

Abstract
d-Alanine:d-alanine ligase (EC 6.3.2.4; Ddl) catalyzes the ATP-driven ligation of two d-alanine (d-Ala) molecules to form the d-alanyl:d-alanine dipeptide. This molecule is a key building block in peptidoglycan biosynthesis, making Ddl an attractive target for drug development. d-Cycloserine (DCS), an analog of d-Ala and a prototype Ddl inhibitor, has shown promise for the treatment of tuberculosis. Here, we report the crystal structure of Mycobacterium tuberculosis Ddl at a resolution of 2.1 Å. This structure indicates that Ddl is a dimer and consists of three discrete domains; the ligand binding cavity is at the intersection of all three domains and conjoined by several loop regions. The M. tuberculosis apo Ddl structure shows a novel conformation that has not yet been observed in Ddl enzymes from other species. The nucleotide and d-alanine binding pockets are flexible, requiring significant structural rearrangement of the bordering regions for entry and binding of both ATP and d-Ala molecules. Solution affinity and kinetic studies showed that DCS interacts with Ddl in a manner similar to that observed for d-Ala. Each ligand binds to two binding sites that have significant differences in affinity, with the first binding site exhibiting high affinity. DCS inhibits the enzyme, with a 50% inhibitory concentration (IC50) of 0.37 mM under standard assay conditions, implicating a preferential and weak inhibition at the second, lower-affinity binding site. Moreover, DCS binding is tighter at higher ATP concentrations. The crystal structure illustrates potential drugable sites that may result in the development of more-effective Ddl inhibitors.
Tuberculosis (TB), caused by Mycobacterium tuberculosis, remains a leading factor in worldwide morbidity and mortality. Recent trends indicate that TB cases due to multidrug-resistant (MDR) M. tuberculosis and extensively drug-resistant (XDR) M. tuberculosis are continuously increasing, with estimates that at least 490,000 cases of MDR M. tuberculosis infections occur each year (16). Current therapies not only are ineffective against MDR and XDR M. tuberculosis but also require long treatment courses (16). Unfortunately, new anti-TB drugs are not being developed at a suitable rate to keep pace with the rising spread of drug-resistant M. tuberculosis. Thus, there is an urgent need for timely development of novel anti-TB compounds to effectively treat drug-resistant cases and shorten treatment protocols.
The pathways involved in bacterial cell wall biosynthesis are key targets for novel antibiotic design (35). The M. tuberculosis cell wall is a lipid rich structure with a rigid peptidoglycan backbone (2). Peptidoglycan is a branched polymer consisting of β-(1,4)-linked N-acetyl- or N-glycolyl-muramic acid and N-acetyl-glucosamine moieties (27, 42). Small peptide chains consisting of d-alanine (d-Ala), d-glutamate, and meso-diaminopimelate extend from the N-glycolyl or N-acetylmuramic acid component and cross-link adjacent N-glycolyl/acetylmuramic chains via peptide bridges (17). The peptidoglycan backbone confers the cell wall tensile strength; thus, inhibiting this cross-linking leads to extensively weaker cell walls and cell death. d-Ala plays a key role in peptidoglycan cross-linking (31, 37). Therefore, enzymes involved in the d-Ala pathway of peptidoglycan biosynthesis are attractive targets for novel antibiotic design.
d-Alanine:d-alanine ligase, also denoted d-alanyl:d-alanine (d-Ala:d-Ala) synthetase, Ddl, and EC 6.3.2.4, catalyzes the reaction
Ddl enzymes from various microbial sources have multiple ligand binding sites relevant to their catalytic activity: d-Ala1 binds the first monomer of d-Ala with high affinity, and d-Ala2 binds the second d-Ala with lower affinity (13, 39). In addition, there is a distinct ATP binding site (13). The proposed mechanism for the reaction involves binding of d-Ala to the d-Ala1 site, phosphorylation of the amino acid's carboxylate by the γ-phosphate of ATP, and subsequent binding to the d-Ala2 site. Ligation is completed upon the nucleophilic attack of the amine at the d-Ala2 site on the phosphorylated d-Ala at the d-Ala1 site to yield d-Ala:d-Ala (13, 39). Inhibitors mimicking d-Ala or d-Ala:d-Ala have been discovered, including d-cycloserine (DCS), cis- and trans-dl-cyclothreonine, and compounds related to reaction transition state intermediates, such as phosphinate and phosphonate derivatives (32, 34).
DCS has been used as an anti-TB agent and is the only Ddl inhibitor currently used in the treatment of bacterial infections (46). DCS inhibits Ddl by competitive inhibition, but the relative contribution of each d-Ala site to this process remains unknown (19, 31, 32, 40). DCS also inhibits d-alanine racemase, the enzyme in the d-Ala synthesis pathway that catalyzes the interconversion of l-Ala and d-Ala. d-Alanine racemase provides a substrate to Ddl, and its overexpression in Mycobacterium smegmatis leads to resistance by target overproduction (4). It is unclear whether d-alanine racemase, Ddl, or both are the target(s) of DCS bactericidal action in M. tuberculosis. There are side effects to DCS treatment, largely due to lack of specificity, which have led to its limited use (46). Structure-based drug design using DCS as a scaffold could aid in developing Ddl inhibitors with greater specificity and attenuated severity of side effects. Unfortunately, no atomic level structural information regarding M. tuberculosis Ddl has been reported, although Ddl structures from other organisms are available (13, 23, 25, 45).
To gain insights into the mechanism of action of DCS in M. tuberculosis, we have solved the crystal structure of M. tuberculosis Ddl to a 2.1-Å resolution. The overall structure correlates with previously reported Ddl structures from other microbial species. However, divergences in the primary and tertiary structures of M. tuberculosis Ddl, compared to the levels for other organisms, show key differences in the overall folding and active-site structure. We describe the presence of novel pockets in the M. tuberculosis Ddl structure and present fluorescence quenching binding affinity studies, isothermal calorimetry titration data, and enzymatic assays. This combined analysis results in a more complete picture of how M. tuberculosis Ddl interacts with ATP, d-Ala, and DCS.
MATERIALS AND METHODS
Cloning, protein expression, and purification.
M. tuberculosis H37Rv genomic DNA was used as a template to PCR amplify the Ddl gene (Rv2981c accession no. P95114), yielding a 1,122-nucleotide DNA fragment using primers 5′-GCATATGAGTGCTAACGACCG-3′ and 5′-GCTAAGTGCCGATCGCAAG-3′. NdeI and HindIII were used to digest the fragment for subsequent ligation into the pET28b vector (Novagen) modified to contain an N-terminal His tag followed by a tobacco etch virus (TEV) cleavage site for removal of the His tag during protein purification. The Ddlb-pET28b vector was transformed into BL21(DE3) Escherichia coli cells. For protein expression, a 10-ml starter culture was grown from a single colony overnight at 37°C. The starter culture was used to inoculate a 1-liter culture. LB medium supplemented with 30 μg/ml kanamycin was used in all cell growths. Cells were grown to an optical density of 0.5 and then induced with 0.5 mM isopropyl 1-thio-β-d-galactopyranoside (IPTG). Ddl was expressed at 16°C for 16 h. Cells were pelleted and stored at −80 to −120°C. Cells were resuspended in buffer A (20 mM Tris-HCl [pH 8.0], 500 mM NaCl, 10 mM imidazole [pH 8.0], and 2 mM β-mercaptoethanol [BME]) and subjected to two passes through a French press for lysis. Lysed cells were then clarified by centrifugation at 15,000 × g for 1 h. The supernatant was applied to a 5 ml HisTrap crude His column (GE Healthcare), washed, and eluted with a gradient of buffer B (20 mM Tris-HCl [pH 8.0], 500 mM NaCl, 500 mM imidazole [pH 8.0], and 2 mM BME). Ddl was then dialyzed in 2 liters of buffer A while being subjected to TEV proteolysis (at a ratio of 30:1 Ddl/TEV by weight) for removal of the His tag. The cleaved protein was passed over another HisTrap crude His column (GE Healthcare) equilibrated in buffer A to remove uncleaved His-Ddl and His-tagged TEV protease. Ddl was dialyzed to 20 mM Tris HCl (pH 8.0), 20 mM NaCl, and 2.0 mM dithiothreitol (DTT) for storage. Purified protein was concentrated in a Centriprep 15-ml centrifugal concentrator (Millipore) with a 30,000 molecular weight cutoff. The final protein concentration was approximately 10 mg/ml.
Crystallization.
Crystallization conditions were obtained by exploring several commercially available sparse matrix screens, with hits arising almost exclusively in the polyethylene glycol (PEG)/ion screen (Hampton Research). Crystals for data collection were produced by mixing either a 1-μl/1-μl or a 1-μl/1.5-μl ratio of well solution to protein solution, with the well solution (0.5 ml) consisting of 20% PEG 3350 and 0.2 M KNO3, using the vapor diffusion method in a hanging-drop 24-well Limbro plate at 20°C. Crystals grew to full size within 1 week (100 to 300 μm on each side) and were of a cubic shape.
Data collection, structure determination, and refinement.
Crystals were transferred to Paratone-N for cryoprotection and flash cooled to 100 K by submersion into liquid nitrogen. All data collection was carried out at 100 K at beamline 23ID at the Advanced Photon Source (Argonne National Laboratory, Argonne, IL). Data were indexed, integrated, and scaled with the HKL-2000 program in space group P21 to a resolution of 2.1 Å (33). The phase problem was solved by molecular replacement using the program Phaser (28). No homologues of Ddl (including E. coli, Staphylococcus aureus, and Thermus thermophilus) yielded suitable cross-rotation or translation function solutions. With the use of the program Chainsaw, a homology model was made using the S. aureus structure (Protein Data Bank [PDB] accession code 2i87) stripped of all water and ligands (25). A strong solution using the homology model was then obtained in Phaser. The Phaser molecular replacement solution was then refined in Phenix using simulated annealing at 5,000 K. The subsequent model was then subjected to multiple rounds of rebuilding in Coot followed by B-factor and positional refinement in Phenix and CCP4 until R factors converged in the low 20s (1, 20). Stereochemistry was checked by MolProbity, and the results are presented in Table 1, along with data processing and refinement statistics (12).
TABLE 1.
Data processing and refinement statistics
| Parameter | Value for apo Ddla |
|---|---|
| Data collection | |
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 51.7, 108.3, 69.1 |
| α, β, γ (°) | 90.0, 99.9, 90.0 |
| Resolution (Å) | 20.0 (2.10) |
| Rmerge | 0.052 (0.406) |
| I /σI | 24.6 (2.7) |
| Completeness (%) | 93.9 (84.9) |
| Redundancy | 3.6 (3.1) |
| Refinement | |
| Resolution (Å) | 20.0-2.1 |
| No. of reflections | 38,758 |
| Rwork/Rfree | 17.34/22.10 |
| No. of atoms | |
| Protein | 4,557 |
| Water | 383 |
| B factors (Å2) | |
| Protein | 44.7 |
| Water | 47.7 |
| Ramachandran statistics | |
| Most favored | 96.4% |
| Outliers | 0.3% |
| RMSDs | |
| Bond lengths (Å) | 0.004 |
| Bond angles (°) | 0.622 |
Numbers in parentheses represent the highest-resolution shell.
Fluorescence quenching affinity experiments.
The protein concentration was 3.0 μM for all experiments in a total starting volume of 0.5 ml. Experiments were carried out with 20 mM Tris-HCl (pH 8.0), 50 mM NaCl, 1 mM DTT. The DCS titrations were 0.5 μl each and ranged from 1.0 nM to 10.0 mM. d-Ala binding affinity was also determined in a similar manner, but assays were performed from a range of 1.0 nM to 5.0 mM. ATP- and ATP-γS-saturated Ddl fluorescence quenching was achieved by bringing the Ddl protein solution to 5.0 mM ATP and 1.0 mM MgCl2. Experiments using DCS were buffered with 20 mM Tris-HCl (pH 7.0) to ensure that DCS was present in a zwitterionic form shown to give maximum binding affinity with Ddl, as demonstrated by previous studies (30). Fluorescence was achieved by excitation at 295 nm, and the emission was measured at 338 nm. The data were fit using GraphPadPrism5 (GraphPad Software, Inc.), using nonlinear regression.
Isothermal titration calorimetry.
Because the inner filter effect of ATP could not be overcome using intrinsic fluorescence quenching, the affinity of Ddl for ATP was monitored using isothermal titration calorimetry (Microcal). Ddl was thermostated at 30°C in the cell at a concentration of 40 μM, ATP was injected stepwise from a stock concentration of 400 μM, and respective heat changes were measured. Due to the large heat of hydration of ATP, ATP was first injected into buffer and heat changes were subtracted from ATP injected to the protein prior to analysis. Data were analyzed and fit by using Origin 5 software (MicroCal).
Ddl enzyme assay.
Ddl endpoint assays by the thin-layer-chromatography (TLC)-based method were carried out by a modified procedure as described previously (15). Briefly, reaction mixtures were incubated at 37°C for various times in the presence of 50 mM Tris-HCl (pH 7.8), 10 mM MgCl2, 10 mM KCl, different concentrations of d-alanine and ATP, 2.5 mM glutathione, and 5.0 μCi [1-14C]d-alanine (100 μCi/ml; ICN Biochemicals, Inc., Costa Mesa, CA) for radiometric assays. The Ddl enzyme at a final concentration of 10 μg/ml was added to start the reactions. Assays were also run in the presence of inhibitors added at appropriate concentrations, with adjustment of the pH of the final solution to 7.8 as needed. For negative enzyme activity, controls were inactivated by addition of SDS to give a final concentration of 0.25%. Subsequently, 10 μl of the reaction mixture was applied to cellulose-backed TLC plates, and ascending chromatography was developed in n-butanol-acetic acid-water (12:3:5) until the solvent reached the top of the plate. For quantitative analysis, plates were directly dried at 100°C for 5 min and the radioactivity corresponding to the position of d-Ala and d-Ala:d-Ala spots was measured with a Bio-Rad Molecular Imager FX system (Bio-Rad). Alternatively, Ddl activity was assayed by measuring the release of inorganic phosphate (Pi), using the commercial continuous monitoring test for Pi release (EnzChek phosphate assay kit; Invitrogen).
Protein structure accession number.
The atomic coordinates of Ddl have been deposited in the Protein Data Bank under accession code 3LWB.
RESULTS
Ddl binding affinity for d-Ala, DCS, and ATP.
We performed assays to determine the binding affinities of DCS, d-Ala, and ATP as well as a kinetic assay to determine the inhibitory concentrations of DCS (Fig. 1; see also Fig. S1 and S2 in the supplemental material). Two binding sites for DCS and one binding site for ATP per monomer were assumed for the binding assays, as reported by previous studies (44, 47):
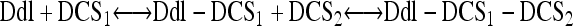 |
(1) |
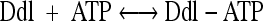 |
(2) |
Intrinsic tryptophan fluorescence quenching was used to determine the affinity of Ddl for d-Ala in the absence of ATP and the affinity of nucleotide-bound Ddl for d-Ala. Ddl binding events for d-Ala were a Kd1 of 95 nM and a, Kd2 of 1.2 mM, where Kd1 and Kd2 are the dissociation constants at d-Ala sites 1 and 2, respectively (see Fig. S1A in the supplemental material). Hence, ATP is not required for d-Ala binding, and ATP binding may not be the first event in the overall reaction. To further investigate the role of nucleotide binding in the affinity of Ddl for d-Ala, we measured the binding affinity of ATP-γS-saturated Ddl for d-Ala (Fig. S1B). The nucleotide-bound enzyme showed, a Kd1 of 62 nM and a Kd2 of 0.97 mM. Nucleotide binding is responsible for a decrease in the Kd1 value of 30 nM (i.e., corresponding to a 1.5-fold increase in affinity) and a decrease of about 0.2 mM (i.e., corresponding to a 1.2-fold increase in affinity) for the second binding event. Thus, d-Ala affinity is not largely dependent on nucleotide binding.
FIG. 1.

M. tuberculosis Ddl inhibition and binding assays. Inhibition assays were carried out with purified Ddl as described in Materials and Methods. Enzyme activity was assayed by either the radiometric method (A) or the continuous Pi assay (B to E). IC50s were defined as the inhibitor concentration that decreased the enzyme activity by 50% and were calculated using regression and graphic analyses, collecting data points (each point represents the mean ± the standard error of the mean [SEM]; n ≥ 3) obtained at specified inhibitor concentrations (A), spanning the range from 1 nM DCS to 5 mM DCS (B), or observed in the linear range of the DCS dose-response curves (C to E).
Intrinsic tryptophan and tyrosine fluorescence quenching was used to measure the binding affinity of Ddl in the absence of ATP for DCS, which gave a Kd1 of 79 nM and a Kd2 of 0.99 mM (see Fig. S1C in the supplemental material), an almost nonspecific binding event. Likewise, fluorescence quenching gave a Kd1 value of ATP-γS-saturated Ddl for DCS of 47 nM (1.7-fold increase in affinity); the value for Kd2 was found to be 0.53 mM (1.9-fold increase in affinity) (Fig. S1D). Thus, the effects of nucleotide binding are comparatively similar in both DCS sites. When d-Ala and DCS binding were compared, nucleotide binding was found to have a moderately greater effect at the low-affinity site, making DCS a slightly better inhibitor for the second binding event. To determine the affinity of apo Ddl for ATP, isothermal titration calorimetry was carried out (Fig. S2). The affinity of apo M. tuberculosis Ddl for ATP yielded a Kd of 14 μΜ. The data were fit with one binding event and showed no cooperativity across the dimer interface. The affinity of ATP for M. tuberculosis Ddl is similar to the values for similar Ddl enzymes from many other species, which have affinities of 38 to 60 μM (25, 47).
DCS inhibition of Ddl.
To determine how the enzyme activity is inhibited by DCS, inhibition parameter values (50% inhibitory concentrations [IC50s]) were determined by kinetic assays (Fig. 1A). An endpoint assay that monitors the progress of the Ddl reaction by the formation of d-Ala:d-Ala as [14C]d-Ala was provided as a substrate (15) was used first. Likewise, the nonhydrolyzable substrate analog β,γ-methylene-adenosine displayed approximately 60% inhibition as added in equimolar amounts to ATP (i.e., both at 6.0 mM in the assay mixture). Thus, inhibitors of both the d-Ala and the ATP binding sites inhibited the Ddl reaction.
To obtain an IC50 for DCS inhibition, we used a continuous assay that monitors the release of Pi as the result of ATP hydrolysis in the presence of d-Ala (1.0 or 20 mM), ATP (0.5 or 6.0 mM), and various concentrations of DCS (Fig. 1B to E) to determine an approximate inhibition value of 0.37 mM (Fig. 1B). This value is similar to the IC50 for E. coli DdlB at 0.32 mM (21). The value of the second binding event for DCS is similar to that of the M. tuberculosis IC50 and published IC50s for the E. coli Ddl (21, 45).
In general, IC50s are dependent upon the concentration(s) of the substrate(s) {i.e., IC50 = Ki [1 + (substrate concentration/Km)] for simple competitive inhibition}. Thus, we determined the effect of varying d-Ala and ATP concentrations on Ddl inhibition by DCS. Raising the d-Ala concentration 20-fold (to 20 mM) while maintaining the ATP concentration at 6.0 mM resulted in competition, increasing the IC50 to 1.0 mM (Fig. 1C). However, this value is less than the 10-fold effect expected under the assumption of simple competitive inhibition (i.e., with a Ki approximately equal to the Kd2 for DCS and a Km approximately equal to the Kd2 for d-Ala) (38). Thus, the low-affinity site may significantly contribute to DCS inhibition. Lowering the ATP concentration approximately 10-fold (to 0.5 mM) while maintaining d-Ala at 1.0 mM results in an increase of the IC50 for DCS to 0.8 mM (Fig. 1D). This is consistent with the affinity data, but the effect at the kinetic level seems more pronounced, reminiscent of potential heterotropic ligand binding site interactions. The effect of ATP on the IC50 for DCS is also observed at higher ATP concentrations (0.5 to 6.0 mM) than those predicted by the affinity data. Thus, the kinetically relevant binding of ATP may occur at higher ATP concentrations. When both the d-Ala and the ATP concentrations were varied together (to final values of 20 and 0.5 mM, respectively), the IC50 increased to 2.5 mM (Fig. 1E), indicating that DCS, under these conditions, cannot significantly inhibit Ddl. Moreover, as observed at 6.0 mM ATP, the effect of a 20-fold increase in the concentration of d-Ala had a minor effect on the IC50, less than expected from classical competitive inhibition. The binding and kinetic parameter values are summarized in Table 2.
TABLE 2.
Kinetic, binding, and inhibition parameter values for the M. tuberculosis Ddl enzyme
| Parameter (ligand) | Value (mM) | Conditionsa |
|---|---|---|
| Kd1 (d-Ala) | 9.49 × 10−6 | E |
| Kd1 (d-Ala) | 6.15 × 10−6 | E + (5 mM) ATP-γS |
| Kd2 (d-Ala) | 1.17 | E |
| Kd2 (d-Ala) | 9.70 × 10−1 | E + (5 mM) ATP-γS |
| Kd1 (DCS) | 7.86 × 10−6 | E |
| Kd1 (DCS) | 4.71 × 10−6 | E + (5 mM) ATP-γS |
| Kd2 (DCS) | 9.90 × 10−1 | E |
| Kd2 (DCS) | 5.30 × 10−1 | E + (5 mM) ATP-γS |
| Kd (ATP) | 1.43 × 10−2 | E |
| IC50 (DCS) | 3.70 × 10−1 | E + 1.0 mM d-Ala + 6.0 mM ATP |
| IC50 (DCS) | 8.0 × 10−1 | E + 1.0 mM d-Ala + 0.5 mM ATP |
| IC50 (DCS) | 1.00 | E + 20.0 mM d-Ala + 6.0 mM ATP |
| IC50 (DCS) | 2.50 | E + 20 mM d-Ala + 0.5 mM ATP |
E (Ddl enzyme) was used at 3 μM for all fluorescence binding assays, 40 μM for the isothermal titration calorimetry, and 0.25 μM (10 μg/ml) in the kinetics assays as described in Materials and Methods.
Structure determination and overall structure.
Ddl was crystallized in the space group P21, with one dimer in the asymmetric unit. The structure was solved by molecular replacement using diffraction data to a resolution of 2.1 Å. Crystallographic data processing and refinement statistics are shown in Table 1. Both subunits of the final model show high structural similarity, with a core root mean square deviation (RMSD) of 1.1 Å2.
Previous crystal structures of several species of Ddl have revealed that it is a homodimer (13, 14, 25). The nucleotide binding site and two d-Ala binding sites form a large cavity spanning the centrally located boundaries of all three domains in the protein. Conserved glutamate, glycine, and arginine residues have been shown to comprise the first d-Ala binding site, positioning d-Ala for nucleophilic attack on the γ-phosphate of ATP, which forms a covalent phosphate-d-Ala intermediate. The second d-Ala molecule is held in place by hydrogen bonds from conserved serine and leucine residues as well as a conserved tyrosine residue located on the ω loop. The binding sites position the d-Ala molecules in close proximity to each other, allowing for covalent linkage.
Each Ddl monomer can be subdivided into three basic subdomains: an N-terminal domain (NTD) consisting of residues from the N terminus extending to residue 140 comprising β-sheet regions 1 to 5 and α-helices 1 to 3, a central domain (MD) consisting of residues 141 to 232 encompassing α-helices 4 to 7 and β-sheet regions 6 to 10, and a C-terminal domain (CTD) consisting of residues 233 to 373 and encompassing α-helices 8 to 11 and β-sheet regions 11 to 17 (Fig. 2A). The ATP catalytic cavity is centrally located within the protein and is conjoined by the NTD, MD, and CTD and formed by H7, B13, and H9, which comprise a loop region consisting of residues 196 to 204, and a loop region consisting of amino acids 23 to 26 (Fig. 2B; see also Fig. S3 in the supplemental material). The secondary and tertiary folds of the protein show high similarity to those observed for other species, providing evidence that the protein has been well conserved evolutionarily among a diverse range of species, including Helicobacter pylori, S. aureus, T. thermophilus, and E. coli. Percents identity range from 26% identity for H. pylori to 41 to 42% identity for both Thermus caldophilus and T. thermophilus (see Table S1 in the supplemental material). In spite of moderate percent identity, the RMSDs for these structures range from 1.5 Å2 for S. aureus to 2.4 Å2 for H. pylori; clearly, the M. tuberculosis structure is most similar to the S. aureus structure (25). A superimposition of the M. tuberculosis apo monomer with Ddl structures from other species can be observed in Fig. S3A in the supplemental material.
FIG. 2.

Overall structure of M. tuberculosis Ddl and domain definition. (A) Ribbon diagram of the Ddl monomer, with helices (H) as well as β-sheet regions (B) defined. Domains are colored blue (CTD), green (MD), and purple (NTD). (B) Transparent space-filling representation of the Ddl dimer, with a ribbon diagram shown beneath the surface. ADP and d-Ala:d-Ala dipeptide are shown as spheres colored by atom type, which were docked in manually, using the ligand-bound form of E. coli Ddlb (PDB accession code 1IOV) as a guide (14). Monomers are denoted by different colors (green and purple).
The NTD (residues 1 to 140) consists of 4 β-strand regions sandwiched between 2 α-helices which are conjoined by several loop regions. The NTD also forms the upper portion of the ligand cavity, which makes contacts with both d-Ala molecules in the active site. Among all three regions of Ddl, the NTD shows the most diversity in large-scale changes, with RMSDs of 1.8 and 1.9 Å2 compared to the levels for E. coli and S. aureus. The M. tuberculosis NTD structure for residues 64 to 93 could not be modeled, due to disorder. The S. aureus and M. tuberculosis structures have an insertion in this region not found in the E. coli structure. This region in S. aureus is a loop that joins the most solvent accessible portion of the β-sheet region to the adjoining helix. Superimposition of the S. aureus and M. tuberculosis structures would also place the missing residues in this region. Another difference between the structures involves the loop region corresponding to residues 118 to 123 and the loop region corresponding to residues 18 to 25. All Ddl structures solved to date have the loop region corresponding to M. tuberculosis residues 18 to 25 protruding into the active site near the first d-Ala site. In the M. tuberculosis structure this has been partially displaced by the loop region corresponding to residues 118 to 123. The loop region corresponding to residues 18 to 25 is positioned outwards toward the solution, contributing to disorder in the electron density profile (Fig. 3).
FIG. 3.

The Ddl active site requires conformational changes. Shown is a superimposition of the ligand-bound form of E. coli Ddlb (PDB accession code 1IOV) with apo M. tuberculosis Ddl. Residues shown as sticks are residues predicted to be involved in the d-Ala:d-Ala ligation reaction (14). Red arrows indicate structural rearrangements that would need to occur for apo M. tuberculosis Ddl to form a ligand-bound conformation such as the E. coli conformation depicted.
The MD (residues 141 to 232) consists of a four-stranded antiparallel β-sheet region which separates a pair of α-helices at each side. The MD flanks the nucleotide binding site, forming half of the sandwich which holds the nucleotide in place along with the CTD. The nucleotide rests flush against the β-sheet region of the MD (see Fig. S3B and C in the supplemental material). The loop region corresponding to M. tuberculosis residues 196 to 203 is inserted into the active site, making contact with the nucleotide phosphate region. The MD domain shows very few global differences with other Ddl structures, with RMSDs of 1.1 and 1.2 Å2, respectively. The region with the most striking difference in the MD is the random coil region corresponding to residues 196 to 204, which is displaced approximately 4.5 Å inward toward the center of the protein away from the bulk solvent region compared to the levels for other species.
The CTD (residues 233 to 373) is formed by a core of six antiparallel β strands and by the ω loop, which extends into the ligand binding pocket and is known to be important in catalysis. In addition, the β-sheet region is capped by 3 α-helices, one of which, H10, forms the interface with the NTD. The core of the CTD is globally well conserved among species, with M. tuberculosis having core RMSDs of 1.6 Å2 compared to the levels for E. coli and S. aureus. The most striking area of divergence in the CTD in M. tuberculosis compared to the levels for other species is localized to the ω loop (Fig. 3). Prior to our M. tuberculosis structure, apo structures were available for the species H. pylori, S. aureus, and T. thermophilus. The M. tuberculosis apo structure has the ω loop pulled into the ATP binding site partially overlapping this site. No other apo Ddl structure has the ω loop in this type of conformation. To accommodate the nucleotide, this region must flex outwards toward the solution. In contrast, structures from H. pylori and T. thermophilus have the ω loop in a conformation that forms an antiparallel β-sheet capped by a loop which extends 180 degrees in the opposite direction of the ATP binding site directly into the bulk solvent. The apo structure from S. aureus does not contain the ω loop, as the residues were too disordered in the electron density to be modeled, implying they were highly mobile. The flexibility of this region has also been confirmed previously for DdlA and DdlB from E. coli and the VanA vancomycin-resistant determinant of Enterococcus faecium by limited proteolysis (44). Therefore, apo M. tuberculosis Ddl has a conformation distinct from that of other apo structures in that Ddl directly blocks the ATP binding site. In other Ddl structures from other bacteria, the ATP binding site is in an open conformation where the ω loop is either highly mobile or locked in a position protruding into the solvent region.
The dimer interface is disrupted by an M. tuberculosis-specific insertion.
The M. tuberculosis dimer interface is almost completely defined by contacts of the α helices between subunits extending from the MD domain (Fig. 4A). Three α helices are predominantly involved in the dimer interface interactions: H4, H5, and H3. Subunit-subunit α-helical packing is such that H3 of one monomer packs against H3 of its dimer partner and H4 of one monomer packs against H4 of its dimer partner; both α helices pack against their partners, with an offset angle of approximately 180 degrees (Fig. 4A and B). In contrast to the Ddl structures of other species which report primarily hydrophobic interactions at the dimer interface (13, 23), M. tuberculosis Ddl includes a variety of interactions, including hydrophobic, electrostatic, and van der Waals interactions. Interactions of the dimer interface can be divided into 3 basic groups: hydrophobic and van der Waals interactions of the H4/H5-H4/H5 interface (Fig. 4A and B), hydrophobic and van der Waals interactions of the H3/H3 interface (Fig. 4A, B, and C), and a variety of electrostatic interactions (Fig. 4D). H4 and H5 of one monomer pack against H4 and H5 of the dimer partner to create a hydrophobic environment in which residues Ala147, Leu158, Phe154, and Val143 of both monomers are all involved in hydrophobic interactions (Fig. 4B). In addition, H3 of one monomer packs against H3 of its dimer partner, with a hydrophobic interaction occurring between the side chains of Leu130 of both monomers (Fig. 4C). Lastly, two electrostatic interactions tie the monomers together as seen in Fig. 4D, the Lys157 side chain of H5 forms an electrostatic interaction with the side chain of Asp162 of H5 on the dimer partner, and the side chain of Asp310 at the base of H9 forms an electrostatic interaction with the side chain of Arg173 (Fig. 4D). Electrostatic interactions of this nature are not observed in the dimer interfaces of Ddl molecules from other species.
FIG. 4.

Ddl dimer interface. (A) Helix packing at the dimer interface. H5, H9, and H4 all pack against their dimer partners. H4 packs against the dimer partner H4 at an offset of almost 180o, and H5 packs against the dimer partner H5 at a similar offset. (B) Hydrophobic cleft of the dimer interface. Residues Leu158, Phe154, Ala147, Leu144, and Val143 all form hydrophobic interactions at the dimer interface. (C) Hydrophobic packing of H3/H3 via residue Leu130. (D) Electrostatic interactions at the dimer interface. Arg173 forms an electrostatic interaction with Asp310, and Lys157 forms an electrostatic interaction with Asp162. (E) Superimposition of M. tuberculosis Ddl (monomer A in green ribbons and monomer B in gray ribbons) with S. aureus Ddl (monomer A in yellow ribbons and monomer B in magenta ribbons) (PDB accession code 2I87), with the M. tuberculosis-specific insertion shown in purple (25). The M. tuberculosis-specific insertion allows for shifts in helices 2 and 3 away from the dimer interface compared to the levels for the S. aureus structure.
A comparison of multiple Ddl sequences reveals that the M. tuberculosis structure contains an insertion in the amino acid sequence not found in most other species, as indicated in the multiple sequence alignment displayed in Fig. S4 in the supplemental material. This insertion consists of residues 95 to 107 compared to the S. aureus sequence and residues 81 to 107 compared to the E. coli sequence. This region forms a small helix surrounded by a coil tertiary structure just upstream of helix 2. This coil region is largely hydrophobic, creating steric hindrance which prevents helix 2 from packing directly against helix 3 as seen in other species (Fig. 4E). Helix 2 is found in the M. tuberculosis structure packed against the β-sheet cluster, allowing the coiled insertion to pack against helix 3, forming largely hydrophobic interactions. This alternative conformation allows helix 3 to flex nearly 5.0 Å away from the dimer interface compared to the dimer interfaces of other species, specifically at the helix 3-helix 3 dimer interface interactions. The altered packing induced by the hydrophobic coil has an impact not only on the dimer interface but on the juxtaposed β-sheet region and the loops gating the active site. The β-sheet region composed of β strands 1 to 4 is shifted as far as 2.0 Å in certain regions compared to the levels for other species. Most intriguing, the loop region upstream of helix 3 overlaps, by the altered dimer interface, with the d-Ala binding sites displacing the loop bearing the catalytic Glu23.
M. tuberculosis Ddl active site.
Despite exhaustive cocrystallization and soaking attempts, we were unable to obtain ligand-bound structures for ADP, ATP, or DCS. Thus, we manually docked ADP into the active site of our apo structure using superimposed structures from E. coli (PDB accession no. 1iow) and S. aureus (PDB accession no. 2i8c) (13, 25). Many, if not most, of the residues spanning the active site are well conserved among Ddl proteins. The purine ring is sandwiched between Phe192 and Phe320, making stacking interactions with both residues (see Fig. S3C in the supplemental material). In addition, three nitrogen atoms of the purine ring participate in hydrogen bonds: N6 is a donor to the oxygen of the side chain of Glu230 as well as the main chain oxygen Ala231, N1 hydrogen bonds with the backbone carbonyl of Ile233, and N7 hydrogen bonds with the Lys194 side chain nitrogen (Fig. S3C). Only one oxygen atom from the ribose sugar participates in protein interactions; the O2′ oxygen atom hydrogen bonds with Glu237 (Fig. S3C). Both phosphate moieties are involved in electrostatic interactions (Fig. S3C and D). The α-phosphate forms two electrostatic interactions, one with the side chain nitrogen of Lys152 and the other with the side chain of Asn329. The β-phosphate forms only one hydrogen bond with the side chain oxygen of Ser200.
Inspection of the binding site in comparison to the E. coli d-Ala:d-Ala dipeptide intermediate mimic allowed us to define the portion of the active site encompassing both d-Ala binding sites (Fig. 3 and 5A) (14). The first d-Ala binding site, the N-terminal site, is defined by three highly conserved residues: His118, Gly336, and Arg316. The carbonyl group of d-Ala is positioned to hydrogen bond with the backbone nitrogen of Gly336 as well as the NH2 moiety of the Arg316 side chain. The M. tuberculosis Arg residue is very well conserved, with RMSDs over all atoms of 1.0 and 0.9 Å for S. aureus and E. coli, respectively. The glycine residue of the d-Ala1 site is moderately conserved, with RMSDs over all atoms of 1.5 and 2.5 for M. tuberculosis compared to the levels for S. aureus and E. coli, respectively. All structural reports among species thus far have identified an N-terminal glutamic acid as a key residue of the N-terminal d-Ala site, with the side chain acting as a hydrogen bond donor to the d-Ala amino group. Interestingly, in the M. tuberculosis structure this residue, Glu23, is part of a disordered loop that, based on the location of the base of each end of the loop, we infer extends away from the globular region of the protein. The active-site region, filled by this catalytic glutamate residue in other species, is instead occupied by Glu123 in the M. tuberculosis structure (Fig. 3). This is made possible by the complete repositioning of this region of the active site compared to homologous structures. The loop region between B4 and H3 fills this side of the active site, allowing Glu123 to be positioned next to d-Ala, whereas in homologous structures, this side of the active site is formed by the loop adjoining H2 and B1. In addition, the conserved histidine (His118), shown to pack against the first d-Ala molecule in other species, is also found on a loop that would require significant remodeling to pack against the d-Ala1 site (Fig. 3). The second C-terminal d-Ala2 site is also defined by highly conserved residues among species and includes residues Tyr277, Ser341, and Met342. The apo structure shows that the end of H10 and the beginning of H11, bearing the ω loop and conserved Tyr277, require a shift away from the ligand cavity of approximately 5 to 7 Å to make contact with d-Ala2 (Fig. 3). This outward shift also allows for the ATP site to open and be freed from the occupying Ddl residues. Upon the helix shift, the hydroxyl group of Tyr277 is in place to be a hydrogen bond acceptor to the d-Ala amino group. While this residue is absent due to disorder in the S. aureus structure, the E. coli structure has an RMSD of 6.2 Å2 compared to the level for the M. tuberculosis structure. In addition, H10 is positioned such that the Ser341 hydroxyl and Met342 main chain nitrogen both require a small shift to be in position to hydrogen bond with the d-Ala carboxyl group (Fig. 3).
FIG. 5.

M. tuberculosis apo Ddl active-site cavity and adjoining clefts. Apo Ddl is shown as a surface representation (purple). Manually docked ADP (colored yellow by atom) and d-Ala:d-Ala (red) are shown as stick representations, using the ligand-bound form of E. coli Ddlb (PDB accession code 1IOV) as a guide (14). The ω loop was removed from all representations to allow for a clear view of the binding pocket. (A) Overview of the apo M. tuberculosis Ddl pocket. (B) A cleft behind the d-Ala binding site extends back and away from the binding pocket. (C) A shallow cleft lies to the right of the ATP sugar group oxygen atoms. (D) A deep cleft lies below the nucleotide binding site.
M. tuberculosis-specific active-site clefts.
Three clefts branching from the active site of apo M. tuberculosis Ddl are not present in other species or are present with larger and more-encompassing volumes. The first cleft is located proximally to the d-Ala binding site, making the design of inhibitors which span from the first d-Ala site as well as into this cleft an attractive possibility (Fig. 5B). The cleft is homologous to a cleft previously reported for the S. aureus structures but absent in all other species to date. This cleft in M. tuberculosis is deeper than the homologous S. aureus cleft extending directly back from the d-Ala binding site nearly 13 Å in length. The opening of the cleft is nearly 7 Å wide and is defined by residues His118, Ser30, Ser341, and Met334. While the opening of the S. aureus cleft shares conserved residues on a primary sequence level and Ser30, Ser341, and Met334 show little variation between the two species spatially, the loop bearing His128 is shifted approximately 6 Å away from the active site in M. tuberculosis compared to the level for S. aureus. This shift in M. tuberculosis compared to the level for S. aureus directly away from the active site not only creates a different environment at the cleft opening but also creates a unique environment along the wall of the cleft. The second cleft of interest, found on the M. tuberculosis active site, is unique to M. tuberculosis compared to other structures (Fig. 5C). The cleft is relatively shallow, 3.5 Å deep, and the opening is formed by residues Asp318, Glu239, Glu237, and Val327, which form a 5-Å-diameter opening. The location of this cleft next to the ATP sugar ring makes this cleft the most suitable site for rational inhibitor design. The cleft lies near the O3′ oxygen atom of the ribose ring of the ATP molecule, making it an appealing target for ATP analogs, with moieties extending from the oxygen atoms of the ribose sugar. While the cleft is shallow, its location proximal to the proposed ATP site would allow for substitution of ATP analogs quite readily, given that the diameter and depth could accommodate a wide variety of different chemical moieties. The last notable cleft of the M. tuberculosis structure is located lining the opposite side of the ATP molecule as the d-Ala sites and the previously mentioned cleft (Fig. 5D). This cleft is also seen in most of the other Ddl structures to date, but not as deep or wide as in the M. tuberculosis apo structure. The cleft opening is approximately 8 Å wide and 12 Å long, formed by residues Asp318, Glu239, Glu237, and Val327, and extends about 12 Å away from the ligand binding pocket. The opening of the cleft is located close to the proposed sites of the ATP phosphate moieties, making this cleft an engaging target for rational inhibitor design of moieties designed from scaffolds based on ATP analogs as well as d-Ala:d-Ala intermediate analogs. Additionally, given that many of the clefts are lined with charged residues, this allows for the introduction of hydrogen bonding networks to improve the affinity of d-Ala and ATP mimics.
DISCUSSION
The genes for d-alanine racemase and Ddl are present as a single copy in all sequenced mycobacterial genomes (http://www.ncbi.nlm.nih.gov/genomes). These enzymes catalyze sequential steps in the d-alanine pathway of peptidoglycan biosynthesis (43). Both enzymes are inhibited by DCS in a concentration-dependent manner (4, 11, 15). In M. smegmatis, d-alanine racemase insertion mutants are viable but hypersusceptible to DCS (5, 6, 18), suggesting that Alr may protect a more fundamental lethal target of lower affinity, such as, for example, Ddl (15, 18). Nonetheless, other studies with M. smegmatis alr deletion mutants using different experimental conditions did not detect viable cells in the absence of d-alanine (29). Ddl essentiality in M. smegmatis is also substantiated by genetic experiments, including one involving the isolation of a mutant whose thermosensitive phenotype was complemented to thermoresistance by supplying a wild-type ddl gene (3). The d-alanine racemase and Ddl genes have both been identified as essential genes in M. tuberculosis by two methods of genome-wide analysis based on transposon mutagenesis at either saturation or subsaturation levels (22, 36). Indeed, the lethal effect of DCS on M. tuberculosis may be due to the inhibition of both Ddl and d-alanine racemase, whose crystal structure has been solved (24). The essentiality of Ddl in M. tuberculosis is strengthened by the involvement of Ddl in the second step of d-Ala:d-Ala biosynthesis. Microorganisms such as Listeria monocytogenes possess alternative pathways for synthesizing d-alanine without d-alanine-racemase (41). However, Ddl or a highly homologous variant of Ddl (whose presence would be easily detectable in the M. tuberculosis genome sequence, as indicated by the high homology among d-Ala ligases from widely divergent species [http://www.ncbi.nlm.nih.gov/sutils/blink.cgi?mode=query]) is required for the synthesis of d-Ala:d-Ala or another dipeptide (e.g., d-Ala-X) that can be used instead. Ddl function is also essential in microorganisms with two Ddl enzymes, as demonstrated by the thermosensitive E. coli mutant ST640, with thermosensitive and null mutations in the corresponding ddl genes (26, 47). Thus, there are no known alternative pathways that could bypass the requirement for a dipeptide containing d-Ala. Nonetheless, there are no studies that provide a formal proof of d-alanine racemase or Ddl essentialities in M. tuberculosis, and further studies are needed to define the lethal target(s) of DCS action.
Association/dissociation equilibrium constants have not been reported for Ddl of any species; however, many Km values have (47). Our binding data are consistent with previous kinetic studies from other species which show both high-affinity and low-affinity events. Here, we demonstrate by binding data that apo Ddl has high affinity for d-Ala in the absence of nucleotide and that the addition of nucleotide confers little added affinity for d-Ala. Likewise, the binding assays suggest direct binding of DCS to apo and nucleotide-bound forms of Ddl. DCS also exhibits high- and low-affinity binding events. The value for the low-affinity constant more closely resembles the IC50s of the inhibitor in a kinetic assay. Furthermore, the MIC values of DCS for M. tuberculosis are also consistent with the IC50 for Ddl inhibition by DCS. Moreover, if a mechanism of competitive inhibition is involved, these data indicate that DCS has an affinity just slightly greater than that of d-Ala at both d-Ala sites, but with a greater difference at the low-affinity site. In this situation, it is important to underscore that IC50s are dependent not only on the absolute affinity of the inhibitor but also on the relative affinities of the inhibitor and the substrate, the substrate concentration, and the assay system (7). Nonetheless, our IC50 data, binding data, and previous in vivo data seem to rule out the hypothesis that the high-affinity site alone is sufficient for inhibition. However, the Ddl reaction is more complex than simple competitive inhibition, as the overall reaction follows a ter-ter mechanism (8-10) with two substrates, one with two binding sites. Thus, a full kinetic analysis, now in progress, is needed to further substantiate these arguments. Thus, the M. tuberculosis Ddl enzyme has high affinity for both d-Ala and DCS at both sites, with a greater difference at site 2 (acceptor) than at site 1 (donor).
The crystal structure of M. tuberculosis Ddl indicates that, in contrast to what is observed for other species, the apo protein exists in a closed conformation and contains many cavities adjacent to the active site. Because many unique pockets exist in the M. tuberculosis Ddl active site, we propose that this allows for guided design of M. tuberculosis-specific inhibitors. The M. tuberculosis Ddl ligand binding pocket appears to be flexible in nature, requiring several structural rearrangements to be in position to bind ATP and both d-Ala molecules (indicated by arrows in Fig. 3). The two conformations may play a role in the complex kinetics of the ter-ter mechanism described above. Moreover, the M. tuberculosis Ddl structure is markedly different from previously solved structures from other species in that the M. tuberculosis apo form must undergo significant rearrangement for ligand binding and catalysis. While other species, such as S. aureus, do undergo small rearrangements, these rearrangements are not as profound as those that we propose for the M. tuberculosis model, as seen in Fig. S3B in the supplemental material and Fig. 3. The flexibility of the ligand binding pocket of M. tuberculosis Ddl is made possible because most of the conserved catalytic residues lie on loop regions which cap the catalytic site. Because ATP is unlikely to bind apo M. tuberculosis Ddl in its current conformation, due to clashing, and drawing from comparisons to ligand-bound forms of Ddl from other species, we propose that there are two distinct conformations of M. tuberculosis Ddl. The more rigid and closed ligand-bound conformation will be referred to as the closed conformation, and the more flexible conformation we shall denote the open conformation. The transition from the closed to the open conformation requires three distinct events: (i) the outward movement of the ω loop from the ligand binding cleft toward the solution, (ii) the displacement of the loop bearing Glu123 with the loop bearing Glu23, and (iii) an inward movement toward the active site of the loop containing the catalytic residues, including Gly336, as well as the loop bearing the conserved residue Ser201. The movement of the ω loop not only allows for the space required for nucleotide binding but also places the conserved catalytic residue Tyr277 into the d-Ala2 active site so that this residue is capable of hydrogen bonding with d-Ala2 via the d-Ala molecule's amino group. The displacement of the loop bearing Glu123 with that bearing Glu23 is an event that places the conserved Glu23 and His118 residues from their position distant from the active site to a position at the d-Ala1 site, allowing Glu23 to hydrogen bond with the amino group of d-Ala1. The movement of the loop containing Ser201 inward would allow for a hydrogen bond with Glu23 of approximately 2.8 Å, as seen in Ddl structures from other species, and van der Waals packing of the Tyr277side chain nearly 4 Å from the Ser side chain, allowing for a more rigid conformation. Finally, in the open conformation, Tyr277 is positioned such that it forces the loop containing the conserved catalytic residues Met342, Ser341, and Gly336 in an outward position; once the ω loop is positioned more outwardly, Tyr277 is repositioned, and the loop containing Gly336, Ser341, and Met342 move inward toward the d-Ala binding sites, allowing for catalysis.
In summary, known Ddl inhibitors, such as DCS, directly compete with one or both of the d-Ala binding sites or are allosteric inhibitors which overlap with the d-Ala active sites. Because in the M. tuberculosis Ddl structure the M. tuberculosis-specific cavities overlap with the active site, we propose that this allows for the design of specific inhibitors, but additional modeling and experimentation are needed to determine the suitability of these sites for targeting. Nonetheless, we predict that specific Ddl inhibitors, especially the allosteric kind, will be bactericidal agents in their own right that could be combined with specific inhibitors of other essential functions of M. tuberculosis. In summary, the crystal structure of the M. tuberculosis Ddl enzyme and the kinetic and binding studies presented can be used as a guide for targeting new inhibitors by rational drug design.
Supplementary Material
Acknowledgments
GM/CA CAT has been funded in whole or in part with federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Science (Y1-GM-1104). Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under contract no. DE-AC02-06CH11357. This work was supported by National Institutes of Health grant P0168135 (J.C.S.), funds from the Robert A. Welch Foundation (grant no. A-0015) (J.C.S.), National Institutes of Health grant R03AI051176 (R.G.B.), USDA Cooperative State Service Project NEB 14-141 (R.G.B.), and the School of Veterinary Medicine and Biomedical Sciences (R.G.B.).
We thank Robert J. Fenton and Harshdeep Dogra for technical assistance at the University of Nebraska.
Footnotes
Published ahead of print on 18 October 2010.
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1.Adams, P. D., R. W. Grosse-Kunstleve, L. W. Hung, T. R. Ioerger, A. J. McCoy, N. W. Moriarty, R. J. Read, J. C. Sacchettini, N. K. Sauter, and T. C. Terwilliger. 2002. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58:1948-1954. [DOI] [PubMed] [Google Scholar]
- 2.Alderwick, L. J., H. L. Birch, A. K. Mishra, L. Eggeling, and G. S. Besra. 2007. Structure, function and biosynthesis of the Mycobacterium tuberculosis cell wall: arabinogalactan and lipoarabinomannan assembly with a view to discovering new drug targets. Biochem. Soc. Trans. 35:1325-1328. [DOI] [PubMed] [Google Scholar]
- 3.Belanger, A. E., J. C. Porter, and G. F. Hatfull. 2000. Genetic analysis of peptidoglycan biosynthesis in mycobacteria: characterization of a ddlA mutant of Mycobacterium smegmatis. J. Bacteriol. 182:6854-6856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caceres, N. E., N. B. Harris, J. F. Wellehan, Z. Feng, V. Kapur, and R. G. Barletta. 1997. Overexpression of the d-alanine racemase gene confers resistance to d-cycloserine in Mycobacterium smegmatis. J. Bacteriol. 179:5046-5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chacon, O., L. E. Bermudez, D. K. Zinniel, H. K. Chahal, R. J. Fenton, Z. Feng, K. Hanford, L. G. Adams, and R. G. Barletta. 2009. Impairment of d-alanine biosynthesis in Mycobacterium smegmatis determines decreased intracellular survival in human macrophages. Microbiology 155:1440-1450. [DOI] [PubMed] [Google Scholar]
- 6.Chacon, O., Z. Feng, N. B. Harris, N. E. Caceres, L. G. Adams, and R. G. Barletta. 2002. Mycobacterium smegmatis d-alanine racemase mutants are not dependent on d-alanine for growth. Antimicrob. Agents Chemother. 46:47-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng, Y., and W. H. Prusoff. 1973. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22:3099-3108. [DOI] [PubMed] [Google Scholar]
- 8.Cleland, W. W. 1963. The kinetics of enzyme-catalyzed reactions with two or more substrates or products. I. Nomenclature and rate equations. Biochim. Biophys. Acta 67:104-137. [DOI] [PubMed] [Google Scholar]
- 9.Cleland, W. W. 1963. The kinetics of enzyme-catalyzed reactions with two or more substrates or products. II. Inhibition: nomenclature and theory. Biochim. Biophys. Acta 67:173-187. [DOI] [PubMed] [Google Scholar]
- 10.Cleland, W. W. 1963. The kinetics of enzyme-catalyzed reactions with two or more substrates or products. III. Prediction of initial velocity and inhibition patterns by inspection. Biochim. Biophys. Acta 67:188-196. [DOI] [PubMed] [Google Scholar]
- 11.David, H. L., K. Takayama, and D. S. Goldman. 1969. Susceptibility of mycobacterial d-alanyl-d-alanine synthetase to d-cycloserine. Am. Rev. Respir. Dis. 100:579-581. [DOI] [PubMed] [Google Scholar]
- 12.Davis, I. W., A. Leaver-Fay, V. B. Chen, J. N. Block, G. J. Kapral, X. Wang, L. W. Murray, W. B. Arendall III, J. Snoeyink, J. S. Richardson, and D. C. Richardson. 2007. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35:W375-W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan, C., P. C. Moews, C. T. Walsh, and J. R. Knox. 1994. Vancomycin resistance: structure of d-alanine:d-alanine ligase at 2.3 A resolution. Science 266:439-443. [DOI] [PubMed] [Google Scholar]
- 14.Fan, C., I. S. Park, C. T. Walsh, and J. R. Knox. 1997. d-alanine:d-alanine ligase: phosphonate and phosphinate intermediates with wild type and the Y216F mutant. Biochemistry 36:2531-2538. [DOI] [PubMed] [Google Scholar]
- 15.Feng, Z., and R. G. Barletta. 2003. Roles of Mycobacterium smegmatis d-alanine:d-alanine ligase and d-alanine racemase in the mechanisms of action of and resistance to the peptidoglycan inhibitor d-cycloserine. Antimicrob. Agents Chemother. 47:283-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Future Medicine, Ltd. 2008. WHO report on extent of drug resistance in global TB infections. Future Microbiol. 3:125-127. [Google Scholar]
- 17.Ghuysen, J. M. 1968. Use of bacteriolytic enzymes in determination of wall structure and their role in cell metabolism. Bacteriol. Rev. 32:425-464. [PMC free article] [PubMed] [Google Scholar]
- 18.Halouska, S., O. Chacon, R. J. Fenton, D. K. Zinniel, R. G. Barletta, and R. Powers. 2007. Use of NMR metabolomics to analyze the targets of d-cycloserine in mycobacteria: role of d-alanine racemase. J. Proteome Res. 6:4608-4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoeprich, P. D. 1965. Alanine: cycloserine antagonism. VI. Demonstration of d-alanine in the serum of guinea pigs and mice. J. Biol. Chem. 240:1654-1660. [PubMed] [Google Scholar]
- 20.John Wiley & Sons, Inc. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50:760-763. [DOI] [PubMed] [Google Scholar]
- 21.Kovac, A., V. Majce, R. Lenarsic, S. Bombek, J. M. Bostock, I. Chopra, S. Polanc, and S. Gobec. 2007. Diazenedicarboxamides as inhibitors of d-alanine-d-alanine ligase (Ddl). Bioorg. Med. Chem. Lett. 17:2047-2054. [DOI] [PubMed] [Google Scholar]
- 22.Lamichhane, G., M. Zignol, N. J. Blades, D. E. Geiman, A. Dougherty, J. Grosset, K. W. Broman, and W. R. Bishai. 2003. A postgenomic method for predicting essential genes at subsaturation levels of mutagenesis: application to Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 100:7213-7218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee, J. H., Y. Na, H. E. Song, D. Kim, B. H. Park, S. H. Rho, Y. J. Im, M. K. Kim, G. B. Kang, D. S. Lee, and S. H. Eom. 2006. Crystal structure of the apo form of d-alanine: d-alanine ligase (Ddl) from Thermus caldophilus: a basis for the substrate-induced conformational changes. Proteins 64:1078-1082. [DOI] [PubMed] [Google Scholar]
- 24.LeMagueres, P., H. Im, J. Ebalunode, U. Strych, M. J. Benedik, J. M. Briggs, H. Kohn, and K. L. Krause. 2005. The 1.9 A crystal structure of alanine racemase from Mycobacterium tuberculosis contains a conserved entryway into the active site. Biochemistry 44:1471-1481. [DOI] [PubMed] [Google Scholar]
- 25.Liu, S., J. S. Chang, J. T. Herberg, M. M. Horng, P. K. Tomich, A. H. Lin, and K. R. Marotti. 2006. Allosteric inhibition of Staphylococcus aureus d-alanine:d-alanine ligase revealed by crystallographic studies. Proc. Natl. Acad. Sci. U. S. A. 103:15178-15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lugtenberg, E. J., and A. van Schijndel-van Dam. 1973. Temperature-sensitive mutant of Escherichia coli K-12 with an impaired d-alanine:d-alanine ligase. J. Bacteriol. 113:96-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mahapatra, S., H. Scherman, P. J. Brennan, and D. C. Crick. 2005. N glycolylation of the nucleotide precursors of peptidoglycan biosynthesis of Mycobacterium spp. is altered by drug treatment. J. Bacteriol. 187:2341-2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCoy, A. J. 2007. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63:32-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Milligan, D. L., S. L. Tran, U. Strych, G. M. Cook, and K. L. Krause. 2007. The alanine racemase of Mycobacterium smegmatis is essential for growth in the absence of d-alanine. J. Bacteriol. 189:8381-8386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neilands, J. B. 1956. Metal and hydrogen-ion binding properties of cycloserine. Arch. Biochem. Biophys. 62:151-162. [DOI] [PubMed] [Google Scholar]
- 31.Neuhaus, F. C. 1962. The enzymatic synthesis of d-alanyl-d-alanine. II. Kinetic studies on d-alanyl-d-alanine synthetase. J. Biol. Chem. 237:3128-3135. [PubMed] [Google Scholar]
- 32.Neuhaus, F. C., and J. L. Lynch. 1964. The enzymatic synthesis of d-alanyl-d-alanine. 3. On the inhibition of d-alanyl-d-alanine synthetase by the antibiotic d-cycloserine. Biochemistry 3:471-480. [DOI] [PubMed] [Google Scholar]
- 33.Otwinowski, Z., and W. Minor. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307-326. [DOI] [PubMed] [Google Scholar]
- 34.Parsons, W. H., A. A. Patchett, H. G. Bull, W. R. Schoen, D. Taub, J. Davidson, P. L. Combs, J. P. Springer, H. Gadebusch, B. Weissberger, et al. 1988. Phosphinic acid inhibitors of d-alanyl-d-alanine ligase. J. Med. Chem. 31:1772-1778. [DOI] [PubMed] [Google Scholar]
- 35.Sacchettini, J. C., E. J. Rubin, and J. S. Freundlich. 2008. Drugs versus bugs: in pursuit of the persistent predator Mycobacterium tuberculosis. Nat. Rev. Microbiol. 6:41-52. [DOI] [PubMed] [Google Scholar]
- 36.Sassetti, C. M., D. H. Boyd, and E. J. Rubin. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48:77-84. [DOI] [PubMed] [Google Scholar]
- 37.Schleifer, K. H., and O. Kandler. 1972. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol. Rev. 36:407-477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Segel, I. H., and R. L. Martin. 1988. The general modifier (“allosteric”) unireactant enzyme mechanism: redundant conditions for reduction of the steady state velocity equation to one that is first degree in substrate and effector. J. Theor. Biol. 135:445-453. [DOI] [PubMed] [Google Scholar]
- 39.Shi, Y., and C. T. Walsh. 1995. Active site mapping of Escherichia coli d-Ala-d-Ala ligase by structure-based mutagenesis. Biochemistry 34:2768-2776. [DOI] [PubMed] [Google Scholar]
- 40.Strominger, J. L., E. Ito, and R. H. Threnn. 1960. Competitive inhibition of enzymatic reactions by oxamycin. J. Am. Chem. Soc. 82:998-999. [Google Scholar]
- 41.Thompson, R. J., H. G. Bouwer, D. A. Portnoy, and F. R. Frankel. 1998. Pathogenicity and immunogenicity of a Listeria monocytogenes strain that requires d-alanine for growth. Infect. Immun. 66:3552-3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vollmer, W., D. Blanot, and M. A. de Pedro. 2008. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 32:149-167. [DOI] [PubMed] [Google Scholar]
- 43.Walsh, C. 2003. Antibitocs: actions, origins, resistance. ASM Press, Washington, DC.
- 44.Wright, G. D., and C. T. Walsh. 1993. Identification of a common protease-sensitive region in d-alanyl-d-alanine and d-alanyl-d-lactate ligases and photoaffinity labeling with 8-azido ATP. Protein Sci. 2:1765-1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu, D., L. Zhang, Y. Kong, J. Du, S. Chen, J. Chen, J. Ding, H. Jiang, and X. Shen. 2008. Enzymatic characterization and crystal structure analysis of the d-alanine-d-alanine ligase from Helicobacter pylori. Proteins 72:1148-1160. [DOI] [PubMed] [Google Scholar]
- 46.Yew, W. W., and C. C. Leung. 2008. Management of multidrug-resistant tuberculosis: update 2007. Respirology 13:21-46. [DOI] [PubMed] [Google Scholar]
- 47.Zawadzke, L. E., T. D. Bugg, and C. T. Walsh. 1991. Existence of two d-alanine:d-alanine ligases in Escherichia coli: cloning and sequencing of the ddlA gene and purification and characterization of the DdlA and DdlB enzymes. Biochemistry 30:1673-1682. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.