

Abstract
IDX184 is a nucleotide prodrug designed to enhance formation in the liver of the active triphosphate of 2′-methylguanosine (2′-MeG), a potent and specific polymerase inhibitor of the hepatitis C virus (HCV). In the present study, single ascending oral doses of 5, 10, 25, 50, 75, and 100 mg IDX184 were administered sequentially to cohorts of 8 healthy subjects, randomized 6:2, active/placebo. Plasma and urine pharmacokinetic sampling was performed over a period of 120 h after dosing. Upon absorption, IDX184 rapidly disappeared from plasma, with a mean half-life (t1/2) of approximately 1 h, while plasma concentrations of 2′-MeG gradually increased. Consistent with a liver-targeting approach, plasma exposure of IDX184 and 2′-MeG was low and was also dose related: the mean maximum concentrations ranged from 1.1 to 17 ng/ml for IDX184 and 1.7 to 19 ng/ml for 2′-MeG, and the respective mean total area under the curve ranged from 1.2 to 22.7 and 17.3 to 334 ng·h/ml. Mean 2′-MeG plasma concentrations 24 h after dosing were 0.6 to 3 ng/ml for the 25- to 100-mg doses. Mean 2′-MeG t1/2 values ranged from 18 to 43 h for doses of 25 mg and above. Mean cumulative urine excretion was 0.2% and 12 to 20% of administered doses for the unchanged IDX184 and 2′-MeG, respectively. IDX184 was safe and well tolerated; no serious adverse events (SAEs), dose-dependent adverse events (AEs), or dose-limiting toxicities were observed. The incidence of AEs and laboratory abnormalities was low and was similar among subjects receiving IDX184 or a placebo. All AEs were mild to moderate and resolved at the end of study. The favorable safety and pharmacokinetic profiles support further clinical evaluation of IDX184 in HCV-infected patients.
Hepatitis C virus (HCV) infection is a global public health problem. The global prevalence of hepatitis C infection is estimated to average 3%, resulting in approximately 170 million HCV-infected persons worldwide (8).
The current standard of care for patients with chronic hepatitis C and compensated liver disease is the combination of parenterally administered pegylated interferon and orally administered ribavirin. However, sustained virologic response rates are less than 50% in treatment-naive patients with genotype 1 infection (2, 4). In addition, many patients are not good candidates for treatment with these agents due to advanced liver disease or concurrent medical conditions, and many patients show poor tolerance of these treatments. Therefore, there is a need for new anti-HCV therapies, particularly for patients with genotype 1 HCV or those who have failed to respond to the available treatment options.
While nucleoside HCV polymerase inhibitors have a higher genetic barrier to resistance and have demonstrated clinical anti-HCV activity, early nucleoside analogs have wide systemic exposure and inefficient phosphorylation of their 5′-monophosphate (MP) metabolite, resulting in limited formation of the active 5′-triphosphate (TP) in the liver, the site of HCV replication (1, 3, 5). In that context, IDX184 (Fig. 1) is a novel liver-targeted nucleotide prodrug designed to selectively deliver the 5′-MP of 2′-methylguanosine (2′-MeG) in hepatocytes. By bypassing the rate-limiting monophosphorylation process, 2′-MeG-MP is then efficiently converted to its active 5′-TP within hepatocytes by cellular kinases. The complete metabolic pathway of IDX184 is yet to be fully elucidated. In vitro experiments showed that the metabolism of IDX184 to 2′-MeG-MP takes place predominantly in liver cells and involves both cytochrome P450 (CYP450)-dependent and -independent processes (S. Good, unpublished data). 2′-MeG-TP is a potent inhibitor of the HCV polymerase NS5B, and IDX184 has demonstrated antiviral activity against HCV in vitro and in chimpanzees (1, 6, 7).
FIG. 1.
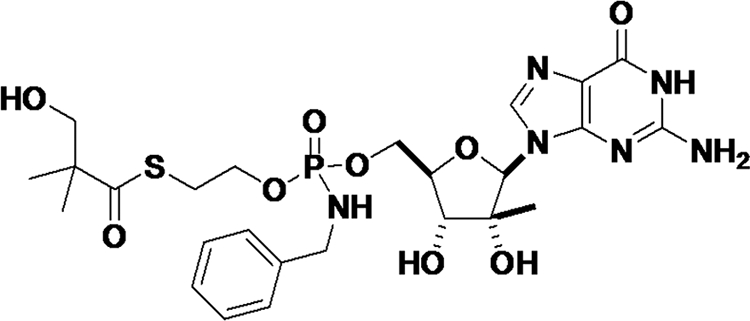
Chemical structure of IDX184.
The enhanced formation of active intracellular 2′-MeG-TP with IDX184 was demonstrated in vitro. In primary human and animal hepatocytes exposed to IDX184 and the nucleoside 2′-MeG, the formation of 2′-MeG-TP was about 100-fold higher with IDX184 than with 2′-MeG (S. Good, unpublished data). The enhanced formation of intracellular 2′-MeG-TP with IDX184 was associated with increased anti-HCV activity. In the HCV replicon assay system, IDX184 was about 10-fold more potent than 2′-MeG in inhibiting HCV replication (D. Standring, unpublished data).
The liver-targeting delivery approach may result in a favorable therapeutic index by increasing the levels of active 2′-MeG-TP at the site of HCV replication while lowering systemic exposure of the nucleoside, potentially minimizing the risk for systemic side effects. In vivo studies in rats and monkeys have shown that, as anticipated, oral IDX184 was largely extracted by the liver (approximately 95%), with low plasma levels of the nucleoside metabolite 2′-MeG (1).
The objective of this first-in-human study was to evaluate the safety, tolerability, and pharmacokinetics of single doses of IDX184, escalating from 5 mg to 100 mg in healthy subjects.
MATERIALS AND METHODS
This study was conducted in accordance with Good Clinical Practice procedures, the principles of the Declaration of Helsinki, and U.S. Food and Drug Administration regulations. Approval for the study was obtained from an independent institutional review board (Lincoln NE).
Clinical conduct for the study took place at MDS Pharma Services in Lincoln, NE. The first subject was dosed on 27 July 2008, and the last subject completed the study on 10 October 2008.
Study population.
All subjects voluntarily gave written informed consent after the nature of the study was fully explained. Eligible subjects were healthy adult nonsmoking male or female subjects from the general population. They were 19 to 65 years of age, with body mass indices between 18 and 32 kg/m2 and no evidence of clinically significant abnormalities in medical history, physical examination, 12-lead electrocardiogram (ECG), or clinical laboratory testing during screening. Subjects of childbearing potential were required to use an acceptable double-barrier method of birth control during the study and for up to 90 days after the last dose of the study drug. Female subjects were excluded if they were pregnant or breastfeeding. Subjects were excluded if they had donated blood or had a significant blood loss within 56 days or donated plasma within 7 days of screening or had used a known inhibitor and/or inducer of CYP450 3A4 (CYP 3A4) within 30 days of reporting to the clinic. Subjects were excluded if they received any chronic prescription medications within 3 months, acute prescription drugs within 14 days, or systemic over-the-counter medications (including aspirin, vitamins, and herbal supplements) within 7 days of reporting to the clinic. Subjects were excluded if currently abusing alcohol or illicit drugs or having a history of alcohol or illicit drug abuse within the preceding 2 years. Subjects were also excluded if they tested positive for HIV, hepatitis C virus, or hepatitis B virus, tested positive for drugs of abuse or alcohol; or participated in a clinical study within 30 days prior to screening.
Study design.
The trial was a single-center, randomized, double-blind, placebo-controlled, single-dose-escalation study in healthy male or female subjects.
IDX184 and matching placebo were supplied as white opaque capsules. Both a 5-mg and a 25-mg capsule were provided. The single-dose escalation started at 5 mg, which escalated to 10, 25, 50, 75, and 100 mg. The 5- and 10-mg cohorts used the 5-mg capsules, while other doses employed the 25-mg capsules. Eight subjects were randomly assigned at a 6:2 active/placebo ratio within each cohort. Safety and plasma pharmacokinetic evaluations were performed before escalation to the next dose level.
The study drug was given on an empty stomach after a fasting period of approximately 10 h prior to dosing and for an additional 4 h postdosing. At least 240 ml (8 fluid oz.) of water was given with the study dose. A standard lunch was served no less than 4 h after dosing. Meals and beverages served within 24 h prior to and after dosing were free of xanthine and caffeine. Grapefruit juice was not allowed within 24 h prior to and after dosing. Water was permitted ad libitum except for 1 h prior to and after dosing. Subjects were asked to remain upright (sitting or standing) for the first 3 h following dosing and not to engage in any strenuous activity during their stay at the study center.
Safety and tolerability evaluation.
Safety assessments consisted of collecting all adverse events (AEs) and serious adverse events (SAEs), along with determining their severity and relationship to the study drug. Safety assessments also included regular monitoring of hematology, blood chemistry, and urinalysis, as well as vital signs, 12-lead ECG, and physical examination. Concomitant medications were also collected.
Pharmacokinetic sampling.
Intensive plasma pharmacokinetic sampling was performed over a period of 120 h after dosing at the following time points: 0 (predose) and 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, 48, 72, 96, and 120 h. Blood samples were collected in Vacutainer tubes containing EDTA as an anticoagulant at the time points specified above within a time window of up to ±10 min. Plasma was obtained by centrifugation at 1,500 × g for 10 min at 4°C and stored frozen at a nominal temperature of −80°C until analysis.
Urine samples were collected over 120 h according to the following time intervals: −2 to 0 (predose), 0 to 4, 4 to 8, 8 to 12, 12 to 24, 24 to 48, 48 to 72, 72 to 96, and 96 to 120 h. Urine samples were stored frozen at a nominal temperature of −80°C until analysis.
Plasma and urine IDX184 and 2′-MeG have been shown to remain stable during storage and assay. The short-term stability of both analytes has been documented when spiked samples were subjected to 6 freeze and thaw cycles (−80°C to 8°C in plasma and −20°C to ambient temperature in urine), storage for at least 24 h at 8°C in plasma and at ambient temperature in urine, and postpreparative for 153 h for plasma samples and at least 108 h for urine samples. Long-term storage stability was 441 days in plasma at −80°C and 86 days in urine under −20°C. Study samples were analyzed without exceeding freeze-thaw/postpreparative stability or long-term/short-term stability.
Sample analysis.
Plasma and urine samples were analyzed for IDX184 and 2′-MeG using validated high-performance liquid chromatography (HPLC) and tandem mass spectrometry (MS/MS) methodologies. Concentrations of the two analytes in a sample were simultaneously measured in a single run. For plasma samples, an aliquot of 25 μl of an internal standard working solution containing 16 ng/ml 13CD3-IDX184 and 8 ng/ml D3-2′-MeG was added to 200 μl of calibration standards (0.1 to 20 ng/ml for IDX184 and 0.2 to 40 ng/ml for 2′-MeG), quality controls (QCs) (0.3 to 15 ng/ml for IDX184 and 0.6 to 30 ng/ml for 2′-MeG), and unknown plasma samples. For urine samples, an aliquot of 25 μl of an internal standard working solution containing 40 ng/ml 13CD3-IDX184 and D3-2′-MeG was added to 100 μl of calibration standards (1 to 200 ng/ml), QCs (3 to 150 ng/ml), and unknown urine samples. Plasma and urine samples were then diluted with 1 ml and 0.5 ml, respectively, with a 100 mM ammonium acetate solution. After mixing, diluted samples (approximately 600 μl) were subjected to solid-phase extraction on a 96-well plate (Bond Elut PBA, 100 mg; Varian, Palo Alto, CA). The plate was preconditioned and thereafter successively washed after sample loading with acetonitrile and water. Analytes were eluted with 0.8 ml 5% formic acid in methanol. Eluent was evaporated to dryness under a gentle nitrogen flow at 40°C. The residual was reconstituted with 200 μl acetonitrile, and a 25-μl volume of the final prepared sample was analyzed via HPLC with MS/MS detection. Under these conditions, average extraction recovery from plasma and urine was 76% and 59%, respectively, for IDX184 and 79% and 69%, respectively, for 2′-MeG. For both matrices, chromatography was performed on a Zorbax 300-SCX column (50 mm by 3 mm; particle size, 5 μm; Agilent Technologies, Santa Clara, CA) preceded by a SecurityGuard ODS C18 guard column (4 mm by 3 mm; particle size, 5 μm; Phenomenex, Torrance, CA). Elution was carried out isocratically at a constant flow rate of 1.5 ml/min with a mobile phase of 95:5 (vol/vol) acetonitrile/ammonium formate (100 mM, pH 3.8). Under these conditions, the retention times were approximately 1.4 and 1.0 min for IDX184 and 2′-MeG, respectively. The analytes were monitored using a PE Sciex API 5000 triple quadrupole mass analyzer at a mass transition of 627.2 → 316.1 m/z and 631.2 → 320.1 m/z for IDX184 and 13CD3-IDX184, respectively, and 298.2 → 152.1 m/z and 301.2 → 152.1 m/z for 2′-MeG and D3-2′-MeG, respectively. The mass analyzer was operated in positive ion mode using electrospray ionization. This assay has a lower limit of quantitation (LOQ) of 0.1 ng/ml for IDX184 and 0.2 ng/ml for 2′-MeG in plasma and 1 ng/ml for both analytes in urine. The intra- and interday precisions (coefficient of variation) and accuracies (percent deviation) for plasma and urine assays were from 2.5 to 16.1% and −13.3 to 10.3%, respectively, for IDX184 and from 0.8 to 6.6% and −7.2 to 5.0%, respectively, for 2′-MeG.
Pharmacokinetic and statistical analyses.
The plasma concentration-time data for IDX184 and 2′-MeG were analyzed using noncompartmental methods. The maximum drug concentration in plasma (Cmax), time to Cmax (Tmax), and concentration 24 h after dosing (C24) were obtained directly from the plasma concentration-time profiles. The area under the plasma concentration-time curve (AUC) from time zero to time t (AUC0-t), where t is the time at which the last sample with a measurable concentration was obtained, was calculated according to the linear trapezoidal rule. The AUC from time zero to infinity (AUC0-∞) was estimated as AUC0-t + Ct/ke, where Ct is the concentration in the last plasma sample in which a measurable concentration was obtained and ke is the slope of the linear portion of the natural log-transformed postpeak plasma drug concentration-time curve estimated by liner regression. The terminal half-life (t1/2) over the sampling period was calculated as 0.693/ke. For IDX184, apparent total plasma clearance (CL/F) was calculated as dose/AUC0-∞, and the apparent total volume of distribution (Vd/F) was calculated as CL/ke. Both parameters were further normalized to body weight.
Principal parameters underlying plasma drug exposure, including Cmax and AUC0-∞, were assessed for dose proportionality in the 5- 100-mg dose range by using the following log-linearized power model: log (Yij) = a + b × log(Dj) + eij, where Dj is the dose at level j, Yij is the pharmacokinetic parameter for subject i at dose level j, a and b are the mean intercept and slope, respectively; and eij is the residual error for subject i at dose level j. Fitting was carried out by linear regression using the GLM procedure in SAS (version 9.2; SAS Institute Inc., Cary, NC). A dose-proportional relationship is concluded if the 95% confidence interval (CI) of the mean slope (b) includes unity and was contained within a critical range of 0.70 to 1.30.
Cumulative urine excretion (Au0-t) was calculated as the sum of the amounts excreted during each interval and expressed as a percentage of the administered dose. Renal clearance (CLR) was calculated as Au0-t/AUC0-t, where the time interval 0-t was the same for the numerator and the denominator.
RESULTS
Subject characteristics and disposition.
Treatment cohorts were generally comparable with respect to baseline characteristics. Subjects enrolled were mostly Caucasians. While male subjects represented most of the study population, female subjects were enrolled in all cohorts. Table 1 summarizes baseline characteristics.
TABLE 1.
Subject characteristics
| Characteristic | Value for subjects receiving placebo or indicated dose (mg) |
Total | ||||||
|---|---|---|---|---|---|---|---|---|
| Placebo | 5 | 10 | 25 | 50 | 75 | 100 | ||
| No. of subjects | 12 | 6 | 6 | 6 | 6 | 6 | 6 | 48 |
| Age (yr)a | 35.0 ± 15.1 | 27.8 ± 6.4 | 33.7 ± 17.6 | 28.8 ± 6.4 | 37.0 ± 14.8 | 24.3 ± 2.6 | 29.5 ± 10.7 | 31.4 ± 12.2 |
| Wt (kg)a | 73.5 ± 11.9 | 80.5 ± 23.4 | 79.6 ± 7.2 | 79.9 ± 11.8 | 76.9 ± 10.1 | 78.5 ± 16.1 | 79.3 ± 24.6 | 77.7 ± 14.9 |
| BMIb (kg/m2)a | 26.4 ± 3.8 | 25.6 ± 5.0 | 25.8 ± 3.4 | 26.3 ± 3.7 | 25.6 ± 2.8 | 25.1 ± 3.4 | 24.9 ± 3.1 | 25.8 ± 3.5 |
| No. (%) male | 6 (50) | 4 (67) | 5 (83) | 5 (83) | 3 (50) | 5 (83) | 4 (67) | 32 (67) |
| No. (%): | ||||||||
| Caucasian | 11 (92) | 6 (100) | 6 (100) | 4 (67) | 5 (83) | 5 (83) | 6 (100) | 43 (90) |
| Asian | 1 (8) | 0 | 0 | 1 (17) | 0 | 1 (17) | 0 | 3 (6) |
| Black | 0 | 0 | 0 | 1 (17) | 0 | 0 | 0 | 1 (2) |
| Native American | 0 | 0 | 0 | 0 | 1 (17) | 0 | 0 | 1 (2) |
Values are presented as means ± SDs.
BMI, body mass index.
All enrolled subjects completed safety and pharmacokinetic evaluations with no premature discontinuation.
Safety and tolerability.
Single-dose IDX184 was well tolerated. There were no SAEs or dose-limiting toxicities. The most common AE was dizziness, which occurred in 2 of 36 subjects (5.6%) exposed to IDX184 and 3 of 12 subjects (25%) exposed to a placebo. Other, less-frequent AEs observed in both placebo and active groups included dermatitis contact, dysmenorrhoea, fatigue, and headache. AEs reported were all mild or moderate in intensity and resolved by the end of the study. There were no discernible patterns in AEs between treatment groups.
Laboratory parameters were stable over time for all treatment groups. No clinically meaningful changes were observed in vital sign measurements, physical examination findings, or ECG parameters.
Plasma pharmacokinetics.
IDX184 is a prodrug that delivers the monophosphate of the nucleoside 2′-MeG in the liver. Both the parent, IDX184, and its nucleoside metabolite, 2′-MeG, were monitored in the blood. Mean (+standard deviation [SD]) plasma concentration-time curves of IDX184 (left panel) and 2′-MeG (right panel) after administration of single doses of IDX184 escalating from 5 to 100 mg are depicted in Fig. 2. Summary pharmacokinetic parameters are presented in Table 2. Following oral administration of a single dose under fasted conditions in healthy subjects, IDX184 was rapidly absorbed, and the Tmax corresponded in most subjects to the first sampling time, with a cohort median value of 0.25 to 0.49 h regardless of the administered doses. The level of plasma exposure to IDX184 was low and proportional to administered doses, with a cohort mean Cmax and AUC0-∞ ranging from 1.12 to 17.3 ng/ml and 1.19 to 22.7 ng·h/ml, respectively, as doses escalated from 5 to 100 mg. The pharmacokinetic dose proportionality of IDX184 in the studied dose range was evaluated by regression analyses of log-transformed parameters of exposure and doses. The model estimate of the slope was close to unity for Cmax (b = 0.98; 95% CI = 0.76 to 1.21) and AUC0-∞ (b = 1.01; 95% CI = 0.85 to 1.17). The plasma kinetic profile of IDX184 exhibited a steep disposition phase, with plasma concentrations quickly falling below the limit of quantitation (0.1 ng/ml) with a short yet consistent mean t1/2 of 0.58 to 1.06 h across doses. Cohort mean CL/F and Vd/F, normalized to body weight, were 49.0 to 92.1 liters/h/kg and 67.8 to 122 liters/kg, respectively, and were independent of administered doses of IDX184.
FIG. 2.

Plasma concentration-time profiles of IDX184 (left panel, time axis truncated to 8 h for better clarity) and 2′-MeG (right panel) after administration of 5 to 100 mg of single doses of IDX184 in healthy subjects (n = 6 per dose). The mean (+SD) is shown.
TABLE 2.
Plasma pharmacokinetic parameters of IDX184 and the nucleoside metabolite 2′-MeGa
| Drug or metabolite | Dose (mg) | Cmax (ng/ml) | Tmax (h) | AUC0-∞ (ng·h/ml) | AUC ratio (%)b | t1/2 (h) | CL/Fe (liters/h/kg) | Vd/Fe (liters/kg) | C24 (ng/ml) |
|---|---|---|---|---|---|---|---|---|---|
| IDX184 | 5 | 1.12 ± 0.70 | 0.38 (0.23-0.98) | 1.19 ± 0.48 | 3.8 ± 1.4 | 0.58 ± 0.14 | 69.4 ± 45.6 | 67.8 ± 28.9 | |
| 10 | 1.39 ± 1.15 | 0.37 (0.25-1.00) | 2.22 ± 1.15 | 2.2 ± 0.9 | 1.06 ± 0.37 | 68.6 ± 28.0 | 122 ± 49.6 | ||
| 25 | 4.93 ± 4.40 | 0.25 (0.25-2.00) | 5.53 ± 4.25 | 2.9 ± 1.8 | 0.82 ± 0.23 | 83.3 ± 49.3 | 94.8 ± 31.0 | ||
| 50 | 10.9 ± 6.52 | 0.26 (0.23-0.50) | 15.2 ± 6.25 | 2.4 ± 1.1 | 0.80 ± 0.16 | 49.0 ± 16.7 | 88.4 ± 21.4 | ||
| 75 | 12.4 ± 11.4 | 0.49 (0.27-1.50) | 16.1 ± 10.6 | 2.3 ± 1.3 | 0.90 ± 0.22 | 92.1 ± 64.1 | 98.7 ± 11.8 | ||
| 100 | 17.3 ± 5.55 | 0.25 (0.25-0.50) | 22.7 ± 4.29 | 3.7 ± 1.2 | 0.92 ± 0.19 | 63.8 ± 25.2 | 103 ± 27.6 | ||
| 2′-MeG | 5 | 1.74 ± 1.19 | 4.00 (3.00-6.00) | 17.3 ± 11.2 | 5.42 ± 1.65c | 0.25 ± 0.06 | |||
| 10 | 4.82 ± 1.62 | 6.00 (4.00-6.00) | 53.2 ± 25.5 | 12.2 ± 7.88c | 0.40 ± 0.21 | ||||
| 25 | 5.93 ± 2.30 | 6.00 (3.00-8.00) | 81.9 ± 23.2 | 18.0 ± 11.8 | 0.58 ± 0.17 | ||||
| 50 | 18.6 ± 5.63 | 6.00 (4.00-6.00) | 318 ± 139 | 22.4 ± 6.72 | 2.88 ± 1.39 | ||||
| 75 | 15.5 ± 6.81 | 6.00 (4.00-6.00) | 311 ± 78.9 | 42.5 ± 45.7d | 2.31 ± 1.46 | ||||
| 100 | 14.6 ± 3.28 | 5.00 (3.00-6.00) | 334 ± 161 | 19.9 ± 4.72 | 2.23 ± 0.88 |
All parameters except Tmax are presented as means ± SDs; for Tmax, values are presented as medians (ranges).
Ratio (expressed as a percentage) of AUC00-∞ of IDX184 to 2′-MeG on a molar basis.
t1/2 of 2′-MeG was underestimated for the 5-mg and 10-mg dose cohorts due to a partially observed terminal phase.
Cohort mean value for t1/2 was inflated by a subject whose t1/2 value was 122 h; group median value for t1/2 was 26.5 h.
Parameters normalized to body weight.
The rapid elimination of IDX184 from plasma was accompanied by the gradual appearance of 2′-MeG. Similar to results for the parent drug, the plasma concentrations of 2′-MeG were also low and reached a maximum at a cohort median Tmax of 4.00 to 6.00 h across doses. While mean Cmax and mean C24 values of 2′-MeG increased 10-fold, from 1.74 to 18.6 ng/ml and 0.25 to 2.88 ng/ml, respectively, with doses in the 5- to 50-mg range, these parameters became less dose proportional at higher doses (Table 2). The values of slope b (95% CI) resulting from dose-proportionality analyses were 0.76 (0.60 to 0.92) for Cmax and 0.84 (0.65 to 1.03) for C24. Mean Cmax and mean C24 values of 2′-MeG with 50- to 100-mg doses of IDX184 were 14.6 to 18.6 ng/ml and 2.23 to 2.88 ng/ml, respectively. In contrast, the mean AUC0-∞ of 2′-MeG had a 20-fold increase, from 17.3 to 334 ng·h/ml, as the dose increased 20-fold, and the values were shown to be dose proportional in the studied dose range (b = 1.04; 95% CI = 0.88 to 1.19). The mean elimination t1/2 of 2′-MeG, whose estimation depends on the actual observed portion of the elimination phase (concentration > LOQ), ranged from 5.42 to 12.2 h for doses up to 10 mg and from 18.0 to 42.5 h for doses of ≥25 mg.
The cohort mean ratios of IDX184 to 2′-MeG AUC0-∞ on a molar basis were low (<4%) and remained independent of the amount of dose administered, indicating a near-complete and unsaturated biotransformation of IDX184 in the studied dose range.
Urine excretion.
The amounts of parent IDX184 and its nucleoside metabolite 2′-MeG excreted in urine were monitored during a 120-h interval after administration in all subjects. Urine IDX184 could only be measured up to 12 h postdose except for one sample, where IDX184 was observed up to 24 h. Cumulative urine excretion of IDX184, which increased with increasing doses, remained consistently low across cohorts, representing only 0.2% of administered doses (Table 3). In contrast, urine 2′-MeG was quantifiable in all fractions during the entire 120-h postdose collection period. In general, the cumulative amount of 2′-MeG excreted in urine increased as the dose was escalated (Table 3). The cumulative amount of 2′-MeG excreted in urine was higher than that of IDX184, representing, on a molar basis, 12 to 20% of administered doses. Near-maximum cumulative urine excretion of 2′-MeG was reached between 48 and 72 h, and more than half of the total amount of 2′-MeG excreted was recovered within the first 24 h. Renal clearance (CLR) was independent of the amount of dose administered and remained consistent across cohorts, with a cohort mean of 123 to 177 ml/min for IDX184 and 271 to 322 ml/min for 2′-MeG.
TABLE 3.
Urine pharmacokinetic parameters of IDX184 and the nucleoside metabolite 2′-MeGa
| Drug or metabolite | Dose (mg) | Au0-t (μg) | % Dose Excrb | CLR (ml/min) |
|---|---|---|---|---|
| IDX184 | 5 | 10.2 ± 3.67 | 0.203 ± 0.073 | 165 ± 30.5 |
| 10 | 17.2 ± 3.83 | 0.172 ± 0.038 | 158 ± 54.5 | |
| 25 | 51.0 ± 30.8 | 0.204 ± 0.123 | 177 ± 46.3 | |
| 50 | 109 ± 36.9 | 0.217 ± 0.074 | 128 ± 41.9 | |
| 75 | 123 ± 50.7 | 0.164 ± 0.068 | 153 ± 54.1 | |
| 100 | 166 ± 36.4 | 0.165 ± 0.036 | 123 ± 15.3 | |
| 2′-MeG | 5 | 330 ± 144 | 13.9 ± 6.06 | 289 ± 57.7 |
| 10 | 924 ± 531 | 19.5 ± 11.1 | 272 ± 73.2 | |
| 25 | 1,424 ± 490 | 12.0 ± 4.13 | 322 ± 61.2 | |
| 50 | 4,837 ± 1,706 | 20.4 ± 7.17 | 271 ± 75.7 | |
| 75 | 4,566 ± 1,392 | 12.8 ± 3.89 | 281 ± 36.3 | |
| 100 | 5,701 ± 2,991 | 12.0 ± 6.30 | 286 ± 51.3 |
All parameters are presented as means ± SDs.
Percentage of dose excreted. Based on molar ratio for 2′-MeG.
DISCUSSION
It is desirable for a direct-acting HCV antiviral to concentrate in the liver, the main site of HCV replication. A high hepatic concentration can be achieved through a liver-targeting process. An increased liver drug concentration with a lower systemic drug exposure should lead to improved anti-HCV activity with potentially reduced side effects, typically associated with a wide body drug distribution. In addition, a liver-targeted drug is expected to be administered at much reduced doses, amenable to being combined with other antiviral agents as part of a combination therapy regimen.
IDX184 is a liver-targeted prodrug of the nucleotide 2′-MeG-MP. In contrast to first-generation anti-HCV nucleoside analogs, such as valopicitabine, that generated low levels of the active 5′-TP in primary hepatocytes due to the rate-limiting monophosphorylation process, IDX184 is rapidly and extensively converted to 2′-MeG-MP, which is subsequently phosphorylated to yield high levels of intracellular 2′-MeG-TP (1).
In the current study, plasma samples were not assessed for 2′-MeG-MP or other related phosphates since they are charged molecules which cannot diffuse across the cell membrane. In addition, the phosphates are extremely instable in the blood and are expected to instantly be degraded to the corresponding nucleoside by phosphatases. Pharmacokinetic evaluation instead focused on plasma IDX184 and 2′-MeG. While plasma IDX184 represented a small fraction of dose that escaped from hepatic extraction, at least part of the plasma 2′-MeG might come from its intracellular phosphates following degradation. Therefore, plasma 2′-MeG likely reflected intracellular phosphorylated 2′-MeG species.
IDX184 was rapidly absorbed: in most cases, Cmax was observed at the first or second blood collection time (∼0.25 to 0.5 h). Plasma levels of IDX184 were low, due primarily to an extensive hepatic extraction and first-pass effect as reflected by a large apparent oral clearance and volume of distribution. These results are consistent with preclinical findings in monkeys in which approximately 95% of the absorbed fraction was retained by the liver following administration by oral gavage (1). The oral bioavailability of the prodrug IDX184 or its nucleoside metabolite 2′-MeG remains unknown. However, the mean cumulative urinary excretion of 2′-MeG in this study was up to 20% of administered doses, suggesting that the bioavailability of all IDX184-related species would be at least 20%. This estimate is inline with observations in rats treated with radiolabeled IDX184 by both oral and intravenous routes, where the extent of absorption based on total radioactivity was estimated to be at least 22% (S. Good, unpublished data).
In the current study, plasma IDX184 disappeared rapidly with a short and dose-independent half-life of approximately 1 h, suggesting that the parent IDX184 is unlikely to accumulate with a typical oral dosing regimen over time.
Plasma 2′-MeG appeared gradually as IDX184 was quickly eliminated. Plasma exposure of 2′-MeG was also low (Cmax less than 20 ng/ml). In cohorts with plasma 2′-MeG concentrations measurable beyond 48 h (25 mg and higher), the elimination phase was better characterized, with a long half-life of approximately 20 h, which is in the range of the intracellular half-life (15 h) of 2′-MeG-TP in human hepatocytes in vitro (1).
In HCV-infected chimpanzees receiving 10 mg/kg/day of IDX184 as a single agent for 4 days, the viral response at the end of treatment correlated significantly with 2′-MeG exposure but not with IDX184 exposure. Trough concentrations of 2′-MeG ranging from 2 to 8 ng/ml were associated with 1 to 4 log10 reductions in HCV RNA (6, 7). In the current study, plasma concentrations of 2′-MeG remained quite sustained, i.e., still above 2 ng/ml 24 h after a single dose of 50 to 100 mg IDX184. Steady-state trough concentrations were predicted to be around 10 ng/ml after repeat once-per-day (QD) dosing (data not shown). In light of the pharmacokinetic/pharmacodynamic (PK/PD) relationship established in the chimpanzees, a dose-dependent viral response can be anticipated with IDX184 treatment in HCV-infected patients (7).
Currently, ethical concerns and technical challenges prevent direct assessment in humans of hepatic intracellular IDX184 and related phosphorylated forms of 2′-MeG. While liver biopsy specimens were not obtained in this study, the extent of conversion from IDX184 to 2′-MeG-MP and subsequent phosphates may nevertheless be approximated by the ratio of plasma exposure of parent IDX184 to 2′-MeG. Despite a 20-fold change in dose, the mean molar ratio of AUC0-∞ of IDX184 to 2′-MeG, ranging from 2.2 to 3.8%, was low and consistent across doses, suggesting a near-complete conversion which was not saturated at the highest IDX184 dose studied.
Parent IDX184 and its metabolite 2′-MeG were excreted in urine. Although no formal statistical analyses were performed, the amounts of both entities recovered in urine appeared to be dose related. Urinary excretion of IDX184 was limited, whereas cumulative urine excretion of 2′-MeG was substantial, accounting for approximately 12 to 20% of the administered dose. Renal clearance, in particular for 2′-MeG, was greater than the normal glomerular filtration rate of 80 to 120 ml/min, indicating the involvement of active components in renal elimination.
In summary, single oral doses of 5 mg to 100 mg of IDX184 were safe and well tolerated by the healthy subjects in this study. Following oral administration, plasma IDX184 and 2′-MeG concentrations were low, consistent with IDX184 being a liver-targeted prodrug. The favorable safety and pharmacokinetic profiles of IDX184 support further clinical evaluation of IDX184 in HCV-infected patients.
Acknowledgments
We thank the healthy volunteers and the staff of MDS Pharma Services.
Footnotes
Published ahead of print on 8 November 2010.
The authors have paid a fee to allow immediate free access to this article.
REFERENCES
- 1.Cretton-Scott, E., C. Perigaud, S. Peyrottes, L. Licklider, M. Camire, M. Larsson, M. La Colla, E. Hildebrand, L. Lallos, J. Bilello, J. McCarville, M. Seifer, M. Liuzzi, C. Pierra, E. Badaroux, G. Gosselin, D. Surleraux, and D. N. Standring. 2008. In vitro antiviral activity and pharmacology of IDX184, a novel and potent inhibitor of HCV replication. J. Hepatol. 48(Suppl. 2):S220. [Google Scholar]
- 2.Fried, M. W., M. L. Shiffman, K. R. Reddy, C. Smith, G. Marinos, F. L. Gonçales, Jr., D. Häussinger, M. Diago, G. Carosi, D. Dhumeaux, A. Craxi, A. Lin, J. Hoffman, and J. Yu. 2002. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 347:975-982. [DOI] [PubMed] [Google Scholar]
- 3.Godofsky, E., N. Afdhal, V. Rustgi, L. Shick, L. Duncan, X. J. Zhou, G. Chao, C. Fang, B. Fielman, M. Myers, and N. A. Brown. 2004. First clinical results for a novel antiviral treatment for hepatitis C: a phase I/II dose escalation trial assessing tolerance, pharmacokinetics, and antiviral activity of NM283. J. Hepatol. 40(Suppl. 1):35. [Google Scholar]
- 4.Manns, M. P., J. G. McHutchison, S. C. Gordon, V. K. Rustgi, M. Shiffman, R. Reindollar, Z. D. Goodman, K. Koury, M. Ling, and J. K. Albrecht. 2001. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 358:958-965. [DOI] [PubMed] [Google Scholar]
- 5.McCown, M. F., S. Rajyaguru, S. Le Pogam, S. Ali, W. R. Jiang, H. Kang, J. Symons, N. N. Cammack, and I. Najera. 2008. The hepatitis C virus replicon presents a higher barrier to resistance to nucleoside analogs than to nonnucleoside polymerase or protease inhibitors. Antimicrob. Agents Chemother. 52:1604-1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Standring, D. N., R. Lanford, E. Cretton-Scott, L. Licklider, M. Larsson, C. Pierra, G. Gosselin, C. Perigaud, D. Surleraux, B. Mayes, A. Moussa, and J. Selden. 2008. Potent antiviral activity of second generation nucleoside inhibitors, IDX102 and IDX184, in HCV-infected chimpanzees. J. Hepatol. 48(Suppl. 2):S30. [Google Scholar]
- 7.Standring, D. N., R. Lanford, B. Li, R. J. Panzo, M. Seifer, M. Larsson, S. S. Good, and X. J. Zhou. 2009. Antiviral activity of the liver targeted nucleotide HCV polymerase inhibitor IDX184 correlates with trough serum levels of the nucleoside metabolite in HCV-infected chimpanzees. J. Hepatol. 50(Suppl. 1):S37. [Google Scholar]
- 8.World Health Organization. 2002. Hepatitis C. http://www.who.int/csr/disease/hepatitis/whocdscsrlyo2003/en/index.html. World Health Organization, Geneva, Switzerland.