

Abstract
The peptide editor HLA-DM (DM) mediates exchange of peptides bound to major histocompatibility (MHC) class II molecules during antigen processing; however, the mechanism by which DM displaces peptides remains unclear. Here we generated a soluble mutant HLA-DR1 with a histidine-to-asparagine substitution at position 81 of the β-chain (DR1βH81N) to perturb an important hydrogen bond between MHC class II and peptide. Peptide–DR1βH81N complexes dissociated at rates similar to the dissociation rates of DM-induced peptide–wild-type DR1, and DM did not enhance the dissociation of peptide–DR1βH81N complexes. Reintroduction of an appropriate hydrogen bond (DR1βH81N βV85H) restored DM-mediated peptide dissociation. Thus, DR1βH81N might represent a `post-DM effect' conformation. We suggest that DM may mediate peptide dissociation by a `hit-and-run' mechanism that results in conformational changes in MHC class II molecules and disruption of hydrogen bonds between βHis81 and bound peptide.
Shortly after being synthesized in the antigen-presenting cell, major histocompatibility complex (MHC) class II αβ heterodimers form nonameric assemblies with invariant chain (Ii) in the endoplasmic reticulum and are then transported through the Golgi complex to the endocytic pathway1,2. During transport through the endocytic pathway, most Ii is removed from MHC class II molecules by low pH and acid proteases3, leaving a proteolytic fragment of Ii called `CLIP' bound to MHC class II molecule4. CLIP acts as a `placeholder' for the MHC class II groove, inhibiting conformational changes that render the groove closed5–13, and it must be removed to allow binding of exogenous peptides to nascent MHC class II complexes. Human HLA-DM (called `DM' here), or H2-M in mice, is a nonclassical HLA molecule that is critical in the displacement of CLIP14–17. In addition to displacing CLIP, DM transiently interacts with empty MHC class II molecules to generate a peptide-receptive conformation and is active in the selection of specific peptide–MHC class II complexes during antigen processing18–26. The two concurrent hypotheses for the recognition of certain peptide–MHC class II by DM relate to the intrinsic affinity between MHC class II and the peptide22,27,28 or to subtle structural variations among different peptide-MHC complexes25,29–32, whereby structurally flexible complexes are susceptible to DM-induced dissociation, and `rigid' complexes are resistant to DM25. Although those studies may have brought greater understanding of the criteria for the recognition of specific peptide–MHC complexes by DM, the exact mechanism for DM effector function remains unknown.
To address the mechanism of DM effector function on peptide dissociation, we considered altering the hydrogen bonds found between the peptide backbone and conserved amino acids of the MHC class II molecule33–37. Published reports have shown that such hydrogen bonds are crucial for peptide–MHC class II complex stability. For example, substitution of two acidic residues with amides in the core of the hydrogen-bond network in I-Ek enhances the kinetics of peptide exchange at low pH38. Also, substitution of the histidine at position 81 of the β-chain (`βHis81') with arginine reduces the stability of peptide–I-Ad complexes39. The formation of hydrogen bonds between βHis81 of I-Ek and peptides contributes substantially to the thermal stability of the complex40. The possibility that hydrogen bonds could be involved in DM effector function has been posited but not experimentally demonstrated. We hypothesized that after recognizing a suitable MHC class II pocket 1 structure, DM causes conformational changes that result in the breaking of one or more hydrogen bonds formed between the peptide backbone and the MHC class II groove, destabilizing the bound peptide. One of those hydrogen bonds is the short, strong hydrogen bond formed between the conserved βHis81 of HLA–DR1 and a carbonyl group on the main chain of the bound peptide33. This specific hydrogen bond has various attributes that suggest it could represent a potential target for DM effector function. To test the hypothesis, we generated a soluble mutant HLA–DR1 molecule in which βHis81 was substituted with asparagine (DR1βH81N) to perturb that strong hydrogen bond in the pH range in which DM exerts its greatest effect.
DR1βH81N seemed functional for all aspects tested. However, peptide–DR1βH81N complexes dissociated very rapidly and independently of peptide sequence at rates similar to the accelerated dissociation rates of complexes of peptide and wild-type DR1 (peptide–DR1WT) in the presence of DM. Although DM recognized peptide–DR1βH81N complexes, it failed to accelerate the dissociation rate of peptides from DR1βH81N. The `rescue' mutant DR1(βH81NβV85H), with potential reintroduction of an appropriate histidine-mediated hydrogen bond with peptide, restored DM-mediated dissociation of peptide from the MHC class II molecule. Thus, we propose that DR1βH81N represents a `post-DM effect' transitional state. We suggest that DM effects peptide–MHC class II complex dissociation by a `hit-and-run' mechanism in which transient interaction between DM and DR1 causes a conformational change in DR1, leading to the perturbation of the βHis81 hydrogen bond, resulting in destabilization of the bound peptide.
RESULTS
Peptide binding of DR1βH81N
To evaluate the effect of substitution of position β81 on peptide-binding kinetics in a human MHC class II molecule, we produced soluble recombinant DR1βH81N and did basic physical characterization by size-exclusion separation. The profile of DR1βH81N could be superimposed with that of DR1WT (Fig. 1a), suggesting that there were no gross structural abnormalities or aggregation and/or degradation of the mutant αβ heterodimer. We next tested the ability of DR1βH81N to bind peptides and to form SDS-stable complexes in a `gentle' SDS-PAGE assay in which the samples are not boiled. As shown before26, peptides that bind HLA–DR1 and fill the hydrophobic pocket 1 with bulky aromatic residues (Trp, Phe and Tyr) or aliphatic residues (Met, Ile, Val and Leu) form complexes that are resistant to SDS-induced chain dissociation independent of the dissociation rate of the peptide–MHC class II complex12. A DR1 molecule with an empty pocket 1 dissociates into separate α- and β-chains in this assay. To differentiate between SDS-stable and SDS-sensitive conformations of DR1, we allowed DR1WT and DR1βH81N to separately bind the proteolytic fragment of invariant chain (CLIP(81–105), with methionine in pocket 1) or the immunodominant peptide of influenza virus hemagglutinin (HA(306–318), with tyrosine in pocket 1) or its synthetic variant (HA(anchorless), with alanine in pocket 1). Similar to the results obtained with DR1WT, HA(306–318)–DR1βH81N and CLIP(81–105)–DR1βH81N complexes were SDS stable (CLIP–DR1 complexes migrate slightly faster than HA (306–318)–DR1 complexes), whereas both empty DR1βH81N and HA(anchorless)–DR1βH81N were SDS sensitive (Fig. 1b). This finding suggested that the βH81N substitution did not compromise the ability of DR1 to bind peptides and to undergo peptide-induced conformational changes.
Figure 1.

DR1βH81N binds peptides to form complexes similar to peptide–DR1WT. (a) Size-separation profiles of soluble recombinant DR1WT and DR1βH81N molecules in PBS analyzed on a column equilibrated in PBS. The main peak corresponding to the αβ heterodimer (arrow) appears as expected at about 36 min for both molecules. (b) SDS stability of peptide–DR1βH81N and peptide–DR1WT complexes incubated in 0.1% SDS in PBS and separated by 12% SDS-PAGE; gel is silver-stained. HA(anch), HA(anchorless). (c) Kinetics of association of DR1βH81N or DR1WT (2 μM) incubated with excess (60 μM) FITC–labeled HA(306–318) in citrate phosphate buffer, pH 5.5, in the absence of DM; complexes were separated from unbound peptide and fluorescence measured was plotted versus time, then data were fitted to biphasic curves. Data are representative of five or more experiments (a,b) or are from one of three independent experiments (c).
βH81N does not alter the biphasic nature of peptide binding
We compared the formation of fluorescein isothiocyanate (FITC)–labeled peptide–DR1 complexes for DR1βH81N and DR1WT. Comparison of the association rates for DR1βH81N and DR1WT with HA(306–318) showed that the βH81N substitution enhanced the rate of FITC–peptide–DR1 complex formation, but the nature of the curve was biphasic and similar to that of DR1WT (Fig. 1c). The association curve for DR1βH81N had a more prominent first phase, presumably because of the faster dissociation rate of existing peptide–DR1βH81N complexes (discussed below), which would result in an increased molar fraction of molecules in the peptide-receptive conformation during the association reaction. This result was in contrast to the kinetic pattern of the binding of peptide to DR1βG86Y, which is monophasic25, presumably because of the βG86Y substitution that fills pocket 1. That difference is important, as it demonstrated that the DR1βH81N mutant we generated, unlike DR1βG86Y, is not fixed in a peptide-receptive conformation.
The βH81N substitution destabilizes peptide–DR1 complexes
To measure the effect of the βH81N substitution on the stability of peptide–DR1 complexes, we measured the dissociation kinetics of complexes formed with DR1WT or DR1βH81N using FITC–HA(306–318) and FITC–HA(anchorless) peptides. The `off rates' of both peptides from DR1βH81N were accelerated considerably by that single substitution, and both peptides dissociated with similar half-life (t1/2) of about 10 min, in contrast to DR1WT, from which HA(306–318) dissociated with a t1/2 of 6 d and HA(anchorless) peptide dissociated with a t1/2 of about 1 h (Fig. 2a). For the first set of experiments, we used the pH range at which HLA–DM is active (citrate phosphate buffer; pH 5.5). However, because asparagine can theoretically form hydrogen bonds at a range of higher pH values, we repeated the experiments at neutral pH, in phosphate-buffered saline (PBS), pH 7.4 (data not shown). The stability of the peptide–DR1βH81N complexes did not increase at neutral pH, suggesting that strong hydrogen bonds are not formed between asparagine and peptide. That fast `off rate' from the mutant DR1 in the absence of DM was nearly identical to the rate for the dissociation of HA(anchorless) from DR1WT in the presence of DM (Fig. 2b). The similar half-lives of dissociation were not an artifact due to limitations in the sensitivity of the assay and did not represent the fastest possible rate of peptide dissociation from a DR1 molecule. Thus, these results led us to speculate that DM might function by disrupting the formation of the hydrogen bond between βHis81 and peptide backbone.
Figure 2.

Kinetics of dissociation of peptides in the absence of DM are faster for mutant DR1βH81N and resemble DM-mediated peptide dissociation. (a) Dissociation of DR1WT and DR1βH81N in complex with FITC–HA(anchorless) or FITC–HA(306–318) in the presence of a 100× molar excess of relevant unlabeled peptides. The fluorescence of the labeled complex before dissociation is arbitrarily assigned a value of 1.0, and fluorescence after dissociation is expressed as a fraction of fluorescence before dissociation. (b) Dissociation of HA(anchorless)–DR1WT in the absence (t1/2, 53 min) or presence (t1/2, 7.3 min; R2 = 0.997) of DM (DM/DR, 1:1) and HA(308–316)–DR1βH81N (t1/2, 7.4 min, R2 = 0.986) in the absence of DM, produced and dissociated as described in a. Data from one of three or more independent experiments were fitted to a single-exponential curve and approximate t1/2 values were calculated.
Modeling of the DR1β81–peptide main chain distance
We compared 11 crystal structures of various human and mouse peptide–MHC class II complexes using two different modeling software programs to model the substitution of asparagines for histidine at β81 (or its equivalent; Table 1 and Fig. 3). The distance between the carbonyl group on the bound peptide and the amide group on β81Asn increased by an average of about 1.1Å relative to that formed with βHis81. The data represent the closest `allowed' conformation of an asparagine (Table 1) and suggest that such a substitution would substantially weaken any hydrogen bond formed between DR1 and peptide. These data suggested that the rapid dissociation of peptides from DR1βH81N may have been due to a considerably weakened hydrogen bond with the backbone of the bound peptide.
Table 1.
Increase in hydrogen-bonding distance after His → Asn
| PDB ID | Allele–peptide residue | d(His) | d(Asn) (PyMol) | Change |
|---|---|---|---|---|
| 1DLH | DR1–Lys2 | 2.69Å | 3.86Å (3.75) | 1.17Å |
| 1A6A | DR3–Lys2 | 3.03Å | 3.80Å (3.75) | 0.77Å |
| 1AQD | DR1–Asp4 | 2.68Å | 3.67Å (3.48) | 0.99Å |
| 1H15 | DR2–Val3 | 2.77Å | 3.85Å (3.74) | 1.07Å |
| 1HQR | DR2–Phe3 | 3.27Å | 4.14Å (4.10) | 1.17Å |
| 1HXY | DR1–Lys2 | 2.66Å | 3.73Å (3.66) | 1.08Å |
| 1J8H | DR1–Lys2 | 2.57Å | 3.65Å (3.55) | 1.08Å |
| 1KLU | DR1–Leu3 | 3.00Å | 4.17Å (4.13) | 1.17Å |
| 1KT2 | I-Ek–Leu4 | 3.03Å | 4.20Å (4.02) | 1.17Å |
| 1KTD | I-Ek–Leu4 | 2.88Å | 4.04Å (3.86) | 1.16Å |
| 1IEA | I-Ek–Val4 | 2.72Å | 4.11Å (3.98) | 1.39Å |
Modeling of His→Asn substitution with Swiss PDBviewer or PyMol41 for various human and mouse MHC class II molecule crystal structures. All structures have the conserved histidine residue; however, the side chains of the peptides whose carbonyl group potentially form a hydrogen bond with that histidine residue vary (column 2). d(His), distance between the conserved histidine and the oxygen atom of carbonyl group, as in the structure; d(Asn), distance between closest allowed 'conformer' of asparagine (after the substitution) and that same carbonyl group; change, d(Asn) – d(His). The potential hydrogen-bonding distance consistently increases by an average of 1.1Å; hence, the bond is weaker for the asparagine mutant models for all structures analyzed. PDB ID, Protein Data Bank accession number.
Figure 3.
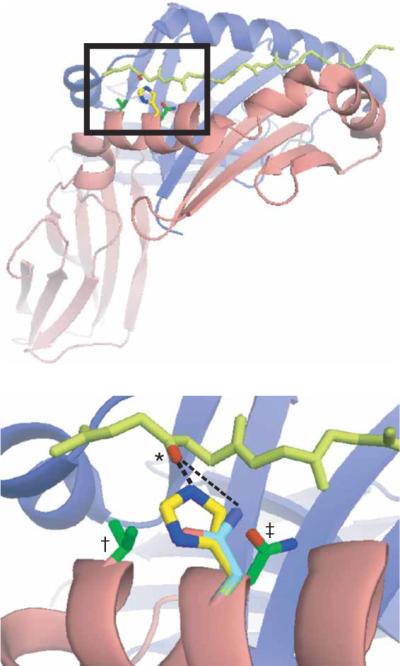
Pocket 1 of HA(306–318)–DR1. Based on modeling programs, βHis81 (yellow; 2.69Å) but not the substituted residue β81Asn (cyan; 3.75Å) of DR1 forms a short, strong hydrogen bond (dashed lines) with the carbonyl group (*) on Lys307 of HA(306–318) (light green). The residues substituted for histidine in the `rescue' mutants, β85Val (†) and β82Asn (‡), are green; α- and β-chains of DR1 are blue and red ribbons, respectively; and acidic and basic groups on the relevant side chains are red and blue, respectively. Bottom, enlargement of boxed area (pocket 1) above. Structure rendered by PyMol based on the crystal structure of HA(306–318)–DR1.
DM does not enhance peptide dissociation from DR1βH81N
To determine if peptide–DR1βH81N complexes that presumably lack the hydrogen bond between βHis81 and the peptide backbone are susceptible to DM-induced dissociation, we measured the dissociation of FITC-labeled peptides from DR1WT or DR1βH81N during a short 10-minute incubation in the presence or absence of DM. Although the addition of DM caused dissociation of about 80% of HA(anchorless)–DR1WT complexes within 10 min at a ratio of 1:1, its effect on the dissociation of peptide–DR1βH81N complexes was minimal both for HA(306–318) and HA(anchorless) (Fig. 4a). We also did the dissociation experiment at a fixed DM concentration (DR/DM = 5:1) over time with similar results (Fig. 4b). FITC–HA(306–318) or FITC–HA or FITC–HA(anchorless) dissociated from DR1βH81N with a t1/2 of about 10 min in the absence or presence of DM.
Figure 4.

DM has a minimal effect on the dissociation of peptide from DR1βH81N. (a) Dissociation of complexes of FITC–HA(anchorless)–DR1WT, FITC–HA(308–316)–DR1βH81N and FITC–HA(anchorless)–DR1βH81N (2.5 μM), allowed to dissociate separately for 10 min as described in Figure 2a in the absence (–DM) or presence of various concentrations of DM. The fluorescence reading of complexes dissociated in the absence of DM was arbitrarily assigned a value of 1.0, and measurements are expressed as fractions of that value. Data are from one of three experiments. (b) Dissociation of complexes of DR1–H81N and FITC–HA(anchorless) or FITC–HA(308–316), allowed to dissociate as described in a in the presence (+ DM) or absence of DM (DM/DR, 1:5). The t1/2 values for all the reactions from the single-exponential curve fits are very similar: without DM, about 10 min; with DM, about 8 min; R2 = 0.959–0.997. Data are from one of at least three experiments. (c) Dissociation of complexes of HA(Y308A)–DR1WT or HA(308–316)–DR1βH81N (2.5 μM) allowed to bind to an HA(anchorless) or HA(308–316) surface, respectively, and then allowed to dissociate for 20 min in the absence of DM and for a further 20 min in the presence of 8 μM DM at a pH of 6.0. Curves were obtained by subtraction of the curve obtained with DM from the curve obtained without DM and assignment an arbitrary value of 0 for the relative units obtained at time 0. Data are from one of two experiments.
Given the fast overall dissociation rates of peptide–DR1βH81N complexes, we attempted a real-time surface plasmon resonance assay as described25 to evaluate DM-mediated changes in the dissociation rates of the peptide–DR1βH81N complexes. We injected solutions of peptide-receptive DR1WT or DR1βH81N over surfaces of BIAcore CM5 chips coupled to HA(306–318) or to HA(anchorless). We then allowed the complexes to dissociate in the absence of DM (wash buffer only) and then in the presence of DM. Using those data, we calculated the net change in the rate of peptide–DR1 dissociation after injection of DM. Although DM efficiently accelerated the dissociation of HA(anchorless) from DR1WT, there was no effect on the dissociation of DR1βH81N regardless of the peptide bound (Fig. 4c). The upward slope in net peptide–DR1βH81N bound caused by the addition of DM was probably due to the ability of DM to mediate the reassociation of freshly dissociated complexes. Some reassociation of peptide–DR complexes occurs on BIAcore chips, and the dissociation curves are thus a net result of both reassociation and dissociation events.
The βH81N substitution does not affect DR1–DM interactions
To ascertain whether the asparagine substitution introduced at position β81 did not alter recognition by DM, we measured the change in the intrinsic tryptophan fluorescence of the two molecules after mixing. Tryptophan residues in proteins can be selectively excited at 295 nm and will emit with a maximum around 350 nm; however, they are exquisitely sensitive to the local environment, such that small changes in ionic polarity can cause a shift in the emission spectrum. Thus, interactions that cause burial of solvent-exposed tryptophan residues at the DM–DR interface might cause an enhancement of overall emission intensity. In the absence of a more specific molecular `readout', this assay can serve as a qualitative measure of transient DM–DR interactions and has been used for wild-type DR1 (ref. 25).
There was a measurable increase in intrinsic tryptophan fluorescence after mixture of DM with any peptide–DR1 complex because of nonspecific protein-protein interactions in solution (Fig. 5a). However, that increase in intrinsic tryptophan fluorescence was greater when the targeted peptide–DR1 complex was `DM sensitive'; that is, a peptide–DR1 complex known to be targeted by DM. We used HA(anchorless), which binds DR1 without filling binding pocket 1, to simulate a DM-sensitive conformation. Tyr308 of the HA(306–318) peptide fills pocket 1, hence HA(306–318)-DR1WT complexes do not interact specifically with DM25. As expected, there was a measurable and significant increase in the interaction of DM with HA(anchorless)–DR1WT compared with that of HA(306–318)–DR1WT or HA(anchorless)–DR1βG86Y (P < 0.000001; Fig. 5a,b). We also noted that trend with peptide–DR1βH81N complexes; DM interacted with HA(anchorless)–DR1βH81N significantly more than with HA(306–318)–DR1βH81N (P < 0.000001). Hence, the recognition of DR1 conformation by DM is not disturbed by the βH81N substitution and only the effector function of DM is compromised. We also measured the intrinsic tryptophan fluorescence of DR1βG86Y and of the double mutant DR1(βH81N βG86Y). Both of those DR1 molecules have a βG86Y substitution at the base of binding pocket 1. Thus, pocket 1 is filled by a tyrosine residue even in the absence of peptide and those molecules are not expected to be recognized by DM. Indeed, DR1βG86Y and DR1(βH81N βG86Y) produced tryptophan fluorescence values similar to those of HA(306–318)–DR1WT and HA(306–318)–DR1βH81N, respectively (Fig. 5b). These data suggested that the lack of DM-mediated dissociation of peptide–DR1βH81N complexes cannot be attributed to lack of recognition of DR1βH81N by DM.
Figure 5.

The βH81N substitution does not alter the interaction of DM with DR. (a) Tryptophan fluorescence of DR1WT or DR1βH81N (0.2 μM) in complex with HA(anchorless) or HA(308–316) incubated in citrate phosphate, pH 5.5, before and after the addition of 0.1 μM DM, monitored from 310 nm to 410 nm after excitation at 295 nm. Raw data here are from one of two experiments after subtraction of `blank' values (citrate phosphate only). (b) Average change in fluorescence (ΔTrp fluorescence) over 310–390 nm obtained from the raw data in a. *, P < 0.000001. (c,d) Fluorescence of 2 μM DR1WT (c) or DR1βH81N (d) incubated for various times at 37 °C with 60 μM FITC–HA(308–316) in citrate phosphate, pH 5.5, in the presence (+ DM) or absence (− DM) of 1 μM DM; complexes were separated from excess fluorescent peptide for fluorescence measurement. Data obtained with DM were fitted to monophasic association curves for both DR1WT and DR1βH81N. Data are from one of at least two independent experiments.
DM can mediate peptide binding to DR1βH81N
We incubated soluble DR1WT or DR1βH81N molecules with FITC–HA(306–318) peptide in the presence of DM for various times at 37 °C at a DR/DM ratio of 5:1 and a pH of 5.5 and analyzed complex formation by fluorimetry. Notably, the presence of DM converted the biphasic binding of FITC–HA(306–318) to DR1βH81N into a monophasic binding curve, similar to that of DR1WT (ref. 25; Fig. 5c,d). A monophasic binding curve is indicative of the conversion of DR1 to a peptide-receptive form. These observations further supported our earlier conclusion that the interaction of DR1βH81N with DM remained intact and that the βH81N substitution did not interfere with conformational changes necessary for the conversion of DR1βH81N to a peptide-receptive form. Thus, the effect of DM in enhancing the association of peptide with an empty DR molecule may be distinct from its effect in mediating peptide dissociation and the former may not involve βHis81. These data collectively suggested that breaking the hydrogen bond between βHis81 and peptide by DM might be a key step for peptide dissociation and that the peptide–DR1βH81N complex may represent a `post-DM effect' transitional state. That could explain our observations that peptide–DR1βH81N complexes dissociated with a `DM-mediated' kinetic pattern and that DM did not further accelerate that dissociation even though it could recognize and interact with the peptide–DR1βH81N complexes.
Reintroduction of histidine in DR1βH81N restores DM function
To ascertain if the hydrogen bond formed between the carbonyl group of the peptide backbone and βHis81 is the main target for DM-induced peptide dissociation, we engineered two additional DR1 constructs. We designed DR1(βH81N bV85H) to reintroduce a histidine residue in the pocket 1 area such that the reactive imidazole side-chain points toward and is within hydrogen-bonding distance of the peptide. In parallel, we designed the `mock rescue' mutant DR1(βH81N bN82H) to introduce a histidine residue in the same region of DR1, but with the reactive group facing away from pocket 1. Based on the crystal structure of HA(306–318)–DR1WT (Protein Data Bank accession number, 1DLH) modeled by PyMol41, the potential minimum hydrogen-bond distances between histidine and the relevant carbonyl group (Lys307, for HA(306–318)) would be about 2.4Å for DR1(βH81NβV85H) and about 5.05Å for DR1(βH81N βN82H) (Fig. 3). Thus, DR1(βH81N βV85H) was designed to present a possible exposed hydrogen-bond target for DM, whereas DR1(βH81N βN82H) presumably does not.
We expressed and characterized each double-mutant DR1 molecule as described above. Size-separation chromatography (Supplementary Fig. 1 online) and reactivity with conformation-specific antibody L243 (data not shown) indicated proper folding of the proteins. A `gentle' SDS-PAGE assay showed that these double-mutant molecules formed either faint or no discernable SDS-stable bands with peptide (Supplementary Fig. 2 online), presumably because of minor conformational changes in the peptide-binding groove that rendered the peptide–DR1 complexes more susceptible to SDS. However, both DR1(βH81N bV85H) and DR1(βH81N bN82H) were capable of binding peptide over time. The association curves for both were biphasic and were converted to monophasic binding curves by DM (Fig. 6a), suggesting that, notably, the ability of DM to interact with and mediate peptide association with these double mutants was not affected. The binding of peptide to DR1(βH81N bV85H) and to DR1(βH81N bN82H) was fast and the resulting complexes dissociated extremely rapidly, with a t1/2 of less than 5 min, as assessed by fluorescence (Fig. 6b) and surface plasmon resonance (Supplementary Fig. 3 online). These results confirmed our earlier conclusion that although βHis81 may not be involved in peptide association to DR in the presence or absence of DM, it is critical for the stability of bound peptide.
Figure 6.

Reintroduction of an appropriate histidine residue partially restores DM-mediated dissociation. (a) Fluorescence of DR1(βH81N βV85H) and DR1(βH81N βN82H) associated with excess FITC–HA(306–318) in citrate phosphate buffer, pH 5.5. in the presence or absence of DM (DM/DR = 1:5) as described in Figure 5c,d; data from one of two experiments are fitted to biphasic or monophasic curves for groups without or with DM, respectively. (b) Dissociation of HA(308–316)–DR1(βH81N βV85H) (t1/2, 4.2 min; R2 = 0.985) and HA(308–316)–DR1(βH81N βN82H) (t1/2, 4.0 min; R2 = 0.999) complexes in the absence of DM, analyzed as described in Figure 2a. Data were fitted to a single-exponential curve and approximate t1/2 values were calculated. (c) Dissociation of FITC–HA(anchorless) in complex with DR1(βH81N βV85H) or DR1(βH81N βN82H), allowed to dissociate separately for 5 minutes as described in Figure 4a, in the absence (–DM) or presence of various concentrations of DM. Fluorescence is expressed as a fraction of complexes dissociated in the absence of DM. (d) Dissociation of HA(anchorless) from double mutants as described in Figure 2a (DM/DR = 1:5). FITC–HA(anchorless) dissociates from DR1(βH81N βN82H) with a t1/2 of about 1 min in the presence (R2 = 0.987) or absence (R2 = 0.998) of DM and dissociates from DR1(βH81N βV85H) with a t1/2 of about 4 min in the presence of DM (R2 = 0.944) and with a t1/2 of about 1 min in the absence of DM (R2 = 0.994). Data for dissociation kinetics are from one of at least four independent experiments.
The reintroduction of histidine in two different positions did have a considerable effect on sensitivity of these DR molecules to DM-mediated peptide dissociation. DM was able to dissociate a third of the `DM-sensitive' peptide HA(anchorless) from DR1 DR1(βH81N bV85H) within just 5 min, but had no effect on DR1(βH81N bN82H) in this experiment (Fig. 6c). A time-course kinetic experiment also showed that DM accelerated the dissociation of HA(anchorless) from DR1(βH81N βV85H) but not from DR1(βH81N βN82H) (Fig. 6d). The lack of a DM-mediated effect on peptide–DR1(βH81N bN82H) complex dissociation cannot be solely due to the rebinding of dissociated peptides, as the experiments were done in the presence of an approximately 50-molar excess of unlabeled HA(306–318) competitor peptide to reduce the possibility of rebinding of the FITC–labeled peptide.
In summary, comparison of two to three independent experiments for each group at a DM/DR ratio of 1:5 showed that DM was unable to catalyze the dissociation of peptide–DR1βH81N complexes within 10 min (Fig. 4a). However, with the `rescue' mutant DR1(βH81N βV85H) but not with the `mock rescue' mutant DR1(βH81N βN82H), DM effector function was restored and DM was able to dissociate on average about half of those complexes by 10 min (Fig. 6c). Overall, these data established that the hydrogen bond formed between βHis81 and the carbonyl group of the bound peptide is a chief target for DM-induced peptide–MHC class II complex dissociation. We propose that DM may act by a `hit-and-run' mechanism in the antigen-presenting cell (Supplementary Fig. 4 online), in which DM perturbs the hydrogen bonds, mainly the βHis81 hydrogen bond between peptide and the MHC class II molecule, to mediate dissociation of the peptide–MHC class II complex.
DISCUSSION
Published reports have defined conformational differences between peptide–MHC class II complexes as being a dominant factor in recognition by HLA-DM25. Here we have presented evidence that DM dissociates the peptide–MHC class II complexes that it recognizes by perturbing a critical hydrogen bond between a conserved histidine residue on the β-chain of the MHC class II molecule and the peptide backbone. In our experimental design we used substitution on the MHC class II molecule HLA-DR1 (βH81N) to perturb a short, strong hydrogen bond formed between the peptide backbone and DR1, destabilizing the bound peptide. We specifically chose to alter βHis81 for several reasons: βHis81 is a highly conserved residue among different MHC class II alleles, even across species, and is positioned directly above pocket 1 of the peptide-binding groove. That region of DR1 is critical in peptide-MHC complex stability12,42,43 and also maps to the purported interface for DM interaction25,30. A study of all human and mouse peptide–MHC class II crystal structures in the Protein Data Bank has shown that the histidine above pocket 1 forms a strong conserved hydrogen bond with peptide, and modeling of a substitution of that residue with asparagine demonstrated considerable weakening of the putative hydrogen bond in `allowed' models. We appreciate that the modeled substitutions were based on crystal structures and therefore cannot account for possible local or global conformational changes that could alter the hydrogen-bonding distance. Nevertheless, the consistent and substantial increase in the potential hydrogen-bond distance between the carbonyl group of peptide and the substituted β81Asn side chain correlates well with the accelerated dissociation rates observed. Thus, disruption of the conserved hydrogen bond destabilizes the bound peptide, consistent with the observation that disruption of the orthologous βHis81 hydrogen bond in a mouse mutant MHC class II molecule I-Ek reduces the thermal stability of peptide–I-Ek complexes40.
Introducing the βH81N substitution in DR1 had a profoundly destabilizing effect on the peptide–DR1 complex independent of the peptide sequence, whereas that substitution did not affect the ability of the molecule to bind peptides. Disruption of that specific histidine-mediated hydrogen bond rendered the other hydrogen bonds that line the peptide-binding groove almost irrelevant, suggesting that perturbing that single bond of an MHC class II molecule may be an energetically efficient way to destabilize the bound peptide. Thus, it seems plausible that the βHis81 hydrogen bond could be targeted by DM to dissociate peptides from DR1.
Indeed, in the absence of DM, peptides dissociated from DR1βH81N molecules with kinetics very similar to those of the DM-mediated peptide dissociation from DR1WT, and DM could not accelerate the dissociation of peptides from DR1βH81N. Those results suggest that the conformation of the mutant DR1βH81N may closely resemble an intermediate conformation of DR1 immediately after interaction with DM. Thus, HLA-DM mediates the dissociation of `DM-sensitive' peptides from DR1 by inducing a conformational change in DR1 that perturbs the βHis81 hydrogen bond.
The validity of the model described above was strengthened by the finding of partial reconstitution of DM-mediated peptide dissociation in the double mutant DR1(βH81N βV85H), presumably because of the reappearance of an appropriate histidine-mediated hydrogen bond with peptide in the vicinity of pocket 1. The accessibility and position of the histidine is critical, as substitution of a buried residue in the same region of DR1 (DR1βH81N βN82H) did not restore DM effector function. Modeling a histidine at β82 in the HA(306–318)–DR1 structure would allow that residue to be in close proximity to the Lys307 carbonyl group of the bound peptide. However, any hydrogen bond formed is likely to be considerably `kinked' because of the position of the reactive groups and is therefore likely to be much weaker than the β81His hydrogen bond. The rapid dissociation of peptides from the double mutants and the less-than-complete reconstitution of DM function could be attributed to slight conformational changes in those molecules. Nonetheless, DM-mediated dissociation of peptides from DR1 was partially restored in the presence of an appropriate hydrogen bond, suggesting that DM indeed targets the hydrogen bond formed by βHis81 in DR1WT to mediate peptide dissociation.
A mutant form of DR1, DR1βG86Y, has been reported that does not interact with DM and remains refractory to DM-induced peptide–DR1 complex dissociation. Here we have demonstrated that the new mutant DR1βH81N did interact with DM but was still refractory to DM-induced dissociation. We propose that those two mutant DR1 molecules allow categorization of the interaction of DM with MHC class II into two distinct steps: first, recognition of the peptide–MHC ligand, which is conformation dependent, followed by an effector function that results in conformational changes in DR that perturb a highly conserved hydrogen bond between βHis81 and the peptide main chain.
A `hit-and-run' mechanism might explain those observations. In our model, DM interacts transiently and perhaps repetitively with HLA-DR1 to induce conformational changes that lead to the disruption of hydrogen bonds between peptide and MHC class II. A `hit-and-run' mechanism implying transient macromolecular interactions has been reported for several biological systems, especially those involving enzymatic reactions44,45. That proposed mechanism can explain observations that complexes of DM–MHC class II are not readily detectable46 and might explain the lack of success in obtaining DM–MHC class II `cocrystals' in all conditions reported so far.
In the low-pH compartments of an antigen-presenting cell, a DR1 molecule that is empty has a closed peptide-binding groove and the molecule is in a conformation that is DM sensitive. A DM `hit' rescues the molecule from denaturation and generates an open conformation that is peptide receptive. The βHis81 residue presumably does not form an energetically important bond in empty DR; hence, DM can mediate peptide association with the mutant DR1βH81N. If peptide is not available in the milieu, the DR molecule collapses again, as the lifetime of the peptide-receptive conformation is very short11,32,47. However, if peptide binds DR1 without filling pocket 1, the DR1 molecule remains in a `floppy' conformation. DM can recognize and interact with such a complex to induce conformational changes that result in perturbation of the hydrogen bond between DR1 and the bound peptide. We suspect that the destabilization of that hydrogen bond might be the effect rather than the cause of the conformational change induced by the DM `hit'. As a result, the bound peptide is released, yielding an empty and peptide-receptive DR1 that is now ready to bind another peptide. Finally, if DR1 binds a peptide that fills its pocket 1, the complex acquires a DM-insensitive conformation. Because DM recognition is now abolished, the DR1 molecule remains bound to the peptide and is presumably exported to the cell surface.
Thus, whereas the recognition function of DM on peptide–MHC class II complexes is selective and depends on the sequence of the peptide bound, its ability to dissociate peptide is sequence independent and targets a conserved feature of the peptide–MHC class II complex (that is, hydrogen bonds that stabilize peptides in the groove). Notably, those intricacies can be reduced to a single action that can be assigned to DM: the generation of a peptide-receptive conformation of MHC class II by opening of the peptide-binding groove of the molecule. Furthermore, as that conformation by itself is DM sensitive, various peptides can be screened in succession until the DM-insensitive peptide-DR complex is identified. Thus, the newly generated pool of peptides in the antigen-presenting cell undergo selection by a process of elimination by HLA–DM before being expressed on the cell surface.
METHODS
Construction of soluble DR1 mutants
Mutant molecules DR1βH81N, DR1(βG86Y βH81N) (filled pocket 1), and DR1(βH81N βV85H) and DR1(βH81N βN82H) (`rescue' mutants) were generated as follows. The cDNA encoding the extracellular domain of DRB1*0101 was mutated to change β81His to β81Asp. Wild-type genes (DRA and DRB1*0101) were cloned into a dual-promoter pAcUW51 vector (Pharmingen). The HLA-DRA and G86Y mutant DRB1*0101 was cloned into pAcUW51 vector in a similar way43. The desired substitution was introduced into β-chains of DR1 in both constructs through site-directed mutagenesis (Stratagene) with two synthetic primers complementary to opposite strands containing the nucleotides to be changed. The sequence of oligonucleotide used was 5'-CGGTGGACACCTACTGCAGAAACAACTACGGGG-3′ (coding strand; altered nucleotides underlined). For the `rescue' mutants, the mutations were introduced into the β-chain of DRB1*0101H81N with the primers 5′-GGACACCTACTGCAGAAACCACTACGGGGTTGG-3′ for DR1(βH81N βN82H) and 5′-CCTACTGCAGAAACAACTACGGGCATGGTGAGAGCTTC-3′ for DR1(βH81N βV85H) (coding strand; altered nucleotides underlined). Clones containing the mutated genes were screened by sequencing and transfer vectors containing the desired mutations were used for transfecting Sf 9 cells to generate recombinant baculovirus and were then used for protein production43.
Production of recombinant soluble DR1 and DM proteins
Soluble DR1 proteins were expressed and purified as originally described48. Baculovirus DNA (BaculoGold; PharMingen) and transfer vectors carrying wild-type or mutated genes were transfected together into Sf 9 insect cells to produce recombinant viruses. Hy5 cells were infected with the recombinant viruses and DR1 proteins were purified from culture supernatants with immunoaffinity chromatography columns with monoclonal antibody L243 to DR1 (purified from HB-55 hybridoma; American Type Culture Collection). Wild-type and mutant DR1 molecules migrated similarly by SDS-PAGE with the expected sizes of α- and β-subunits when samples were boiled before electrophoresis.
Soluble HLA-DM was expressed by Hy5 cells transduced with recombinant baculovirus containing extracellular domains of the genes encoding the α- and β-chains of human HLA-DM. The truncated DM α- and β-chains were genetically modified to contain the Flag epitope (DYKDDDDK) and the c-Myc epitope (EQKLISEEDL) respectively, at their C termini. Protein was purified from culture supernatants with monoclonal antibody to M2 (anti-Flag) sepharose resin (Sigma), was eluted with 0.1 mg/ml of Flag peptide in 0.05% (weight/volume) sodium azide in PBS, was further purified by gel-filtration chromatography (Superdex 200 HR 10/30 column; Amersham Pharmacia) and was stored at −80 °C at a concentration of about 1 mg/ml in 0.05% (weight/volume) sodium azide in PBS.
SDS-PAGE
SDS-PAGE was done essentially as described6,43. DR1WT or DR1βH81N (1 μM) was incubated for 24 h at 37 °C without any additional peptide or with various peptides (100 μM) in either PBS, pH 7.4, or citrate phosphate buffer, pH 5.5. In the latter pH condition, reaction samples were neutralized before being mixed with equal volumes of SDS-PAGE sample buffer containing 0.1% (weight/volume) SDS (final concentration) and were incubated for 10 min at 25 °C. Samples were then separated by 12% SDS-PAGE and the gels were silver-stained according to standard protocols.
Peptide synthesis and labeling
HA(306–318) (PKYVKQNTLKLAT) and the variants HA(Y308A) (PKAVKQNTLKLAT) and HA(anchorless) (PKAVKANGAKAAT) (substituted amino acids underlined) were purified to apparent homogeneity of over 95% by reverse-phase preparative high-performance liquid chromatography and their identities were confirmed by mass spectrometry. Then, a 0.15-mM solution of Cys–HA(306–318) or Cys–HA(anchorless) in 10 ml PBS was incubated for 1 h at 25 °C with 25 μl of 75 mM fluorescein-5-maleimide (Molecular Probes) in N,N-dimethylformamide. Samples were concentrated to 0.1 ml in a SpeedVac (Savant Instruments). Excess free fluorescent label was removed by passage of the sample through a Sephadex G-10 column (Amersham Pharmacia Biotech). Concentrations were determined by spectrophotometry according to the extinction coefficient of fluorescein-5-maleimide (83 mM−1 cm−1).
Peptide association and dissociation assays
Purified DR1WT or DR1βH81N (2.4 μM) was incubated for various times at 37 °C in the presence or absence of 1 μM DM with 100 μM fluorescence-labeled peptides in 0.15 M citrate phosphate buffer, pH 5.5. After removal of free peptides by a Sephadex G-50 spin column equilibrated with PBS, pH 7.4, fluorescence emission of the FITC–peptide–DR complexes was measured at 25 °C and514–516 nm with excitation at 492 nm on a Fluoromax3 spectrofluorometer (Horiba Jobin-Yvon) with a slit width of 2 nm. For dissociation assays, DR1 was incubated at 37 °C with fluorescent peptides in 0.15 M citrate phosphate buffer, pH 5.5, for at least 48 h to yield maximal loading. For double mutants, that incubation was done overnight because of quick association. Samples were then spun through a Sephadex G-50 spin column equilibrated with 0.15 M citrate phosphate, pH 5.5, for removal of excess unbound peptides. Samples were then incubated for the required length of time at 37 °C with excess unlabeled competitor peptide (usually 50-molar excess) in the presence or absence of various concentrations of DM. After one more spin through a Sephadex G-50 spin column for removal of dissociated fluorescent peptide, fluorescence was measured as described above.
Intrinsic tryptophan fluorescence measurements
Complexes of DR1WT, DR1βG86Y, DR1βH81N or the double-mutant DR1 (0.2 μM) with HA(306–318) or HA(anchorless) were incubated at 37 °C and the intrinsic fluorescence due to surface tryptophan residues of peptide–DR1 complexes alone or of complexes immediately after the addition of DM (0.1 μM) was measured. Care was taken to use protein solutions of the exact same concentration in different runs. The buffer in all cases was 0.15 M citrate phosphate, pH 5.5. Samples were excited at 295 nm (to minimize interference from tyrosine residues) and emission was monitored in a range of 310–430 nm on a temperature-controlled Fluoromax-3 fluorometer with a slit width of 5 nm.
Surface plasmon resonance measurement of dissociation rates
Cys–HA(306–318) or Cys–HA(anchorless) peptides were immobilized on an sulfosuccinimidyl4-(p-maleimidophenyl)-butyrate (SMPB)–activated CM5 chip in a BIAcore 2000, and HA(306–318)–DR1βH81N or HA(Y308A)–DR1WT complexes (about 2.5 μM) were allowed to bind to the peptide surface for 4 min (ref. 20). Complexes were allowed to dissociate for about 20 min, after which a solution of DM (8 μM in citrate phosphate, pH 6.0) was injected over the surface and dissociation was monitored for another 20 min. The running buffer was citrate phosphate, pH 6.0, and the flow speed was maintained at 10 μl/min. As a negative control, DM was injected over a HA(306–318)–DR1WT surface; that allowed a surface plasmon resonance measurement of nonspecific DM interaction with the surface. That `readout' was then factored into the raw data for complex dissociation obtained with DM to obtain the dissociation of DR1 only from the surface after the addition of DM.
Data analysis
Grafit or Origin 6.1 software was used for all kinetic analyses. All raw association data were fitted into single- or double-exponential association equations as follows: Y = Y0 + A1 (1 − e−x/t1), and Y = Y0 + A1 (1 − e−x/t1) + A2 (1 − e−x/t2) where `Y0' is offset, `A1' and `A2' are the amplitudes, and `t1' and `t2' are the width constants (for association curves) or decay constants (for dissociation curves) for phase 1 and phase 2 of the biphasic reactions, respectively. All raw dissociation data were fitted into single-exponential dissociation equations as follows: Y = Y0 + A1e−x/t1. In the surface plasmon resonance experiments, the raw dissociation in the absence of DM best fitted a biphasic curve: Y = Y0 + A1e−x/t1 + A2e−x/t2 (calculations, Supplementary Note online).
Supplementary Material
ACKNOWLEDGMENTS
We thank L.J. Stern and E.D. Mellins for discussions; K. Su for help with the experimental setup, C.-H. (Bear) Huang for help with modeling; and Erika Darrah for critical reading of the manuscript. P. Roche (National Cancer Institute) donated cDNA constructs for the HLA-DM α-and β-chains. K.N. dedicates dedicate this work to the memory of Varun Shankar, who passed away during the preparation of this manuscript. Supported by the National Institutes of Health (R01AI063764 and R01GM53549 to S.S.-N.).
Footnotes
Note: Supplementary information is available on the Nature Immunology website.
COMPETING INTERESTS STATEMENT The authors declare that they have no competing financial interests.
References
- 1.Neefjes JJ, Stollorz V, Peters PJ, Geuze HJ, Ploegh HL. The biosynthetic pathway of MHC class II but not class I molecules intersects the endocytic route. Cell. 1990;61:171–183. doi: 10.1016/0092-8674(90)90224-3. [DOI] [PubMed] [Google Scholar]
- 2.Peters PJ, Neefjes JJ, Oorschot V, Ploegh HL, Geuze HJ. Segregation of MHC class II molecules from MHC class I molecules in the Golgi complex for transport to lysosomal compartments. Nature. 1991;349:669–676. doi: 10.1038/349669a0. [DOI] [PubMed] [Google Scholar]
- 3.Blum JS, Cresswell P. Role for intracellular proteases in the processing and transport of class II HLA antigens. Proc. Natl. Acad. Sci. USA. 1988;85:3975–3979. doi: 10.1073/pnas.85.11.3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cresswell P. Assembly, transport, and function of MHC class II molecules. Annu. Rev. Immunol. 1994;12:259–293. doi: 10.1146/annurev.iy.12.040194.001355. [DOI] [PubMed] [Google Scholar]
- 5.Sadegh-Nasseri S, McConnell HM. A kinetic intermediate in the reaction of an antigenic peptide and I-Ek. Nature. 1989;337:274–276. doi: 10.1038/337274a0. [DOI] [PubMed] [Google Scholar]
- 6.Sadegh-Nasseri S, Germain RN. A role for peptide in determining MHC class II structure. Nature. 1991;353:167–170. doi: 10.1038/353167a0. [DOI] [PubMed] [Google Scholar]
- 7.Sadegh-Nasseri S, Germain RN. How MHC class II molecules work: peptide-dependent completion of protein folding. Immunol. Today. 1992;13:43–46. doi: 10.1016/0167-5699(92)90131-P. [DOI] [PubMed] [Google Scholar]
- 8.Sadegh-Nasseri S, Stern LJ, Wiley DC, Germain RN. MHC class II function preserved by low-affinity peptide interactions preceding stable binding. Nature. 1994;370:647–650. doi: 10.1038/370647a0. [DOI] [PubMed] [Google Scholar]
- 9.Castellino F, Zhong G, Germain RN. Antigen presentation by MHC class II molecules: invariant chain function, protein trafficking, and the molecular basis of diverse determinant capture. Hum. Immunol. 1997;54:159–169. doi: 10.1016/s0198-8859(97)00078-5. [DOI] [PubMed] [Google Scholar]
- 10.Romagnoli P, Germain RN. The CLIP region of invariant chain plays a critical role in regulating major histocompatibility complex class II folding, transport, and peptide occupancy. J. Exp. Med. 1994;180:1107–1113. doi: 10.1084/jem.180.3.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Natarajan SK, Assadi M, Sadegh-Nasseri S. Stable peptide binding to MHC class II molecule is rapid and is determined by a receptive conformation shaped by prior association with low affinity peptides. J. Immunol. 1999;162:4030–4036. [PubMed] [Google Scholar]
- 12.Sato AK, et al. Determinants of the peptide-induced conformational change in the human class II major histocompatibility complex protein HLA-DR1. J. Biol. Chem. 2000;275:2165–2173. doi: 10.1074/jbc.275.3.2165. [DOI] [PubMed] [Google Scholar]
- 13.Carven GJ, Stern LJ. Probing the ligand-induced conformational change in HLA-DR1 by selective chemical modification and mass spectrometric mapping. Biochemistry. 2005;44:13625–13637. doi: 10.1021/bi050972p. [DOI] [PubMed] [Google Scholar]
- 14.Denzin LK, Robbins NF, Carboy-Newcomb C, Cresswell P. Assembly and intracellular transport of HLA-DM and correction of the class II antigen-processing defect in T2 cells. Immunity. 1994;1:595–606. doi: 10.1016/1074-7613(94)90049-3. [DOI] [PubMed] [Google Scholar]
- 15.Fung-Leung WP, et al. Antigen presentation and T cell development in H2-M-deficient mice. Science. 1996;271:1278–1281. doi: 10.1126/science.271.5253.1278. [DOI] [PubMed] [Google Scholar]
- 16.Denzin LK, Cresswell P. HLA-DM induces CLIP dissociation from MHC class II αβ dimers and facilitates peptide loading. Cell. 1995;82:155–165. doi: 10.1016/0092-8674(95)90061-6. [DOI] [PubMed] [Google Scholar]
- 17.Green JM, DeMars R, Xu X, Pierce SK. The intracellular transport of MHC class II molecules in the absence of HLA-DM. J. Immunol. 1995;155:3759–3768. [PubMed] [Google Scholar]
- 18.Kropshofer H, Arndt SO, Moldenhauer G, Hammerling GJ, Vogt AB. HLA-DM acts as a molecular chaperone and rescues empty HLA-DR molecules at lysosomal pH. Immunity. 1997;6:293–302. doi: 10.1016/s1074-7613(00)80332-5. [DOI] [PubMed] [Google Scholar]
- 19.Kropshofer H, et al. Editing of the HLA-DR-peptide repertoire by HLA-DM. EMBO J. 1996;15:6144–6154. [PMC free article] [PubMed] [Google Scholar]
- 20.Miyazaki T, et al. Mice lacking H2-M complexes, enigmatic elements of the MHC class II peptide-loading pathway. Cell. 1996;84:531–541. doi: 10.1016/s0092-8674(00)81029-6. [DOI] [PubMed] [Google Scholar]
- 21.Martin WD, et al. H2-M mutant mice are defective in the peptide loading of class II molecules, antigen presentation, and T cell repertoire selection. Cell. 1996;84:543–550. doi: 10.1016/s0092-8674(00)81030-2. [DOI] [PubMed] [Google Scholar]
- 22.Weber DA, Evavold BD, Jensen PE. Enhanced dissociation of HLA-DR-bound peptides in the presence of HLA-DM. Science. 1996;274:618–620. doi: 10.1126/science.274.5287.618. [DOI] [PubMed] [Google Scholar]
- 23.Ullrich HJ, et al. Interaction between HLA-DM and HLA-DR involves regions that undergo conformational changes at lysosomal pH. Proc. Natl. Acad. Sci. USA. 1997;94:13163–13168. doi: 10.1073/pnas.94.24.13163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vogt AB, Moldenhauer G, Hammerling GJ, Kropshofer H. HLA-DM stabilizes empty HLA-DR molecules in a chaperone-like fashion. Immunol. Lett. 1997;57:209–211. doi: 10.1016/s0165-2478(97)00061-8. [DOI] [PubMed] [Google Scholar]
- 25.Chou CL, Sadegh-Nasseri S. HLA-DM recognizes the flexible conformation of major histocompatibility complex class II. J. Exp. Med. 2000;192:1697–1706. doi: 10.1084/jem.192.12.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doebele RC, Busch R, Scott HM, Pashine A, Mellins ED. Determination of the HLA-DM interaction site on HLA-DR molecules. Immunity. 2000;13:517–527. doi: 10.1016/s1074-7613(00)00051-0. [DOI] [PubMed] [Google Scholar]
- 27.Lazarski CA, et al. The kinetic stability of MHC class II:peptide complexes is a key parameter that dictates immunodominance. Immunity. 2005;23:29–40. doi: 10.1016/j.immuni.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 28.Lazarski CA, Chaves FA, Sant AJ. The impact of DM on MHC class II-restricted antigen presentation can be altered by manipulation of MHC-peptide kinetic stability. J. Exp. Med. 2006;203:1319–1328. doi: 10.1084/jem.20060058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Belmares MP, Busch R, Mellins ED, McConnell HM. Formation of two peptide/MHC II isomers is catalyzed differentially by HLA-DM. Biochemistry. 2003;42:838–847. doi: 10.1021/bi020466p. [DOI] [PubMed] [Google Scholar]
- 30.Pashine A, et al. Interaction of HLA-DR with an acidic face of HLA-DM disrupts sequence-dependent interactions with peptides. Immunity. 2003;19:183–192. doi: 10.1016/s1074-7613(03)00200-0. [DOI] [PubMed] [Google Scholar]
- 31.Stratikos E, Wiley DC, Stern LJ. Enhanced catalytic action of HLA-DM on the exchange of peptides lacking backbone hydrogen bonds between their N-terminal region and the MHC class II α-chain. J. Immunol. 2004;172:1109–1117. doi: 10.4049/jimmunol.172.2.1109. [DOI] [PubMed] [Google Scholar]
- 32.Pu Z, Lovitch SB, Bikoff EK, Unanue ER. T cells distinguish MHC-peptide complexes formed in separate vesicles and edited by H2-DM. Immunity. 2004;20:467–476. doi: 10.1016/s1074-7613(04)00073-1. [DOI] [PubMed] [Google Scholar]
- 33.Stern LJ, et al. Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature. 1994;368:215–221. doi: 10.1038/368215a0. [DOI] [PubMed] [Google Scholar]
- 34.Murthy VL, Stern LJ. The class II MHC protein HLA-DR1 in complex with an endogenous peptide: implications for the structural basis of the specificity of peptide binding. Structure. 1997;5:1385–1396. doi: 10.1016/s0969-2126(97)00288-8. [DOI] [PubMed] [Google Scholar]
- 35.Fremont DH, Hendrickson WA, Marrack P, Kappler J. Structures of an MHC class II molecule with covalently bound single peptides. Science. 1996;272:1001–1004. doi: 10.1126/science.272.5264.1001. [DOI] [PubMed] [Google Scholar]
- 36.Fremont DH, Monnaie D, Nelson CA, Hendrickson WA, Unanue ER. Crystal structure of I-Ak in complex with a dominant epitope of lysozyme. Immunity. 1998;8:305–317. doi: 10.1016/s1074-7613(00)80536-1. [DOI] [PubMed] [Google Scholar]
- 37.McFarland BJ, Katz JF, Sant AJ, Beeson C. Energetics and cooperativity of the hydrogen bonding and anchor interactions that bind peptides to MHC class II protein. J. Mol. Biol. 2005;350:170–183. doi: 10.1016/j.jmb.2005.04.069. [DOI] [PubMed] [Google Scholar]
- 38.Wilson N, Fremont D, Marrack P, Kappler J. Mutations changing the kinetics of class II MHC peptide exchange. Immunity. 2001;14:513–522. doi: 10.1016/s1074-7613(01)00140-6. [DOI] [PubMed] [Google Scholar]
- 39.McFarland BJ, Beeson C, Sant AJ. Cutting edge: a single, essential hydrogen bond controls the stability of peptide-MHC class II complexes. J. Immunol. 1999;163:3567–3571. [PubMed] [Google Scholar]
- 40.Saito K, Oda M, Sarai A, Azuma T, Kozono H. Contribution of a single hydrogen bond between bHis81 of MHC class II I-Ek and the bound peptide to the pH-dependent thermal stability. Microbiol. Immunol. 2004;48:53–57. doi: 10.1111/j.1348-0421.2004.tb03487.x. [DOI] [PubMed] [Google Scholar]
- 41.DeLano WL. PyMol. DeLano Scientific; San Carlos, California: 2002. [Google Scholar]
- 42.Jardetzky TS, et al. Peptide binding to HLA-DR1: a peptide with most residues substituted to alanine retains MHC binding. EMBO J. 1990;9:1797–1803. doi: 10.1002/j.1460-2075.1990.tb08304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Natarajan SK, Stern LJ, Sadegh-Nasseri S. Sodium dodecyl sulfate stability of HLA-DR1 complexes correlates with burial of hydrophobic residues in pocket 1. J. Immunol. 1999;162:3463–3470. [PubMed] [Google Scholar]
- 44.McNally JG, Muller WG, Walker D, Wolford R, Hager GL. The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science. 2000;287:1262–1265. doi: 10.1126/science.287.5456.1262. [DOI] [PubMed] [Google Scholar]
- 45.Rigaud G, Roux J, Pictet R, Grange T. In vivo footprinting of rat TAT gene: dynamic interplay between the glucocorticoid receptor and a liver-specific factor. Cell. 1991;67:977–986. doi: 10.1016/0092-8674(91)90370-e. [DOI] [PubMed] [Google Scholar]
- 46.Zwart W, et al. Spatial separation of HLA-DM/HLA-DR interactions within MIIC and phagosome-induced immune escape. Immunity. 2005;22:221–233. doi: 10.1016/j.immuni.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 47.Rabinowitz JD, et al. Formation of a highly peptide-receptive state of class II MHC. Immunity. 1998;9:699–709. doi: 10.1016/s1074-7613(00)80667-6. [DOI] [PubMed] [Google Scholar]
- 48.Stern LJ, Wiley DC. The human class II MHC protein HLA-DR1 assembles as empty αβ heterodimers in the absence of antigenic peptide. Cell. 1992;68:465–477. doi: 10.1016/0092-8674(92)90184-e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.