

Abstract
We discovered the acute inhibition of myocardial phospholipase A2 activity by micromolar concentrations of tert-butyl hydroperoxide and hydrogen peroxide. Specifically, freshly isolated adult rat cardiomyocytes were treated with the oxidants for 30 min, and phospholipase A2 activity was assessed in cell subcellular fractions using (16:0, [3H]18:1) plasmenylcholine and phosphatidylcholine substrates in the absence or presence of calcium. Calcium-independent phospholipase A2 activity was inhibited by approx 50% in both the cytosolic and membrane fractions by the oxidants. The inhibition of the phospholipase A2 activity was concentration dependent and preceded detectable changes in cell viability as assessed by lactate dehydrogenase release and rod-shaped morphology. Taking into account that oxidized sn-2 fatty acyl groups must first be hydrolyzed by phospholipase A2 to be repaired by glutathione peroxidase, we suggest that the observed inhibition of phospholipase A2 activity by oxidants compromises the myocyte’s ability to deal with lipid peroxidation. This conclusion was further validated by the experiments in which pretreatment with the calcium-independent phospholipase A2 inhibitor bromoenol lactone exacerbated cardiotoxicity of tert-butyl hydroperoxide in myocyte cultures.
Keywords: Phospholipases, oxidative stress, lipid peroxidation, hydrogen peroxide, tert-butyl hydroperoxide
Introduction
Phospholipase A2 (PLA2) represents a diverse family of enzymes, which hydrolyze the fatty acyl group from the sn-2 position of glycerophospholipids, releasing a lysophospholipid and a free fatty acid. PLA2s are involved in many biological processes, regulating the release of second messengers (e.g., eicosanoids and lysophospholipids), and growth factors (lysophosphatidic acid), as well as maintaining the composition and hence the physical properties of the membrane (1,2).
Three types of PLA2 have been described in mammalian tissues: secretory (sPLA2), cytosolic Ca2+-dependent (cPLA2), and intracellular Ca2+-independent enzymes (iPLA2). To date, all PLA2 types have been shown to be present in heart tissue and in cardiac myocytes (3–6). Reports from different laboratories, including ours (4,5,7), have shown that the majority of PLA2 in cardiomyocytes is calcium independent and is associated with the membrane fraction.
A number of PLA2s display enhanced catalytic activity in response to cell membrane lipid peroxidation (8–10). The conformational changes imposed by oxidized phospholipids are consistent with reported increases in PLA2 activity at membrane sites that have packing defects and appear to be most prominent for membranes containing phospholipid hydroperoxides (11). Therefore, an increase in PLA2 activity upon treatment with oxidants would be expected. However, it is not known whether imposed oxidative stress affects PLA2 directly.
We have recently shown that doxorubicin, an anti-cancer drug known to increase free-radical formation (12), inhibits intracellular myocardial PLA2 activity both in vivo and in vitro (7). We have also shown that reducing agents, such as dithiothreitol and glutathione, prevent enzyme inhibition. These data suggested that myocardial iPLA2 activity is affected by the status of its essential thiol residues and may be a target of free-radical damage. To test this hypothesis directly, we subjected isolated rat cardiomyocytes to oxidative stress using either hydrogen peroxide (H2O2) or tert-butyl hydroperoxide (t-BOOH) and assayed the activity of PLA2 in both membrane and cytosolic fractions. We found that (1) both cytosolic and membrane iPLA2 are markedly inhibited by micromolar concentrations of H2O2 or t-BOOH and (2) the specific inhibitor of iPLA2, bromoenol lactone (BEL), significantly potentiates oxidant cardiotoxicity.
Materials and Methods
Materials
Collagenase (type II) was purchased from Worthington Biochemical. [3H]Arachidonic acid and [3H] oleic acid were purchased from NEN. Bromoenol lactone (BEL) was a gift from Hoffmann-LaRoche. t-BOOH, minimum essential media (MEM), gentamicin, albumin, and HEPES lactate dehydrogenase (LDH) assay kit and other reagents were purchased from Sigma.
Preparation of Rat Ventricular Cardiomyocytes
Cells were obtained from adult Sprague-Dawley male rats (200–300 g) using retrograde Langendorff perfusion with collagenase. A yield of (5–9) × 106 myocytes per heart was routinely obtained. Myocyte viability was evaluated by the microscopic determination of the number of rod-shaped cells and the number of myocytes that excluded trypan blue.
Short-Term Treatment of Myocytes with Oxidants
Myocytes in suspension (0.25 × 106cell/mL of Tyrode, supplemented with 10 mM HEPES, pH 7.3) were incubated at room temperature with designated oxidant concentrations for 30 min. At the end of incubation, a 100-μL aliquot of myocyte suspension was taken for viability assessment, and the rest of the cells were washed in oxidant-free Tyrode and used for PLA2 assay.
Long-Term Treatment of Myocytes with t-BOOH
Cardiomyocytes were plated onto twelve 12-mm laminin-covered glass cover slips and preincubated for 4–5 h in MEM supplemented with 5 mM HEPES, 10 μg/mL gentamicin, 0.1 μg/mL streptomycin, and 0.1 U/mL penicillin. Each slip was then placed in a separate well of a 32-well plate containing 1 mL MEM. Ten molars of BEL were added, followed by the addition of specified concentrations of t-BOOH, which was done 30 min after BEL application. The same procedure (BEL followed by t-BOOH) was repeated every 12 h without media change. At the end of the 48-h treatment regimen, the viability of each slip was evaluated by LDH assay and morphology. Specifically, after 0.5 mL of media was removed for the assessment of released LDH, cells were fixed by the addition of 0.5% glutaraldehyde-containing medium and counted (at least 200 cells per cover slip).
Preparation of Particulate, Cytosolic, and Membrane Fractions
Myocytes were suspended in an ice-cold buffer containing (mmol/L) 250 sucrose, 10 KCl, 10 imidazole, 5 EDTA, 2 dithiothreitol (DTT) with 10% glycerol, and pH = 7.8 (PLA2 assay buffer). To obtain a particulate fraction (samples used for PLA2 assay in Fig. 1), the suspensions were sonicated six times for 10 s, and the sonicate was centrifuged at 100,000g. The resulting pellet was resuspended in PLA2 assay buffer. To separate membrane and cytosolic fractions (samples used for PLA2 assay in Figs. 2 and 3), cell sonicates were centrifuged at 14,000g for 20 min to remove nuclei and mitochondria, and the resultant supernatant fraction was centrifuged at 100,000g for 1 h. The membrane fraction (pellet) was separated from the cytosolic fraction (supernatant) and resuspended in PLA2 assay buffer.
Fig. 1.

Activity of iPLA2 in the particulate fraction of myocytes treated with oxidants. Freshly isolated cardiomyocytes in suspensions (0.25 × 106cell/mL) were incubated with designated concentrations of oxidants for 30 min, washed with oxidant-free Tyrode, followed by sonication in an ice-cold PLA2 assay buffer. Enzyme activity in particular fractions was measured using (16:0, [3H]18:1) plasmenylcholine in the presence of 4 mM EGTA. *p < 0.05, **p < 0.005 versus corresponding controls. The average from three experiments that had each assay run in duplicate is shown. (A) PLA2 activity incubated with t-BOOH; (B) PLA2 activity incubated with H2O2; (C) PLA2 activity incubated with DOX.
Fig. 2.

Activity of PLA2 in membrane and cytosolic fractions. Freshly isolated cardiomyocytes in suspensions (0.25 × 106cell/mL) were incubated with designated concentrations of oxidants for 30 min, washed with oxidant-free Tyrode, and followed by sonication in ice-cold PLA2 assay buffer. Enzyme activity as measured using (16:0, [3H]18:1) plasmenylcholine in the presence of 4 mM EGTA. The average from three experiments that had each assay run in duplicate is shown. (A) PLA2 activity in the membrane fraction of myocytes treated for 30 min with 25 μM t-BOOH or 25 μM H2O2; (B) PLA2 activity in the cytosolic fraction of myocytes treated for 30 min with 25 μM t-BOOH or 25 μM H2O2; (C) cell viability as assessed by rod-shaped morphology after 30 min of treatment with either 25 μM t-BOOH or 25 μM H2O2.
Fig. 3.

Inhibitory effects of oxidants on membrane-associated PLA2 activity. Freshly isolated cardiomyocytes in suspensions (0.25 × 106cell/mL) were incubated with 25 μM t-BOOH for 30 min, washed with oxidant-free Tyrode, and followed by sonication in an ice-cold PLA2 assay buffer. After cell fractionation, membrane-associated PLA2 activity was assayed using either (16:0, [3H]18:1) plasmenylcholine or (16:0, [3H]18:1) phosphatidylcholine in the absence (4 mM EGTA) or presence of calcium (1 mM). Values are means from three experiments that had each assay run in duplicate.
Assay of PLA2 Activity
Phospholipase A2 activity was quantified by incubating enzyme (8μg particulate protein, 8μg membrane protein, or 200μg cytosolic protein) with 100 μM (16:0, [3H]18:1) plasmenylcholine or phosphatidylcholine in assay buffer containing 100 mM Tris-HCl, 10% glycerol, and pH = 7.0, with either 4 mM EGTA or 1 mM CaCl2 at 37°C for 5 min in a total volume of 200 μL. Reactions were terminated by the addition of 100 μL butanol. Released radiolabeled fatty acid was isolated by thin-layer chromatography on silica G plates, followed by development in petroleum, ether–diethyl acid, and ether–acetic acid (70:30:1 v/v/v), and quantification by liquid scintillation spectrometry. The reaction conditions selected resulted in linear reaction velocities with respect to both time and total protein concentration for each substrate examined. Protein content was determined by the Lowry method.
Statistical Analysis
We used the Student’s t-test for the statistical comparison of values. All results are expressed as means ± SEM. Statistical significance was considered to be p < 0.05.
Results
The Inhibition of Particulate iPLA2 Activity by Oxidants
Suspensions of freshly isolated myocytes were incubated with either H2O2, t-BOOH, or doxorubicin, then washed, sonicated, and assessed for iPLA2 activity, measured using (16:0, [3H]18:1) plasmenylcholine substrate in the absence of calcium (4 mM EGTA). A 0.5-h incubation of myocytes with micromolar concentrations of either H2O2 or t-BOOH (an artificial organic hydroperoxide [HOO–C–(CH3)3]) resulted in a concentration-dependent decrease in iPLA2 activity associated with the particulate fraction (Fig. 1A,B). Doxorubicin (DOX) at a 1-μM concentration also exerted an inhibitory effect (Fig. 1C), in accordance with our previous work (7). The degree of enzyme inhibition by high concentrations of oxidants (500 μM) was only slightly greater than by the one produced by 50 μM of oxidants.
Inhibition of Cytosolic and Membrane-Associated PLA2 Activity by Oxidants
The measurement of iPLA2 activity in cytosolic and membrane fractions from cardiomyocytes treated with 25 μM t-BOOH or 25 μM H2O2 reveals the marked inhibition of enzyme activity in both fractions (Fig. 2A,B). The effect is not caused by the enzyme leakage or proteolytic degradation because short-term treatment did not cause a detectable loss of myocyte viability (Fig. 2C). The 50% decrease in PLA2 activity by either 25 μM t-BOOH or 25 μM H2O2 observed in both cytosolic and membrane fractions was greater than the inhibition of iPLA2 measured in the particulate fraction in Fig. 1. Furthermore, PLA2 activity was inhibited with t-BOOH or H2O2 when measured using both plasmenylcholine and phosphatidylcholine substrates in the presence or absence of calcium. Almost complete inhibition of the membrane activity was observed with phosphatidylcholine substrate in the presence of calcium (Fig. 3).
Cytotoxic Effects of Combined t-BOOH/BEL Treatment
To directly test whether the inhibition of iPLA2 activity impairs the myocyte’s ability to withstand oxidative injury, we performed the following experiment. Isolated cardiomyocytes were cultured in serum-free conditions, which maintain the cell’s rod-shape phenotype for several days (13). Cells were pretreated with the iPLA2 inhibitor BEL (14), followed by the addition of a low concentration of t-BOOH (5–25 μM). This procedure was repeated every 12 h and cell viability was assessed by LDH release 48 h later. As shown in Fig. 4, t-BOOH treatment by itself had little effect on cell viability under these conditions. Likewise, samples treated with only BEL were also not significantly different from controls. However, the pretreatment with BEL significantly decreased viability in samples treated with either 5 or 25 μM t-BOOH. Assessment of viability using rod-shaped morphology confirmed the results obtained with the LDH assay (Fig. 5).
Fig. 4.

Cytotoxicity of combined BEL/t-BOOH treatment: LDH assay. Myocytes were pretreated with 10 μM BEL for 30 min before each t-BOOH application. t-BOOH additions were made every 12 h, and the viability of myocytes was assessed at the end of the 48-h protocol using a LDH release assay. Values represent the mean of four separate preparations. Each preparation consisted of duplicate cover slips for the control and BEL samples and quadruplicates for the t-BOOH and t-BOOH/BEL treatments. **p < 0.01 (versus control samples), #p < 0.05 (versus t-BOOH samples).
Fig. 5.
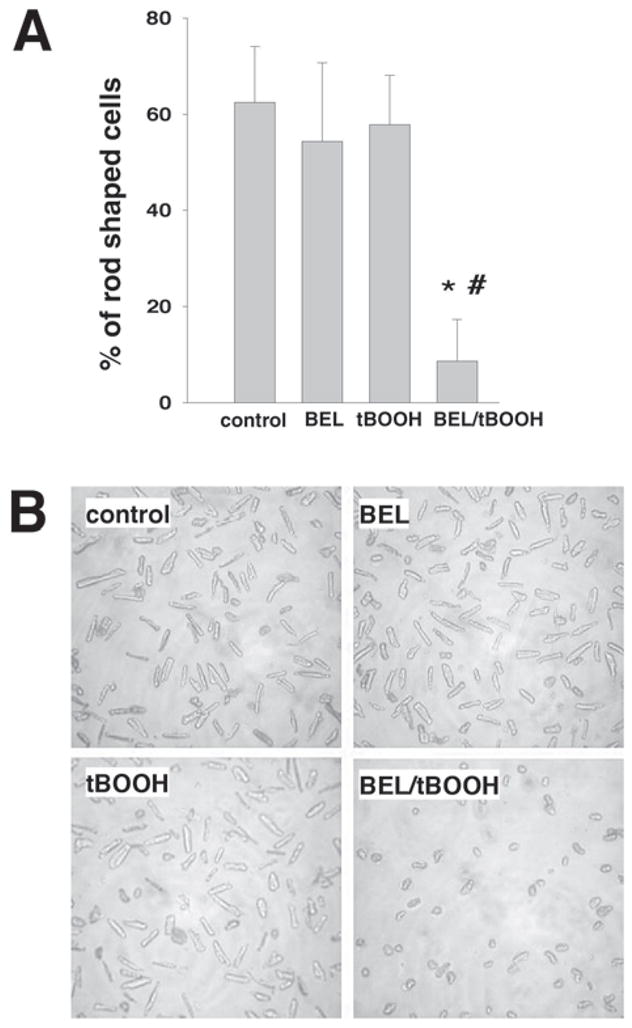
Cytotoxicity of combined BEL/t-BOOH treatment: cell morphology assessment. Myocytes were pre-treated with 10 μM BEL for 30 min before each t-BOOH application. t-BOOH additions were made every 12 h and the viability of myocytes was assessed at the end of the 48-h protocol. Values represent the mean of four separate preparations. (A) Percentage of rod-shaped cells in control, 10 μM BEL, 25 μM t-BOOH, and 10 μM BEL/25 μM t-BOOH samples at the end of the 48-h treatment. (B) Representative images of samples taken at the end of the 48-h treatment.
Discussion
In the first series of experiments, we assessed iPLA2 activity using particulate fractions of oxidant-treated cells. The inhibitory effect on enzyme activity was concentration dependent within a 25- to 50 μM oxidant range (Fig. 1). A 10-fold increase in H2O2 and t-BOOH concentrations only slightly augmented their effect on iPLA2 activity (unavoidable loss of viable cells associated with high oxidant concentrations prevents the use of increased concentrations). The observation that high oxidant concentrations did not completely inhibit the enzyme suggests three possibilities. First, oxidation-mediated changes may not lead to a complete inactivation of the enzyme. Second, a certain portion of the enzyme present in the particulate fraction may be resistant to oxidation-dependent loss of activity. The multiplicity of existent iPLA2 isoforms (1,15,16) makes such an assumption highly plausible. Notably, treatment with doxorubicin also failed to achieve complete enzyme inhibition in homogenates of both rat and rabbit cardiomyocytes and had no effect on cytosolic iPLA2 activity (7). Third, the association of iPLA2 with particular organelles can limit the potency of oxidants resulting from the differences in the redox state of the protein’s immediate environment (17).
The next series of experiments focused on enzyme assessment in cytosolic and membrane fractions. Myocytes incubated with either 25 μM H2O2 or t-BOOH for 30 min demonstrated a 50% inhibition in iPLA2 activity in cytosolic and membrane fractions (Fig. 2). Notably, the degree of iPLA2 inhibition by 25 μM t-BOOH or H2O2 was much less in the particular fraction (approx 20%) than in the membrane fraction. These findings suggest that PLA2 activity associated with other constituents of particular fraction such as nucleus, mitochondria, myofibrils, or small organelles may be less affected by the oxidants.
The magnitude of iPLA2 inhibition varied when different substrates with or without calcium were used (Fig. 3). Substrate-related differences are commonly observed for changes in PLA2 activity induced either by chemicals (4,5) or by ischemia like conditions (18,19). Notably, the magnitude of enzyme inhibition was maximal when phosphatidylcholine in the presence of 1 mM of calcium was used to assess enzyme activity (Fig. 3). The same substrate specificity was observed for doxorubicin-induced PLA2 inhibition (7), suggesting a similar underlying mechanism.
Although transcription of many proteins can be decreased through redox-sensitive pathways such as NFκB/AP-1 (20), an observed rapid loss of PLA2 activity (<30 min) implies a direct effect of oxidants on existing PLA2 protein. Reports by others (21,22) and our recent study of doxorubicin (7) suggest that the oxidation of essential thiols may be a culprit in the observed iPLA2 inhibition. However, the complexity of the pathways ensuing from oxidative injury precludes one from an immediate conclusion, and the exact mechanism behind the H2O2/t-BOOH effect on PLA2 will be addressed in subsequent studies.
Overall, the multiple functions of different PLA2s can be divided into two major categories: signaling and housekeeping (1,15). Calcium-independent iPLA2 that represents the majority of PLA2 activity in the myocardium (15) is believed to play a major role in phospholipid turnover (16), including the repair of lipid peroxides (Fig. 6). The oxidized fatty acid has to be first released from the phospholipid by PLA2 (most unsaturated fatty acids are prone to oxidation and are esterified to the sn-2 position in phospholipids, including arachidonic acid) to be repaired by glutathione peroxidase (GPX) using glutathione (GH) as a reducing agent (23). Subsequent reacylation of the remaining lysophospholipid by acyltransferase and transacylases results in the restoration of intact membrane phospholipids and, consequently, the return of normal membrane function. Although a small portion of oxidized fatty acids can be reduced in situ (24) by phospholipid glutathione peroxidase (PHGPX), the activity of PHGPX in the heart is 100-fold less than that of GPX (25). The iPLA2 activity is, therefore, essential to combat lipid peroxidation occurring even under normal conditions (23).
Fig. 6.
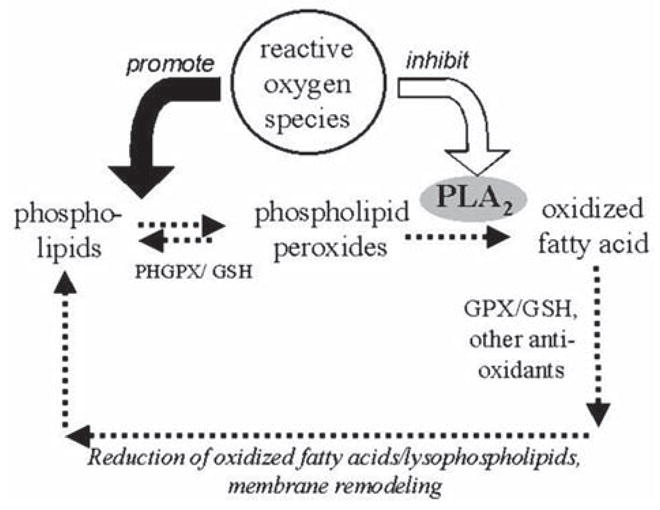
Proposed effect of observed iPLA2 inhibition and its relationship with lipid peroxidation. Abbreviations: GPX—glutathione peroxidase; GH—glutathione; PHGPX —phospholipid glutathione peroxidase.
Oxidative stress such as the treatment with H2O2 or t-BOOH leads to a sharp increase in lipid peroxidation levels (26,27). The fact that oxidative stress also inhibits iPLA2 implies that a cell’s ability to repair oxidized phospholipids is diminished as well. This diminished ability, in turn, may have a negative impact on cell survival based on a variety of pathways attributable to lipid peroxidation (17). We attempted to obtain additional evidence for such a scenario by performing experiments with BEL, a potent inhibitor of iPLA2 (14). The data revealed that the combination of BEL and t-BOOH has a significant cytotoxic effect (Figs. 4 and 5), which suggests the following explanation. Under normal and nonstressed conditions, cells survived even if most of the iPLA2 activity was inhibited by BEL. In the samples treated with t-BOOH only, the remaining iPLA2 or the restoration of membrane enzyme activity between t-BOOH applications, or both, alleviated t-BOOH-induced lipid peroxidation, preserving the cells. However, during t-BOOH/BEL treatment, as the concentration of phospholipid peroxides increased, the inhibition of the detoxification pathway leads to cell death. In other words, treatment with BEL irreversibly inactivated all iPLA2 isoforms, making t-BOOH-induced peroxidation more toxic (in cardiomyocytes, the activities of other PLA2s [e.g., secretory sPLA2 and Ca2+-dependent cPLA2] are minimal when compared to iPLA2 (3–6)).
In summary, using isolated rat cardiomyocytes we have shown for the first time that (1) both cytosolic and membrane iPLA2 are markedly inhibited by micromolar concentrations of H2O2 or tBOOH and that (2) the specific inhibitor of iPLA2, BEL, significantly potentiates oxidant cardiotoxicity. These data have important implications for the role of PLA2 in oxidative injury.
Acknowledgments
The authors thank Ms. Pamela Kell and Mr. Joseph Ugorji for technical assistance and Dr. Arutunyan for valuable discussions. This work was supported by the American Heart Association, Heartland Affiliate to J. McHowat and HL62419 to N. Sarvazyan.
References
- 1.Cummings BS, McHowat J, Schnellmann RG. Phospholipase A(2)s in cell injury and death. J Pharmacol Exp Ther. 2000;294:793–799. [PubMed] [Google Scholar]
- 2.Sevanian A. Lipid damage and repair. In: Davies KJA, editor. Oxidative Damage and Repair: Chemical, Biological, and Medical Aspects. Pergamon; New York: 1988. pp. 543–549. [Google Scholar]
- 3.McHowat J, Creer MH. Calcium-independent phospholipase A2 in isolated rabbit ventricular myocytes. Lipids. 1998;33:1203–1212. doi: 10.1007/s11745-998-0324-5. [DOI] [PubMed] [Google Scholar]
- 4.McHowat J, Creer MH. Thrombin activates a membrane-associated calcium-independent PLA2 in ventricular myocytes. Am J Physiol. 1998;274:C447–C454. doi: 10.1152/ajpcell.1998.274.2.C447. [DOI] [PubMed] [Google Scholar]
- 5.Liu SJ, McHowat J. Stimulation of different phospholipase A2 isoforms by TNF-alpha and IL-1beta in adult rat ventricular myocytes. Am J Physiol. 1998;275:H1462–H1472. doi: 10.1152/ajpheart.1998.275.4.H1462. [DOI] [PubMed] [Google Scholar]
- 6.McHowat J, Tappia PS, Liu S, McCrory R, Panagia V. Redistribution and abnormal activity of phospholipase A(2) isoenzymes in postinfarct congestive heart failure. Am J Physiol Cell Physiol. 2001;280:C573–C580. doi: 10.1152/ajpcell.2001.280.3.C573. [DOI] [PubMed] [Google Scholar]
- 7.McHowat J, Swift LM, Arutunyan A, Sarvazyan N. Clinical concentrations of doxorubicin inhibit activity of myocardial membrane-associated, calcium-independent phospholipase A2. Cancer Res. 2001;61:4024–4029. [PubMed] [Google Scholar]
- 8.Salgo MG, Corongiu FP, Sevanian A. Enhanced interfacial catalysis and hydrolytic specificity of phospholipase A2 toward peroxidized phosphatidylcholine vesicles. Arch Biochem Biophys. 1993;304:123–132. doi: 10.1006/abbi.1993.1330. [DOI] [PubMed] [Google Scholar]
- 9.Burack WR, Biltonen RL. Lipid bilayer heterogeneities and modulation of phospholipase A2 activity. Chem Phys Lipids. 1994;73:209–222. doi: 10.1016/0009-3084(94)90182-1. [DOI] [PubMed] [Google Scholar]
- 10.Rashba-Step J, Tatoyan A, Duncan R, Ann D, Pushpa-Rehka TR, Sevanian A. Phospholipid peroxidation induces cytosolic phospholipase A2 activity: membrane effects versus enzyme phosphorylation. Arch Biochem Biophys. 1997;343:44–54. doi: 10.1006/abbi.1997.0134. [DOI] [PubMed] [Google Scholar]
- 11.Wratten M, van Ginkel G, van’t Veld A, Bekker A, van Faassen E, Sevanian A. Structural and dynamic effects of oxidatively modified phospholipids in unsaturated lipid membranes. Biochemistry. 1992;31:10,901–10,907. doi: 10.1021/bi00159a034. [DOI] [PubMed] [Google Scholar]
- 12.Sarvazyan N. Visualization of doxorubicin-induced oxidative stress in isolated cardiac myocytes. Am J Physiol. 1996;271:H2079–H2085. doi: 10.1152/ajpheart.1996.271.5.H2079. [DOI] [PubMed] [Google Scholar]
- 13.Piper HM, Jacobson SL, Schwartz P. Determinants of cardiomyocyte development in long-term primary culture. J Mol Cell Cardiol. 1988;20:825–835. doi: 10.1016/s0022-2828(88)80007-5. [DOI] [PubMed] [Google Scholar]
- 14.Hazen SL, Zupan LA, Weiss RH, Getman DP, Gross RW. Suicide inhibition of canine myocardial cytosolic calcium-independent phospholipase A2. Mechanism-based discrimination between calcium-dependent and calcium-independent phospholipases A2. J Biol Chem. 1991;266:7227–7232. [PubMed] [Google Scholar]
- 15.Balsinde J, Balboa MA, Insel PA, Dennis EA. Regulation and inhibition of phospholipase A2. Annu Rev Pharmacol Toxicol. 1999;39:175–189. doi: 10.1146/annurev.pharmtox.39.1.175. [DOI] [PubMed] [Google Scholar]
- 16.Ackermann EJ, Dennis EA. Mammalian calcium-independent phospholipase A2. Biochim Biophys Acta. 1995;1259:125–136. doi: 10.1016/0005-2760(95)00143-z. [DOI] [PubMed] [Google Scholar]
- 17.Betteridge DJ. What is oxidative stress? Metabolism. 2000;49(2):3–8. doi: 10.1016/s0026-0495(00)80077-3. [DOI] [PubMed] [Google Scholar]
- 18.McHowat J, Liu S, Creer MH. Selective hydrolysis of plasmalogen phospholipids by Ca2+-independent PLA2 in hypoxic ventricular myocytes. Am J Physiol. 1998;274:C1727–C1737. doi: 10.1152/ajpcell.1998.274.6.C1727. [DOI] [PubMed] [Google Scholar]
- 19.Hazen SL, Ford DA, Gross RW. Activation of a membrane-associated phospholipase A2 during rabbit myocardial ischemia which is highly selective for plasmalogen substrate. J Biol Chem. 1991;266:5629–5633. [PubMed] [Google Scholar]
- 20.Sun Y, Oberley LW. Redox regulation of transcriptional activators. Free Radical Biol Med. 1996;21:335–348. doi: 10.1016/0891-5849(96)00109-8. [DOI] [PubMed] [Google Scholar]
- 21.Li B, Copp L, Castelhano AL, Feng R, Stahl M, Yuan Z, et al. Inactivation of a cytosolic phospholipase A2 by thiol-modifying reagents: cysteine residues as potential targets of phospholipase A2. Biochemistry. 1994;33:8594–8603. doi: 10.1021/bi00194a026. [DOI] [PubMed] [Google Scholar]
- 22.Hazen SL, Gross RW. Identification and characterization of human myocardial phospholipase A2 from transplant recipients suffering from end-stage ischemic heart disease. Circ Res. 1992;70:486–495. doi: 10.1161/01.res.70.3.486. [DOI] [PubMed] [Google Scholar]
- 23.Brigelius-Flohe R. Tissue-specific functions of individual glutathione peroxidases. Free Radical Biol Med. 1999;27:951–965. doi: 10.1016/s0891-5849(99)00173-2. [DOI] [PubMed] [Google Scholar]
- 24.Thomas JP, Maiorino M, Ursini F, Girotti AW. Protective action of phospholipid hydroperoxide glutathione peroxidase against membrane-damaging lipid peroxidation. In situ reduction of phospholipid and cholesterol hydroperoxides. J Biol Chem. 1990;265:454–461. [PubMed] [Google Scholar]
- 25.Bermano G, Nicol F, Dyer JA, Sunde RA, Beckett GJ, Arthur JR, et al. Tissue-specific regulation of selenoenzyme gene expression during selenium deficiency in rats. Biochem J. 1995;311:425–430. doi: 10.1042/bj3110425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Horwitz LD, Wallner JS, Decker DE, Buxser SE. Efficacy of lipid soluble, membrane-protective agents against hydrogen peroxide cytotoxicity in cardiac myocytes. Free Radical Biol Med. 1996;21(6):743–53. doi: 10.1016/0891-5849(96)00177-3. [DOI] [PubMed] [Google Scholar]
- 27.Bhatnagar A. Biochemical mechanism of irreversible cell injury caused by free radical-initiated reactions. Mol Cell Biochem. 1994;137:9–16. doi: 10.1007/BF00926034. [DOI] [PubMed] [Google Scholar]