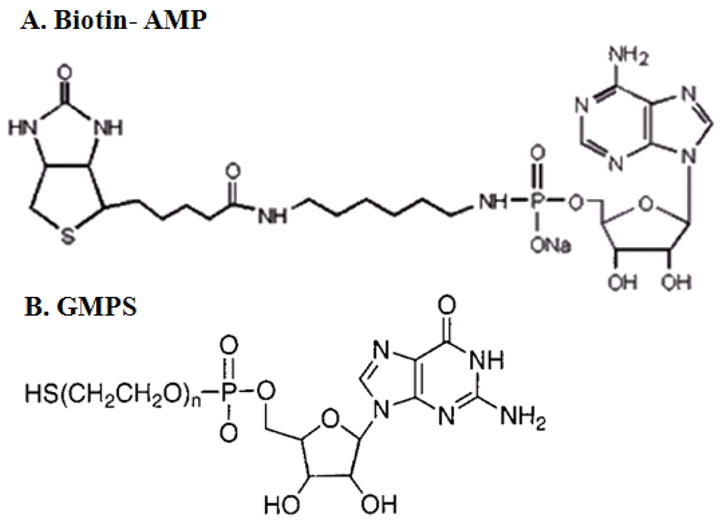
AMP and GMP Derivatives. Biotin-AMP and GMPS can be synthesized by conjugating chemical group with AMP or GMP through the linker molecules [17, 18].
III. CONSTRUCTION OF MONOMERIC pRNA CHIMERA FOR TARGETED DELIVERY
pRNA contains two functional domains (Fig. 2), the procapsid binding domain and the gp16 binding domain[21,22]. The former is located at the central region [23–29] comprising bases 23–97, while the latter is located at the 5′/3′ paired ends [30]. The two domains fold independently of each other and replacement of or insertion to the 5′/3′ helical domain does not interfere with dimer formation. Thus, end conjugation of pRNA with chemical moiety, fusing pRNA with a receptor-binding RNA aptamer, small interfering RNA (siRNA), or ribozyme did not disturb dimer formation or interfere with the function of inserted moieties. Using the circular permutation approach [27–29], almost any nucleotide of the entire pRNA can serves as either the new 5′ or 3′ end of the RNA monomer. With the end labeling technology described above, it was possible to label the pRNA at alternative locations.
Fig. 2.
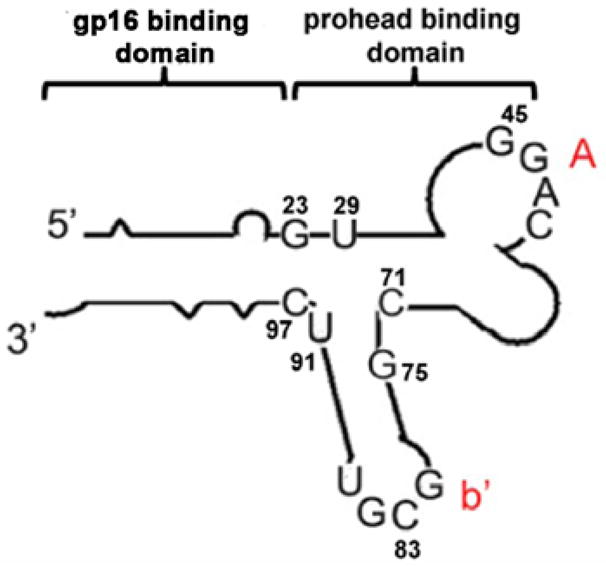
Illustration of pRNA monomer structure. Two domains fold independently of each other. pRNA form multimers via the right hand loop (uppercase) and the left hand loop (lowercase) interaction.
Another approach was to extend the 3′ end of the pRNA with 14–26 nucleotides. A complementary DNA oligo can be annealed to the end of the pRNA. The synthetic DNA oligos which contained different chemical groups were used for therapeutic pRNA construction (Fig. 5A).
Fig. 5.

Illustration of constructing pRNA multimers. A. pRNA chimeric monomer with 3′-end annealed oligo which conjugate the detection molecule and chemical ligand at the same time. B. a. Twin dimer formed through palindrome sequence; b. Dimer formation through loop-loop interaction. C. Tetrameric RNA chimera. D. pRNA Trimer. E. Polyacrylamide gel showing pRNA multimers in native (upper panel) and denatured (lower panel) condition. RNA nomenclature: To simplify the description of RNA construction and multimer assembly, uppercase letters will be used to represent the right hand loop of pRNA and lowercase letters to represent the left hand loop. The same letters in upper and lower cases indicate complementary sequences for loop/loop interaction, while different letters indicate non-complementary loops. For example, pRNA A-b′ represents pRNA where right loop A (5′G45G46A47C48) is complementary to left loop a′ (3′C85C84U83G82) of pRNA B-a′. And left loop b′ (3′U85G84C83G82) of A-b′ pRNA is complementary to the right loop B (5′A45C46G47C48) of pRNA B-a′.
Harboring therapeutic RNA
pRNA double-stranded 5′/3′ end helical domain and intermolecular binding domain fold independently of each other. Complementary modification studies have revealed that altering the primary sequences of any nucleotide of the helical region does not affect pRNA structure and folding as long as the two strands are paired [30]. Extensive studies revealed that siRNA is a double-stranded RNA helix [10–13]. Thus, it was possible to replace the helical region in pRNA with double-stranded siRNA. A variety of chimeric pRNAs with different targets were constructed to carry siRNA connected to nucleotide (nt) 29 and 91 of the pRNA (Fig. 2). Such pRNA/siRNA chimera was proven to be the building block which successfully inhibited target gene expression [31,32] (Fig. 3).
Fig. 3.
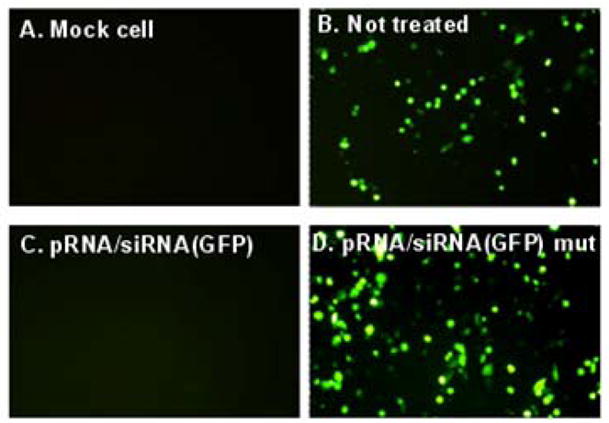
Specific GFP knockdown assay of chimeric pRNA/siRNA (GFP) in KB cells.
In addition, connecting the pRNA 5′/3′ ends with variable sequences did not disturb its folding and function. These unique features, which help prevent two common problems - exonuclease degradation and misfolding in the cell, make pRNA an ideal vector to carry therapeutic RNA. A pRNA-based vector was designed to carry hammerhead ribozymes that cleave the hepatitis B virus (HBV) polyA signal [14]. The pRNA/ribozyme(survivin), which targeted the anti-apoptosis factor survivin to down-regulate genes involved in tumor development and progression, was also shown to suppress survivin expression and initiate apoptosis [33].
Construction of monomeric pRNA chimera for cell targeting
In vitro SELEX [34,35] to screen RNA molecules, or “aptamers”, which bind to specific targets, has become a powerful tool for selecting RNA molecules specific to cell surface receptor. Aptamers were linked to the 3′ and 5′ end of pRNA. To facilitate independent folding, poly U or poly A linkers were placed between the pRNA and the aptamer. The nascent 5′/3′ end of the pRNA were relocated to nt 71 and 75 (Fig. 2) using the circular permutation approach [27,36]. The tightly folded nt 71 and 75 region protected the 5′/3′ end from exonuclease digestion. Folate was also conjugated to pRNA for specific targeting (Fig. 4) [16,37–40].
Fig. 4.
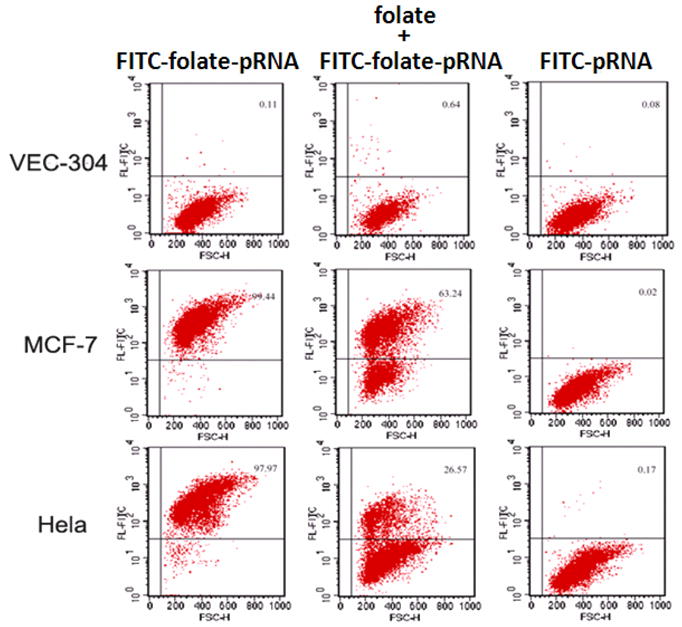
Folate conjugated pRNA showing specific binding to folate positive cancer cell lines (MCF-7 and Hela) while comparing to the folate-free cell line VEC-304.
IV. ASSEMBLY OF DELIVERABLE TETRAMERIC NANOPARTICLES
Dimers were formed by intermolecular interaction of the interlocking right and left loops [41] that are trans-complementary (Fig. 5B.b). Mixing two pRNAs with interlocking loops, for example pRNA A-b′ and B-a′ in a 1:1 molar ratio, resulted in the production of pRNA dimers with close to 100% efficiency [7,8]. One subunit of the dimer was modified to carry RNA aptamer which recognized a specific cell-surface receptor, thereby inducing receptor-mediated endocytosis. The second subunit of the dimer carried therapeutic molecules such as siRNA, ribozyme, or drugs for therapeutic purposes. Dimers were also efficiently self-assembled in solution by introducing a palindrome sequence into the 3′-end of the RNA chimera (Fig. 5B.a).
Tetrameric (Fig. 5C) pRNA nanoparticles were also assembled using the similar principle of intermolecular interlocking loop/loop interactions. To assemble a tetramer, a dimeric pRNA Ab′/Ab′ was first constructed by introducing a six-nucleotide palindrome sequence to the 3′-end of the pRNA. Dimeric pRNA Ba′/Ba′ was also constructed using the same principle. After purification, dimeric pRNA Ab′/Ab′ was mixed with dimeric pRNA Ba′/Ba′ in equal concentrations. Tetramers were self-assembled in the solution with high efficiency.
V. pRNA NANOPARTICLES FOR TARGETED GENE DELIVERY AND THERAPY
The use of small RNA in gene therapy was significantly hampered by the difficulties involved in producing a safe and efficient system by which specific cells could be targeted. The strength of using phi29 pRNA as a delivery vehicle relied on its ability to easily form stable multimers which could be manipulated and sequence-controlled [8,24,42]. This particular system provides unprecedented versatility in constructing polyvalent delivery vehicles by separately constructing individual pRNA subunits with various cargos and mixing them together in any desired combination [9]. For example, the deliverable pRNA complex can be altered to carry therapeutic siRNA, ribozyme RNA, antisense RNA against multiple targets or different regions of one target gene, and RNA aptamer or folic acid for delivery. The other subunits of the RNA complex may carry anti-cancer drugs to enhance the therapeutic effect or to overcome the drug resistance by combination therapy. The therapy and detection of the therapeutics could also be combined into one nanoparticle, making the concomitant therapy and detection of the therapeutics possible with only one administration. Animal trials demonstrated the high efficiency of the polyvalent RNA nanoparticles in the prevention and treatment of cancer.
The homogeneity in size of the pRNA nanoparticles was of extreme importance. Highly efficient and controlled bottom-up self-assembly yielded nanoparticles with well defined structures and stoichiometry. This characteristic was very valuable for the reproducible manufacturing of drugs for the safety issue and will facilitate the FDA approval of the therapeutic reagents. Due to the specific targeting and longer retention time, only low concentration of the nanoparticle is required for administration. In addition, if the RNA particle contained certain toxicity to the cells, the resistant of entry to receptor- free cells due to the larger size of the particle could reduce such effect. The pRNA nanoparticles (dimers, trimers or tetramers) had optimal sizes ranging between 20 and 40 nm. Nanoparticle delivery of siRNA has the potential to improve the pharmacokinetics, pharmacodynamics, biodistribution, and toxicology associated with drug delivery. Furthermore, it can reduce negative side effects such as trapping by Kupffer cells in the liver and macrophages in the lungs.
The incorporation of receptor binding aptamer, folate, or other ligands to the RNA complex with simple procedures ensured the specific binding and targeted delivery to cells. In combination with the advantage of the nano-scale size, the system provides the advantage of high efficiency in gene silencing and low toxicity in treatment.
Acknowledgments
This work was supported by NIH Grants (R01-GM59944, EB003730). Peixuan Guo is the co-founder of Kylin Therapeutics, Inc.
References
- 1.Glotzer SC. Science. 2004;306:419–420. doi: 10.1126/science.1099988. [DOI] [PubMed] [Google Scholar]
- 2.Gates BD, Xu Q, Stewart M, Ryan D, Willson CG, Whitesides GM. Chem Rev. 2005;105:1171–1196. doi: 10.1021/cr030076o. [DOI] [PubMed] [Google Scholar]
- 3.Leontis NB, Lescoute A, Westhof E. Curr Opin Struct Biol. 2006;16:279–287. doi: 10.1016/j.sbi.2006.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaeger L, Chworos A. Curr Opin Struct Biol. 2006;16:531–543. doi: 10.1016/j.sbi.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 5.Jaeger L, Leontis NB. Angew Chem Int Ed Engl. 2000;39:2521–2524. doi: 10.1002/1521-3773(20000717)39:14<2521::aid-anie2521>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 6.Seeman NC. Nature. 2003;421:427–431. doi: 10.1038/nature01406. [DOI] [PubMed] [Google Scholar]
- 7.Shu D, Moll D, Deng Z, Mao C, Guo P. Nano Lett. 2004;4:1717–1724. doi: 10.1021/nl0494497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shu D, Huang L, Hoeprich S, Guo P. J Nanosci Nanotechnol. 2003;3:295–302. doi: 10.1166/jnn.2003.160. [DOI] [PubMed] [Google Scholar]
- 9.Guo P. Journal of Nanoscience and Nanotechnology. 2005;5(12):1964–1982. doi: 10.1166/jnn.2005.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H, Li WX, Ding SW. Science. 2002;296:1319–1321. doi: 10.1126/science.1070948. [DOI] [PubMed] [Google Scholar]
- 11.Brummelkamp TR, Bernards R, Agami R. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 12.Carmichael GG. Nature. 2002;418:379–380. doi: 10.1038/418379a. [DOI] [PubMed] [Google Scholar]
- 13.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 14.Hoeprich S, Hou QZ, Guo S, Qi G, Wang Y, Guo P. Gene Ther. 2003;10:1258–1267. doi: 10.1038/sj.gt.3302002. [DOI] [PubMed] [Google Scholar]
- 15.Kumar M, Carmichael GG. Microbiol Mol Biol Rev. 1998;62:1415–1434. doi: 10.1128/mmbr.62.4.1415-1434.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo S, Huang F, Guo P. Gene Ther. 2006;13:814–820. doi: 10.1038/sj.gt.3302716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang F, Wang G, Coleman T, Li N. RNA. 2003;9:1562–1570. doi: 10.1261/rna.5106403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang L, Sun L, Cui Z, Gottlieb RL, Zhang B. Bioconjug Chem. 2001;12:939–948. doi: 10.1021/bc015504g. [DOI] [PubMed] [Google Scholar]
- 19.Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC. Nucleic Acids Res. 1987;15:8783–8798. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bruce AG, Uhlenbeck OC. Nucleic Acids Res. 1978;5:3665–3677. doi: 10.1093/nar/5.10.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee TJ, Guo P. J Mol Biol. 2006;356:589–599. doi: 10.1016/j.jmb.2005.10.045. [DOI] [PubMed] [Google Scholar]
- 22.Koti JS, Morais MC, Rajagopal R, Owen BA, McMurray CT, Anderson D. J Mol Biol. 2008;381:1114–1132. doi: 10.1016/j.jmb.2008.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reid RJD, Bodley JW, Anderson D. J Biol Chem. 1994;269:5157–5162. [PubMed] [Google Scholar]
- 24.Chen C, Sheng S, Shao Z, Guo P. J Biol Chem. 2000;275(23):17510–17516. doi: 10.1074/jbc.M909662199. [DOI] [PubMed] [Google Scholar]
- 25.Garver K, Guo P. RNA. 1997;3:1068–1079. [PMC free article] [PubMed] [Google Scholar]
- 26.Reid RJD, Zhang F, Benson S, Anderson D. J Biol Chem. 1994;269:18656–18661. [PubMed] [Google Scholar]
- 27.Zhang CL, Tellinghuisen T, Guo P. RNA. 1997;3:315–322. [PMC free article] [PubMed] [Google Scholar]
- 28.Nolan JM, Burke DH, Pace NR. Science. 1993;261:762–765. doi: 10.1126/science.7688143. [DOI] [PubMed] [Google Scholar]
- 29.Pan T, Uhlenbeck OC. Gene. 1993;125:111–114. doi: 10.1016/0378-1119(93)90317-v. [DOI] [PubMed] [Google Scholar]
- 30.Zhang CL, Lee CS, Guo P. Virology. 1994;201:77–85. doi: 10.1006/viro.1994.1267. [DOI] [PubMed] [Google Scholar]
- 31.Khaled A, Guo S, Li F, Guo P. Nano Letters. 2005;5:1797–1808. doi: 10.1021/nl051264s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo S, Tschammer N, Mohammed S, Guo P. Hum Gene Ther. 2005;16:1097–1109. doi: 10.1089/hum.2005.16.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu H, Guo S, Roll R, Li J, Diao Z, Shao N, Riley MR, Cole AM, Robinson JP, Snead NM, Shen G, Guo P. Cancer Biol Ther. 2007;6:697–704. doi: 10.4161/cbt.6.5.3962. [DOI] [PubMed] [Google Scholar]
- 34.Ellington AD, Szostak JW. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 35.Tuerk G, Gold L. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 36.Zhang CL, Trottier M, Guo PX. Virology. 1995;207:442–451. doi: 10.1006/viro.1995.1103. [DOI] [PubMed] [Google Scholar]
- 37.Lu Y, Low PS. J Control Release. 2003;91:17–29. doi: 10.1016/s0168-3659(03)00215-3. [DOI] [PubMed] [Google Scholar]
- 38.Sudimack J, Lee RJ. Adv Drug Deliv Rev. 2000;41:147–162. doi: 10.1016/s0169-409x(99)00062-9. [DOI] [PubMed] [Google Scholar]
- 39.Lee RJ, Low PS. J Biol Chem. 1994;269:3198–3204. [PubMed] [Google Scholar]
- 40.Mathias CJ, Wang S, Lee RJ, Waters DJ, Low PS, Green MA. J Nucl Med. 1996;37:1003–1008. [PubMed] [Google Scholar]
- 41.Hoeprich S, Guo P. J Biol Chem. 2002;277(23):20794–20803. doi: 10.1074/jbc.M112061200. [DOI] [PubMed] [Google Scholar]
- 42.Guo P, Zhang C, Chen C, Trottier M, Garver K. Mol Cell. 1998;2:149–155. doi: 10.1016/s1097-2765(00)80124-0. [DOI] [PubMed] [Google Scholar]