

Abstract
Among the candidate anti-prion chemotherapeutic agents identified to date, complex polyamines constitute the only class of compounds that possess the ability to remove pre-existing PrPSc molecules from infected cells. The potency of branched polyamines such as cationic dendrimers increases with the density of positive charges on their surface. Cationic dendrimers appear to accumulate together with PrPSc molecules in lysosomes, where the acidic environment facilitates dendrimer-mediated PrPSc disaggregation. Dendrimers can disaggregate a range of different amyloid proteins by interacting with specific epitopes on each protein. Studies with model peptides suggest that dendrimers may cause fiber breakage and capping of elongating fibers. Potential limitations to the development of dendrimers as therapeutic compounds for neurodegenerative disorders of protein misfolding such as prion diseases include poor bioavailability, limited spectrum of activity, and detrimental neurological side effects. A related group of compounds, lipopolyamines, are smaller molecules containing a lipophilic tail that may assist membrane targeting. Developing strategies to enable the safe delivery of potent complex polyamines to the central nervous system represents a critical avenue for future research.
Keywords: Prion, PrPSc, branched polyamines, dendrimers
INTRODUCTION
Prion diseases are a group of invariably fatal neurodegenerative diseases that include Creutzfeldt Jacob disease (CJD) in humans, bovine spongiform encephalopathy (BSE) in cattle, scrapie in sheep and goats, and Chronic Wasting disease (CWD) in deer and elk [1]. All known forms of prion diseases are associated with either the mutation or misfolding of an endogenous glycoprotein known as the prion protein (PrP). In normal hosts, PrP molecules adopt a well-folded conformation called “cellular” PrPC, whereas in diseased hosts, PrP molecules adopt an aggregation-prone, protease-resistant “scrapie” conformation termed PrPSc. Several lines of evidence indicate that PrPSc molecules are essential components of infectious prions [2-4].
A striking characteristic of infectious prions is their extreme resistance to inactivation [5]. Effective prion disinfection protocols employ conditions that denature aggregated proteins, such as exposure to >20,000 p.p.m. sodium hypochlorite or 6 M guanidine HCl. Consequently, it is not surprising that the vast majority of anti-prion compounds identified to date act by preventing the conversion of PrPC into PrPSc, rather than by degrading PrPSc molecules. Some of these compounds, such as porphyrins, phthalocyanines, Congo Red, and sulfated polyanions, prolong the lifespan of prion-infected animals, particularly when administered not too long after prion inoculation [6, 7].
To date, there have been no reports showing that any compound can reverse or ameliorate prion disease progression following the onset of neurological symptoms. However, a study using transgenic mice expressing conditionally expressed PrP molecules has demonstrated that reversing the neurological damage caused by prions may be possible [8]. In this study, the gene encoding PrP was deleted by Cre-mediated recombination after establishment of prion infection, causing a halt in PrPSc formation and reversal of early spongiform degeneration [8]. The current therapeutic challenges are to stop the further production of PrPSc molecules, and to remove the PrPSc molecules that accumulated during the pre-symptomatic phase of the disease. Given the extreme chemical and physical resistance of infectious prions to inactivation, accomplishing the latter task without disrupting normal cellular physiology would appear daunting. However, in vitro studies with a number of different complex polyamines suggest that this group of chemical compounds might be able to inactivate PrPSc molecules though unique interactions that do not compromise cell viability [9-14]. The activity, mechanism, and therapeutic potential of these compounds is the subject of this review.
MECHANISM OF PRION FORMATION
The precise mechanism of prion formation in cells is currently unknown. The “protein-only” hypothesis proposes that PrPSc molecules are the only essential components of infectious prions [2, 15, 16]. According to this hypothesis, pre-existing PrPSc molecules induce the conformational change of PrPC molecules into new PrPSc molecules in a self-perpetuating process. Recent studies in which infectious prions were formed de novo from defined substrates lead to the possibility of a modified scenario, in which prions might be composed of several required components, specifically PrP, endogenous polyanions, and lipid molecules [4, 17]. If prions indeed contain essential components other than PrPSc, then a relevant corollary would be the existence of novel non-PrPSc drug targets. For example, it is possible that complex polyamines interact with prion-associated polyanionic molecules rather than PrPSc molecules.
Studies in chronically infected cultured cells have provided insight into the cell biology of prion formation [18-20] (Fig. 1A). Mature PrP molecules contain a C-terminal glycophosphatidylinositol (GPI) anchor, two glycosylation sites at asparagine residues 180 and 196, and a single intramolecular disulfide bond [21-24]. Following biosynthesis, PrPC molecules traffic through the secretory pathway to the extracellular surface of the plasma membrane to which they are attached by the GPI anchor. While localized on the cell surface, PrPC molecules could potentially interact with cofactors such as polyanionic and lipid molecules prior to endocytosis into non-clathrin vesicles (Fig. 1). Although the subcellular compartment in which PrPC to PrPSc conversion occurs has not been precisely determined, immunocytochemical studies indicate that PrPSc molecules eventually accumulate in secondary lysosomes, where they appear to resist degradation by the resident proteases within the acidic environment (Fig. 1A) [25]. Effective removal of PrPSc molecules that accumulated in lysosomes during the pre-symptomatic phase of prion disease would require a therapeutic compound capable of facilitating PrPSc degradation within this compartment. In fact, this appears to be the likely mechanism of action by which branched polyamines act to clear PrPSc from prion-infected cells (Fig. 1B).
Fig. (1).

Hypothetical mechanisms of PrPSc formation and polyamine-mediated clearance in cells (A) Schematic representation of cell biology of PrPSc formation. The biosynthesis of PrPC proceeds through the secretory pathway, which produces mature PrPC molecules that are tethered to the extracellular leaflet of the plasma membrane. Eventually, PrPC molecules are endocytosed through a process apparently mediated by lipoprotein receptor related protein 1 (LRP1) [32, 33]. The pathogenic conformational change of PrPC into PrPSc molecules may require interaction with endogeneous lipid and polyanionic cofactors [4], probably occurs either on the cell surface or within the endocytic pathway, and ultimately results in accumulation of PrPSc molecules in lysosomes [25]. (B) Schematic representation of the cell biology of polyamine-mediated PrPSc clearance. Complex polyamines such as dendrimers appear to be internalized through the endocytic pathway, and eventually become co-localized with PrPSc molecules within lysosomes [10]. The acidic environment of this compartment appears to facilitate polyamine-medicated PrPSc denaturation, and resident hydrolases may also facilitate the clearance of PrPSc molecules from lysosomes.
DENDRIMERS AND OTHER MULTIVALENT POLYAMINES
Dendrimers are branched polyamines manufactured by a repetitive divergent growth technique, allowing the synthesis and isolation of successive, well-defined “generations” of homodisperse structures. Since their discovery, these versatile compounds have been used successfully as catalysts, contrast agents, drug delivery facilitators, and transfection reagents [26]. During the course of a routine transfection assay, one of us (S.S.) observed by chance that Superfect™, a commercially produced transfection reagent composed of a heat-degraded preparation of PAMAM dendrimers, selectively cleared PrPSc molecules from scrapie-infected ScN2a neuroblastoma cells [9]. Subsequent experiments showed that intact dendrimers and other similar branched polyamines, such as polyethyleneimine (PEI), could also clear PrPSc molecules from ScN2a cells, and the strength of this effect was dependent on both polyamine concentration as well as the duration of exposure. At high concentration, PEI removed PrPSc from ScN2a cells rapidly, with t1/2 ~ 4 hrs. At concentrations exceeding those required to clear PrPSc, branched polyamines did not alter N2a cell viability or growth. Currently, complex polyamines are the only class of compounds reported to have the ability to clear pre-existing PrPSc molecules rapidly from prion-infected cells (as opposed to blocking the formation of new PrPSc molecules). The unique ability of complex polyamines to facilitate the clearance of PrPSc molecules is advantageous because endogenous PrPSc clearance mechanisms appear to be relatively inefficient. Bioassays using highly susceptible transgenic mice overexpressing PrPC as recipients confirmed that clearance of PrPSc mediated by branched polyamines also cured ScN2a cells of prion infectivity [10].
Structure-activity studies performed with a variety of polyamines revealed that a high surface density of primary amines and branching architecture were important features of effective anti-prion polyamines (Table 1) [9, 13, 14, 27]. For instance, the potency of PAMAM dendrimers in mediating ScN2a PrPSc clearance increases progressively between generations 0.0 to 4.0, eventually reaching a plateau at generation 5.0 (S.S. unpublished observations). When the 64 terminal amino groups of PAMAM generation 4.0 are replaced with hydroxyl groups, the resulting uncharged compound PAMAM-OH generation 4.0 completely lacks PrPSc clearance activity. Phosphorus-containing dendrimers and tertiary amine terminals (P-dendrimers) also possess the ability to clear PrPSc rapidly from cells [12] (Table 1). The phosphorus atoms in the backbone of P-dendrimers render these molecules more stable against nucleophilic attack and acid-catalyzed hydrolysis, and therefore P-dendrimers may be relatively resistant to degradation. Furthermore, degradation of branched polyamines is not likely to pose an obstacle to therapy, because even a heat-fragmented preparation of PAMAM exhibits potent PrPSc clearance activity in ScN2a cells [9].
Table 1.
Complex Polyamines with Anti-Prion Activity
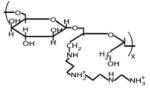
Oligoamine-conjugated polysaccharides were also investigated for their ability to eliminate PrPSc from ScN2a cells [13]. These systematic studies revealed that whereas the structure of the polysaccharide backbone had minimal effects on PrPSc clearance activity, the identity of the oligoamine conjugate was critical; the tetramine compound spermine conferred the highest level of activity among the oligoamines tested (Table 1). Interestingly, shielding the charged oligoamine groups by partial substitution with either methoxypoly (ethylene glycol) or oleic acid reduced the potency of a dextran-spermine conjugate, suggesting that steric accessibility to amino groups may be required for polyamines to interact with PrPSc.
The biochemical mechanism of branched polyamine-mediated PrPSc clearance has been studied with a novel in vitro PrPSc denaturation assay [10]. In this assay, crude prion-infected brain homogenates are mixed with test compounds in various buffers. Following incubation, samples are neutralized, and then subjected to protease digestion to remove endogenous PrPC molecules. The remaining PrPSc molecules are then detected by Western blot. These studies showed that (1) in vitro PrPSc denaturation mediated by dendrimers requires acidic pH; (2) dendrimers can denature purified prion rods in vitro; and (3) the ability of polypropyleneimine (PPI) generation 4.0 to denature PrPSc molecules varies between different prion strains. Each of these findings provides significant information about the mechanism by which dendrimers interact with PrPSc molecules and facilitate their clearance. The pH optimum of dendrimer-mediated PrPSc degradation in an in vitro assay suggested that dendrimers most likely bind to and denature PrPSc molecules in an acidic environment within cells. Consistent with this hypothesis, double-label confocal microscopy indicates that PPI generation 4.0 accumulates specifically in the lysosomes of N2a cells [10]. The ability of dendrimers to denature purified prion rods in vitro showed that these compounds must interact directly either with PrPSc molecules or tightly associated ligands, such as lipids or endogenous polyanionic molecules, to disrupt PrPSc structure (Fig. 1B). This conclusion was further supported by Fourier transform infrared and electron microscopic studies, which showed, respectively, that dendrimer-treated purified prions lost β-sheet structure and became disaggregated [10]. The differential susceptibility of PrPSc molecules derived from various prion strains to degradation in vitro provides both mechanistic and practical insights. From a mechanistic perspective, one can conclude that the efficacy of dendrimer-mediated denaturation depends on the structure of PrPSc itself, reflecting either the specific tertiary conformation or the overall aggregation state associated with each prion strain. It is reasonable to hypothesize that the prion strains resistant to dendrimer-mediated denaturation either possess more stable structures or interact less well with the terminal positive charges of branched polyamines than susceptible strains. From a practical perspective, the in vitro data suggest that branched polyamines may have a limited spectrum of activity against different prion strains.
A similar in vitro PrPSc degradation assay was used to study the ability of a fourth generation P-dendrimer to interact with PrPSc molecules [12]. The results of these studies provided two significant insights into the mechanism of dendrimer action. First, unlike PAMAM and PPI dendrimers, P-dendrimers were able to mediate PrPSc degradation at neutral pH, presumably because their tertiary amine terminals retain significant charge at pH 7. This finding suggests that acidic pH probably does not facilitate PAMAM- and PPI-mediated PrPSc degradation by simply denaturing PrPSc molecules. Instead, acidic pH may promote a more extended conformation of these dendrimers, which may in turn facilitate interaction with PrPSc aggregates [28]. A second significant result was that the spectrum of prion strains susceptible to P-dendrimer-mediated degradation was different than the spectrum of strains susceptible to PPI. This observation offers proof of principle that the spectrum of activity associated with branched polyamines can be altered or extended by changing the chemical structure of the these compounds, and such studies represent an interesting area for future investigation.
Several investigators have studied the effects of dendrimers on the fibrillation of various amyloidogenic proteins and peptides in vitro, including PrP [27, 29, 30]. These interesting studies have provided significant mechanistic insight into the potential molecular interactions between dendrimers and protein fibers. For instance, in an intriguing study using both PrP 185-208 and Alzheimer's peptide Aß 1-28 as target peptides, Klajnert et al. showed that low concentrations of dendrimers unexpectedly accelerate amyloid formation, while high concentrations efficiently disaggregated pre-formed amyloid fibers [27]. These findings support the authors' proposal that dendrimers may be able to break amyloid fibers (accelerating amyloidogenesis by increasing the number of available seeds) while simultaneously capping the elongating ends of growing polymers (an effect that becomes more dominant at higher dendrimer concentrations) [27]. In a separate series of provocative studies, Heegaard et al. showed that dendrimers are able to disaggregate a variety of different amyloid proteins by interacting with individual parts of each protein, rather than acting as general denaturants [28]. These results raise the exciting possibility that dendrimers might eventually find application in a variety of different settings that necessitate protein disaggregation. The recognition that dendrimers can disaggregate both prion and Aß fibrils represent one example in which studies of prion disease can yield results pertinent to more common, non-prion dementias such as Alzheimer's disease (see article by Rodrigo Morales in this issue).
Within cells, PPI dendrimers appear to accumulate in lysosomes, the same compartment that accumulates PrPSc molecules [10]. This fortunate coincidence likely facilitates interaction between dendrimer and PrPSc by local compartmentalization of the reactants and by expansion of the cavitary structure of the dendrimer. Presumably, resident proteases such as cathepsins also facilitate PrPSc clearance within lysosomes by hydrolyzing PrP molecules after dendrimer-induced protein denaturation.
LIPOPOLYAMINES
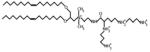
A general limitation of multivalent polyamines is that their large size and highly charged structures tend to limit bioavailability. However, cationic lipopolyamines, smaller compounds with fewer charged groups, have been identified as an alternative class of complex polyamines able to disaggregate and clear PrPSc molecules from cells [11]. These compounds are characterized chemically by the presence of a spermine headgroup, a quaternary ammonium ion linker, and a lipophilic tail. The most potent PrPSc-clearing cationic lipopolysaccharide identified to date is 2,3-dioleyloxy-N-[2(sperminecarboxamido)ethyl]-N,N-dimethyl-1-propanaminium trifluoroacetate (DOSPA) (Table 1). When applied to ScN2a cells, DOSPA liposomes effectively solubilized PrPSc molecules, whereas free spermidine had no apparent effect on PrPSc solubility, indicating that the presence of the lipophilic tail facilitates the interaction between cationic lipopolyamine and PrPSc molecules, possibly by promoting membrane targeting. Like other complex polyamines, DOSPA did not reduce cell viability, and also showed no adverse effects on the biosynthesis or trafficking of PrPC.
CHALLENGES FOR USING BRANCHED POLYAMINES AS THERAPEUTIC AGENTS
The unique ability of complex polyamines to disaggregate prions and other amyloidogenic proteins (without acting as general denaturants) raises the possibility that this class of compounds might become potent therapeutic agents in prion diseases and other neurodegenerative diseases. Because of their unique mechanism of action, complex polyamines might act synergistically with inhibitors of PrPSc conversion. However, several obstacles need to be addressed before the potential clinical utility of complex polayamines can be realized.
One major obstacle is the pharmacologic delivery of relatively large and highly charged polyamines to brain neurons. As they are currently formulated, polyamines are unlikely to cross the blood-brain barrier and distribute throughout the brain [31]. Some possible methods to circumvent potential problems with bioavailability include direct injection into the central nervous system with intraventricular pumps and shielding the high density of positive charges with negatively charged or neutrally charged carrier molecules. One group of complex polyamines that may display superior bioavailability is the lipopolyamines such as DOSPA, due to their small size and lower charge density. Bioavailability is likely to be less problematic outside the central nervous system. For instance, Solassol et al. demonstrated that intraperitoneal injection of P-dendrimers into mice successfully decreased splenic accumulation of PrPSc in vivo [12].
Another potential problem is that in vitro studies suggest that branched polyamines may possess a limited spectrum of activity against various prion strains. For instance, PPI generation 4.0 appears to degrade PrPSc derived from BSE, but not scrapie, in vitro [10]. It is possible that the in vitro studies might underestimate the therapeutic efficacy of these compounds in vivo, but this potential limitation is important to consider. Furthermore, the observation that PPI dendrimers and P-dendrimers can degrade PrPSc molecules derived from different sets of prion strains suggests that chemical modifications could alter or extend the spectrum of activity for branched polyamines [12].
A final consideration is that complex polyamines might cause detrimental side effects in live animals, even though they did not cause detectable cyotoxicity in cultured cells. Some studies have shown that cationic dendrimers are well tolerated systemically, but their potential effects within the central nervous system are not known. A particular concern, given the highly cationic nature of these compounds, is that they could trigger seizures. Another potential problem with larger polyamines is that they might inappropriately cross-link receptors or sequester endogenous molecules that are normally separated spatially (such as neurotransmitters and their inactivating enzymes). Accumulation of branched polyamines in lysosomes could potentially cause disruption of lysosomal hydrolases, or even destruction of the organelle itself. It may be possible to avoid some of these potential problems and simultaneously increase the potency of branched polyamines by selectively targeting of these compounds to specific cellular targets. Such targeting might be achieved by synthesizing dendrimer conjugates, for instance with antibodies to PrPSc molecules.
CONCLUSIONS
Complex polyamines such as cationic dendrimers, other branched polyamines, and lipopolyamines represent a unique class of anti-prion compounds because of their ability to remove pre-existing PrPSc molecules from living cells. Dendrimers appear to accumulate in lysosomes, where PrPSc molecules also accumulate, and the acidic environment within lysosomes facilitates anti-prion activity. Studies with model peptides and proteins suggest that the mechanism of dendrimer-mediated PrPSc denaturation may involve a combination of fiber breakage and polymer capping, and that dendrimers may act on a wide range of amyloid proteins. Current challenges to the development of polyamines into practical therapeutic compounds include bioavailability, spectrum of activity, and potential neurological side effects.
ABBREVIATIONS
- BSE
Bovine spongiform encephalopathy
- CWD
Chronic Wasting disease
- CJD
Creutzfeldt Jacob disease
- DOSPA
Dimethyl-1-propanaminium trifluoroacetate
- GPI
Glycophosphatidylinositol
- PEI
Polyethyleneimine
- PPI
Polypropyleneimine
- PrP
Prion protein
- PrPc
Cellular prion protein
- PrPSc
Scrapie prion protein
REFERENCES
- 1.Prusiner SB. Prions. Proc. Natl. Acad. Sci. USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prusiner SB. Novel Proteinaceous infectious particles cause scrapie. Science. 1982;216(4542):136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 3.Castilla J, Saa P, Hetz C, Soto C. In vitro generation of infectious scrapie prions. Cell. 2005;121:195–206. doi: 10.1016/j.cell.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 4.Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. USA. 2007;104:9741–9746. doi: 10.1073/pnas.0702662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor DM. Inactivation of transmissible degenerative encephalopathy agents: a review. Vet. J. 2000;159:10–17. doi: 10.1053/tvjl.1999.0406. [DOI] [PubMed] [Google Scholar]
- 6.Priola SA, Raines A, Caughey WS. Porphyrin and phthalocyanine antiscrapie compounds. Science. 2000;287:1503–1506. doi: 10.1126/science.287.5457.1503. [DOI] [PubMed] [Google Scholar]
- 7.Ludewigs H, Zuber C, Vana K, Nikles D, Zerr I, Weiss S. Therapeutic approaches for prion disorders. Expert Rev. Anti Infect. Ther. 2007;5:613–630. doi: 10.1586/14787210.5.4.613. [DOI] [PubMed] [Google Scholar]
- 8.Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;302:871–874. doi: 10.1126/science.1090187. [DOI] [PubMed] [Google Scholar]
- 9.Supattapone S, Nguyen HO, Cohen FE, Prusiner SB, Scott MR. Elimination of prions by branched polyamines and implications for therapeutics. Proc. Natl. Acad. Sci. USA. 1999;96:14529–14534. doi: 10.1073/pnas.96.25.14529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Supattapone S, Wille H, Uyechi L, Safar J, Tremblay P, Szoka FC, Cohen FE, Prusiner SB, Scott MR. Branched polyamines cure prion-infected neuroblastoma cells. J. Virol. 2001;75:3453–3461. doi: 10.1128/JVI.75.7.3453-3461.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Winklhofer KF, Tatzelt J. Cationinc lipopolyamines induce degeneration of PrPSc in scrapie-infected mouse neuroblastoma cells. Biol. Chem. 2000;381:463–469. doi: 10.1515/BC.2000.061. [DOI] [PubMed] [Google Scholar]
- 12.Solassol J, Crozet C, Perrier V, Leclaire J, Beranger F, Caminade AM, Meunier B, Dormont D, Majoral JP, Lehmann S. Cationic phosphorus-containing dendrimers reduce prion repliction both in cell culture and in mice infected with scrapie. J. Gen. Virol. 2004;85:1791–1799. doi: 10.1099/vir.0.19726-0. [DOI] [PubMed] [Google Scholar]
- 13.Yudovin-Farber I, Azzam T, Metzer E, Taraboulos A, Domb AJ. Cationic polysaccharides as antiprion agents. J. Med. Chem. 2005;48:1414–1420. doi: 10.1021/jm049378o. [DOI] [PubMed] [Google Scholar]
- 14.Cordes H, Boas U, Olsen P, Heegaard PM. Guanidino- and urea-modified dendrimers as potent solubilizers of misfolded prion protein aggregates under non-cytotoxic conditions, dependence on dendrimer generation and surface charge. Biomacromolecules. 2007;8:3578–3583. doi: 10.1021/bm7006168. [DOI] [PubMed] [Google Scholar]
- 15.Griffith JS. Self-replication and scrapie. Nature. 1967;215:1043–1044. doi: 10.1038/2151043a0. [DOI] [PubMed] [Google Scholar]
- 16.Cohen FE, Pan KM, Huang Z, Baldwin M, Fletterick RJ, Prusiner SB. Structural clues to prion replication. Science. 1994;264:530–531. doi: 10.1126/science.7909169. [DOI] [PubMed] [Google Scholar]
- 17.Geoghegan JC, Valdes PA, Orem NR, Deleault NR, Williamson RA, Harris BT, Supattapone S. Selective incorporation of polyanionic molecules into hamster prions. J. Biol. Chem. 2007;282:36341–36353. doi: 10.1074/jbc.M704447200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caughey B, Raymond GJ, Priola SA, Kocisko DA, Race RE, Bessen RA, Lansbury PT, Jr., Chesebro B. Methods for studying prion proteins (PrP) metabolism and the formation of protease-resistant PrP in cell culture and cell-free systems. An update. Mol. Biotechnol. 1999;13:45–55. doi: 10.1385/MB:13:1:45. [DOI] [PubMed] [Google Scholar]
- 19.Harris DA. Biosynthesis and cellular processing of the prion protein. Adv. Protein Chem. 2001;57:203–228. doi: 10.1016/s0065-3233(01)57023-0. [DOI] [PubMed] [Google Scholar]
- 20.Prusiner SB. Prion Biology and Diseases. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y.: 2000. [Google Scholar]
- 21.Endo T, Groth D, Prusiner SB, Kobata A. Diversity of oligosaccharide structures linked to asparagines of the scrapie prion protein. Biochemistry. 1989;28:8380–8388. doi: 10.1021/bi00447a017. [DOI] [PubMed] [Google Scholar]
- 22.Locht C, Chesebro B, Race R, Keith JM. Molecular cloning and complete sequence of prion protein cDNA from mouse brain infected with the scrapie agent. Proc. Natl. Acad. Sci. USA. 1986;83:6372–6376. doi: 10.1073/pnas.83.17.6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phosphotidylinositol glycolipid. Cell. 1987;51:229–240. doi: 10.1016/0092-8674(87)90150-4. [DOI] [PubMed] [Google Scholar]
- 24.Turk E, Teplow DB, Hood LE, Prusiner SB. Purification and properties of a cellular and scrapie hamster prion proteins. Eur. J. Biochem. 1988;176:21–30. doi: 10.1111/j.1432-1033.1988.tb14246.x. [DOI] [PubMed] [Google Scholar]
- 25.McKinley MP, Taraboulos A, Kenaga L, Serban D, Stieber A, DeArmond SJ, Prusiner SB, Gonatas N. Ultrastructure localization of scrapie prion proteins in cytoplasmic vesicles of infected cultured cells. Lab. Invest. 1991;65:622–630. [PubMed] [Google Scholar]
- 26.Yang H, Kao WJ. Dendrimers for pharmaceutical and biomedical applications. J. Biomater. Sci. Polym. Ed. 2006;17:3–19. doi: 10.1163/156856206774879171. [DOI] [PubMed] [Google Scholar]
- 27.Klajnert B, Cortijo-Arellano M, Cladera J, Bryszewska M. Influence of dendrimer's structure on its activity against amyloid fibril formation. Biochem. Biophys. Res. Commun. 2006;345:21–28. doi: 10.1016/j.bbrc.2006.04.041. [DOI] [PubMed] [Google Scholar]
- 28.Heegaard PM, Boas U, Otzen DE. Dendrimer effects on peptide and protein fibrillation. Macromol. Biosci. 2007;7:1047–1059. doi: 10.1002/mabi.200700051. [DOI] [PubMed] [Google Scholar]
- 29.Heegaard PM, Pedersen HG, Flink J, Boas U. Amyloid aggregates of the prion peptide PrP106-126 are destabilised by oxidation and by the action of dendrimers. FEBS Lett. 2004;577:127–133. doi: 10.1016/j.febslet.2004.09.073. [DOI] [PubMed] [Google Scholar]
- 30.Breydo L, Bocharova OV, Baskakov IV. Semiautomated cell-free conversion of prion protein: applications for high-throughput screening potential antiprion drugs. Anal Biochem. 2005;339:165–173. doi: 10.1016/j.ab.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 31.Boyd BJ, Kaminskas LM, Karellas P, Krippner G, Lessene R, Porter CJ. Cationinc poly-L-lysine dendrimers: pharmacokinetics, biodistribution, and evidence for metabolism and bioresorption after intravenous administration to rats. Mol. Pharm. 2006;3:614–627. doi: 10.1021/mp060032e. [DOI] [PubMed] [Google Scholar]
- 32.Taylor DR, Hooper NM. The low-density lipoprotein receptor-related protein-1 (LRP1) mediates the endocytosis of the cellular prion protein. Biochem. J. 2007;402:17–23. doi: 10.1042/BJ20061736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parkyn CJ, Vermeulen EG, Mootoosamy RC, Sunyach C, Jacobsen C, Oxvig C, Moestrup S, Liu Q, Bu G, Jen A, Morris RJ. LRP1 controls biosynthetic and endocytic trafficking of neuronal prion protein. J. Cell. Sci. 2008;121:773–783. doi: 10.1242/jcs.021816. [DOI] [PubMed] [Google Scholar]