

Abstract
The aim of the present study was to describe the nevirapine (NVP) pharmacokinetics (PK) in pregnant women and their neonates and to evaluate the transplacental drug transfer and administration scheme for the prevention of mother-to-child transmission. Thirty-eight HIV-1-infected pregnant women were administered one tablet of NVP (200 mg) and two tablets of tenofovir-emtricitabine (Truvada) at the initiation of labor. Children were given NVP syrup (2 mg/kg of body weight) as a single dose (sdNVP) on the first day of life. By pair, NVP concentrations were measured in 11 maternal, 1 cord blood, and 2 neonatal plasma samples and analyzed by a population approach. A one-compartment model was used for mothers and neonates; the absorption rate constants for mothers and neonates were 0.95 h−1 (intersubject variability, 111%) and 0.39 h−1, respectively; the apparent elimination clearances were 1.42 liter·h−1 (intersubject variability, 22%) and 0.035 liter·h−1, respectively; and apparent volumes of distribution were 87.3 liters (intersubject variability, 25%) and 5.65 liters, respectively. An effect compartment was linked to maternal circulation by mother-to-cord and cord-to-mother rate constants of 1.10 h−1 and 1.43 h−1, respectively. Placental transfer, expressed as the fetal-to-maternal area under the curve ratio, was 75%. Neonates had a very long half-lives (110 h) compared to adults. In the 38 mothers, the simulated median individual predicted time during which the NVP concentration remained above the half-maximal inhibitory concentration (IC50) was 13.2 days (range, 12 to 19.2 days). Thus, the administration of tenofovir-emtricitabine for at least 3 weeks after delivery should be considered to prevent the emergence of resistant viruses. The neonate must receive sdNVP immediately after birth when the infant is born less than 30 min after maternal drug intake to keep NVP concentrations above the IC50.
Mother-to-child transmission (MTCT) accounts for 20% of all new HIV infections in sub-Saharan Africa (http://data.unaids.org/pub/Report/2009/2009_epidemic_update_en.pdf.). To prevent MTCT of HIV at about the time of delivery, a single dose of nevirapine (sdNVP) is administered at the start of labor and is the most common antiretroviral regimen used in resource-limited settings, as recommended by the World Health Organization (http://www.who.int/hiv/pub/mtct/en/arvdrugswomenguidelinesfinal.pdf). However, the use of sdNVP results in resistance mutations in 15 to 70% of women at 4 to 6 weeks postpartum (2, 10, 14), compromising the success of subsequent treatments with NVP in mother and child (7, 19, 15). The results of a recent clinical trial suggest that adding a single dose of tenofovir (TDF) and emtricitabine (FTC) at delivery may reduce those resistance rates by half (6).
Nevirapine is characterized by rapid and nearly complete absorption, rapid distribution throughout the body, metabolism by the hepatic cytochrome P450 (CYP) 3A4 (CYP 3A4) enzyme (with autoinduction during the first 2 weeks of treatment), and prolonged elimination (8, 22). Emtricitabine and tenofovir are predominantly eliminated by the kidney, and no drug-drug interactions between NVP and tenofovir-emtricitabine (Truvada) are expected.
Physiological changes caused by pregnancy and labor can modify NVP pharmacokinetics at each step. Furthermore, pharmacokinetics in the neonate are different from those in adults; e.g., the low level of expression of the CYP 3A4 enzyme may reduce metabolism.
Placental transfer of NVP has been described by the ratio between the cord blood concentration and the maternal concentration at delivery, and this ratio is highly variable: from 71.9% to 122.8% in the study of Mirochnick et al. (20) and from 57.6% to 93.0% in the study of Musoke et al. (21). A more representative measure of this transfer is proposed in this article.
The objective of this analysis was (i) to describe the pharmacokinetics of NVP after a single dose in the mother, fetus, and neonate, (ii) to estimate the placental transfer of NVP with an exposure ratio, and (iii) to evaluate the actual recommendations of the administration scheme.
MATERIALS AND METHODS
Patients.
The TEmAA (Tenofovir/Emtricitabine in Africa and Asia) ANRS 12109 study was an open, phase I/II trial evaluating the pharmacokinetics, the safety, and the toxicity of the tenofovir-emtricitabine combination in HIV-infected pregnant women and infants. This trial was conducted in Abidjan, Côte d'Ivoire; Phnom Penh, Cambodia; and Soweto, South Africa. Pregnant women (between 28 and 38 weeks of gestation) who were older than 18 years, infected by HIV-1, and naïve to all antiretroviral treatments and who had an indication for antiretroviral prophylaxis for prevention of mother-to-child-transmission (PMTCT) during pregnancy (in line with international or national recommendations, i.e., WHO clinical stage 1 or 2 and a CD4+ count of ≥200 cells/mm3 or stage 3 and a CD4+ count of ≥350 cells/mm3) were eligible. Infants with a gestational age of greater than 37 weeks and a birth weight of greater than 2,500 g were eligible. The study protocol was approved by the national ethics committees of Côte d'Ivoire, Cambodia, and the University of the Witwatersrand, South Africa, and by health authorities and/or medicines regulatory authorities in each country. The mother and, where possible, the father of the child to be born provided signed informed consent in English, French, or their preferred local language.
Treatments.
Mothers were administered one tablet of NVP (200 mg) and two tablets of TDF (300 mg)-FTC (200 mg) at the start of labor, zidovudine (ZDV; 300 mg twice a day) from enrollment to the delivery date, and one tablet of TDF (300 mg)-FTC (200 mg) per day for 7 days postpartum. Children were given NVP syrup (2 mg/kg of body weight) as a single dose on the first day of life and ZDV syrup (4 mg/kg) every 12 h for 7 days.
Sampling.
All women received NVP and underwent blood samplings for pharmacokinetic analysis: at delivery and at 1, 2, 3, 5, 8, 12, 24, 48, 72, and 168 h after administration of sdNVP. A cord blood sample was obtained at delivery; the neonate was sampled on days 1 and 2 of life. The time that elapsed between administrations and the sampling time, maternal body weight, fetal body weight, and gestational age were recorded.
Analytical method.
Plasma NVP concentrations were measured by a validated high-performance liquid chromatography (HPLC)/UV spectrometry assay. The characteristics of the method were as follows: the calibration range was linear from 0.25 to 10 mg/liter, the lower limit of quantification (LOQ) was 0.25 mg/liter, the extraction yield was 88%, the reproducibility evaluated by the coefficient of variation was >93.6%, and the accuracy evaluated by the coefficient of variation was >96%. No analytical interference between the different antiretrovirals was noticed.
Modeling strategy and population pharmacokinetic model.
Data were analyzed using the nonlinear mixed effect modeling software program NONMEM (version VI, level 1.0) with the Digital Fortran compiler (4). The first-order conditional estimation (FOCE) with interaction method was used. The pharmacokinetics of NVP in maternal, cord, and neonatal blood were studied sequentially. First, the concentrations in the mother were analyzed and the parameters of the maternal part of the model were estimated. Then, these parameters were fixed for the estimation of the cord blood parameters, and the cord blood parameters were then also fixed to estimate the parameters for the neonatal part of the model. All the parameters were estimated simultaneously in a final step.
For maternal data, one- and two-compartment models were tested. A model including an autoinduction component on NVP clearance was tested. For cord blood concentrations, two models were tested: an additional compartment linked to the maternal compartment and an effect compartment of negligible volume and negligible drug transfer linked to the maternal circulation. The effect compartment was modeled as a virtual compartment linked to the maternal plasma compartment by first-order processes which do not modify the compartmental model in the mother. After delivery, this fetal compartment is disconnected, the time is reset to zero, and the neonate has his or her own absorption and elimination (Fig. 1). To handle data below the LOQ, the M3 and M6 methods of the Beal classification were tested (11). Several error models (i.e., multiplicative and additive error models) were investigated to describe residual variability. The addition of a correlation between maternal and cord blood residual variabilities using an L2 item was tested. An exponential model was used for intersubject variability (ISV). Only significant ISVs on pharmacokinetic parameters were kept. The effect of each patient covariate was systematically tested via generalized additive modeling on the basic model. Continuous covariates (COs), as body weight, were tested according to the following equation, using clearance (CL) as an example: 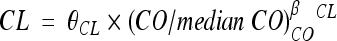 , where θCL is the typical value of clearance for a patient with the median covariate value and
, where θCL is the typical value of clearance for a patient with the median covariate value and 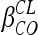 is the estimated influential factor for the continuous covariate. When a covariate was missing, it was set equal to the median value from all the other women. Categorical covariates (CAs; equal to 0 or 1) were tested according to the equation
is the estimated influential factor for the continuous covariate. When a covariate was missing, it was set equal to the median value from all the other women. Categorical covariates (CAs; equal to 0 or 1) were tested according to the equation 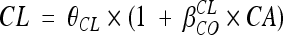 for inducing effect or
for inducing effect or 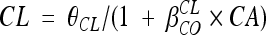 for inhibitory effect. The type of delivery (TD) was tested according to
for inhibitory effect. The type of delivery (TD) was tested according to 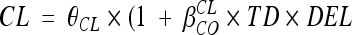 , where DEL is equal to 1 before delivery and 0 after delivery. A covariate was kept if its effect was biologically plausible and it produced a minimum reduction of 6.63 in the objective function value (OFV) and a reduction in the variability of the pharmacokinetic parameter, assessed by the associated intersubject variability. An intermediate model with all significant covariates was obtained. A backward elimination phase was finally performed by deleting each covariate from the intermediate model to obtain the final model, using a likelihood ratio test.
, where DEL is equal to 1 before delivery and 0 after delivery. A covariate was kept if its effect was biologically plausible and it produced a minimum reduction of 6.63 in the objective function value (OFV) and a reduction in the variability of the pharmacokinetic parameter, assessed by the associated intersubject variability. An intermediate model with all significant covariates was obtained. A backward elimination phase was finally performed by deleting each covariate from the intermediate model to obtain the final model, using a likelihood ratio test.
FIG. 1.

Population pharmacokinetic model for the simultaneous prediction of nevirapine concentrations in the mother, the cord (fetus; top), and the neonate (bottom). A one-compartment model with first-order absorption and elimination best described maternal data. For cord concentrations, an effect compartment is modeled as a virtual compartment linked to the maternal plasma compartment by a first-order process. After delivery, the fetal compartment is disconnected and the neonate has his or her own absorption and elimination.
Evaluation and validation.
For evaluation of the goodness of fit, the following graphs were prepared: observed and predicted concentrations versus time, observed concentrations versus population predictions, weighted residuals versus time, and weighted residuals versus predictions. Similar graphs using individual predictive post hoc estimation were displayed. The diagnostic graphs were prepared using the RfN module (S. Urien; http://wfn.sourceforge.net/) with the R program.
Simulated nevirapine profiles and observed data (standardized for a dose of 2 mg/kg in the neonate) were compared by use of a visual predictive check in order to validate the model. The vector of pharmacokinetic parameters from 1,000 patients was simulated using the final model. Each vector parameter was drawn in a log-normal distribution with a variance corresponding to the ISV previously estimated. A simulated residual error was added to each simulated concentration. The simulations were performed using the NONMEM program. The 5th, 50th, and 95th percentiles of the simulated concentrations at each time were then overlaid on the observed concentration data using the R program and a visual inspection was performed.
Maternal and neonatal concentrations after 200 mg NVP administration to the mother at delivery and placental transfer.
After the administration of 200 mg NVP to each pregnant woman, cord (i.e., fetal) and maternal blood NVP concentrations were assessed at delivery. The ratio between the fetal concentration and the maternal concentration was calculated. In order to better evaluate placental transfer for a 200-mg dose administered to the mother, maternal and fetal areas under the curve (AUCs) were estimated and the ratio between fetal AUC and maternal AUC was calculated.
Persistence of NVP in plasma of mothers.
The time during which the nevirapine concentration remained above the half-maximal inhibitory concentration (IC50) of 0.01 mg/liter was simulated for each patient.
Concentration in newborns before administration of sdNVP.
For different time delays between NVP administration to the mother and birth (30 min to 8 h), neonatal concentrations during the first 72 h of life were simulated and compared to the IC50.
RESULTS
Demographic data.
Data from the 38 enrolled women and 30 of their neonates were available for NVP pharmacokinetic evaluation. Table 1 summarizes the patients' characteristics.
TABLE 1.
Characteristics of HIV-infected pregnant women enrolled in the pharmacokinetic study of the TEmAA ANRS 12109 trial, step 1a
| Covariate | Median (min-max) value |
|---|---|
| Maternal body wt at delivery (kg) | 58.3 (46.5-88.1) |
| Gestational age (wk) | 39 (33-42) |
| No. of mothers with vaginal delivery, no. with delivery by cesarean section | 24, 14 |
| Maternal creatinine clearance at enrollment (μmol/liter) | 42.2 (26-88) |
| Neonatal body wt at birth (kg) | 2.7 (2.3-3.6) |
| Neonatal ht at birth (cm) | 48.5 (46.0-53.0) |
| Body surface area at birth (m2) | 0.20 (0.18-0.23) |
Data are for 38 pregnant women.
Population pharmacokinetics.
A total of 405 maternal, 36 cord blood, and 55 neonatal NVP concentrations were used for pharmacokinetic analysis of NVP. Twenty-nine maternal concentrations, two cord blood concentrations, and one neonatal concentration were lower than the LOQ. The M3 method did not improve the estimation of the parameters, so the M6 method was kept: the first LOQ for concentration was set equal to half of the LOQ (3), and the following ones were excluded. Two maternal concentrations and one neonatal concentration were excluded because previous concentrations in these mothers and this neonate were already under the LOQ. A one-compartment model with first-order absorption and elimination best described the maternal data. The autoinduction model did not improve the fit. The effect compartment was kept for cord blood concentrations. Parameters of the model were the maternal absorption rate constant (ka), maternal elimination CL, maternal volume of distribution (V), mother-to-fetus rate constant (k1F), fetus-to-mother rate constant (kF1), neonatal absorption rate constant (kaN), neonatal elimination rate constant (keN), and neonatal volume of distribution (VN). Since NVP was orally administered, only ka, CL/F (where F is the unknown bioavailability), V/F, k1F, kF1, kaN, VN/F, and keN were identifiable. Analytical equations were used in a $PRED section in NONMEM to estimate these pharmacokinetic parameters. The available data were not sufficient to estimate intersubject variability for k1F, kF1, kaN, keN, and VN/F; and fixing the variance of these random effects to zero had no influence on the OFVs. Variabilities were thus estimated for ka, CL/F, and V/F. All residual variabilities were best described by a proportional error model. No significant correlation between maternal and cord blood residual variabilities was found. The effects of maternal body weight, type of delivery, and ethnicity were tested on CL/F, and none of these effects was significant.
Figure 2 displays the observed and predicted NVP plasma concentrations as a function of time for the mother, the cord, and the neonate. To better visualize the neonatal concentrations, cord blood concentrations were reported on the graph at time zero. Table 2 summarizes the final population pharmacokinetic estimates. Final model performance was appreciated by comparing population predicted and individual predicted plasma concentrations to observed plasma concentrations and population weighted residuals versus predicted concentrations and versus time for NVP (data not shown).
FIG. 2.

Observed (○) and model-predicted (lines) nevirapine concentrations versus time in mothers (left), cord (umbilical) blood (upper right), and neonates (bottom right).
TABLE 2.
Population pharmacokinetic parameters of nevirapine from the final modela
| Model | Parameter | Estimate (% RSE) |
|---|---|---|
| Structural model | ka (h−1) | 0.954 (17) |
| CL/F (liters/h) | 1.42 (3.9) | |
| V/F (liters) | 87.3 (4.3) | |
| k1F (h−1) | 1.1 (26) | |
| kF1 (h−1) | 1.43 (26) | |
| kaN (h−1) | 0.39 (27) | |
| keN (h−1) | 0.0064 (38) | |
| VN (liters) | 5.56 (31) | |
| Statistical model | 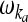 |
1.10 (27) |
| ωCL/F | 0.21 (28) | |
| ω V/F | 0.25 (17) | |
| r (CL − V) | 0.74 (24) | |
| σmother | 0.22 (13) | |
| σcord | 0.29 (37) | |
| σneonate | 0.39 (28) |
The final model for HIV-infected pregnant women (n = 38) enrolled in the TEmAA ANRS 12109 trial, step 1, after they received 200 mg of nevirapine at the start of the labor and for their neonates (n = 30) after they received 2 mg/kg of nevirapine in the 72 h after birth. Abbreviations: RSE, relative standard error [(standard error of estimate/estimate)·100]; σ, residual variability (proportional); ω, intersubject variability estimates. All other abbreviations are defined in the text.
Validation.
A visual predictive check of the final population pharmacokinetic model (Fig. 3) showed the predicted 5th, 50th, and 95th percentiles from the 1,000 simulations and the observed concentrations of nevirapine. The visual predictive check confirmed that the average prediction matched the observed concentrations. The variability was reasonably estimated.
FIG. 3.

Evaluation of the final model: comparison of the 5th (dashed line), 50th (solid line), and 95th (dashed line) percentiles obtained from 1,000 simulations and the observed data (○) for nevirapine concentrations in mother (left), cord blood (middle), and neonate (right).
Maternal and neonatal concentrations after 200 mg NVP administration to the mother at delivery and placental transfer.
The median delay between administration of NVP to the mothers and delivery was 5.1 h (minimum [min] and maximum [max], 0.92 and 20 h). At delivery, the median observed neonatal and maternal concentrations were 1.35 mg/liter (min and max, 0.125 and 2.95) and 1.70 mg/liter (min and max, 0.44 and 3.33), respectively. The median observed cord blood concentration-to-maternal concentration ratio was 71% (min and max, 13 and 140%). This wide range of concentration ratio at delivery suggests that placental transfer depends on the delay between maternal drug intake and delivery and could not be given as a simple percentage. A more representative measure of placental transfer would be the ratio between fetal NVP AUC and maternal NVP AUC for 24 h (12, 13). The predicted fetal AUC0-24-to-maternal AUC0-24 (where AUC0-24 is the AUC from 0 to 24 h) was 75%.
Persistence of NVP in plasma of mothers.
In the 38 mothers, the simulated median individual predicted time during which the NVP concentration remained above IC50 was 13.2 days (range, 12.0 to 19.9 days) (Fig. 4).
FIG. 4.

Simulated concentration-time courses for maternal plasma (semilog scale). Solid vertical lines, shortest and longest delays during which NVP concentrations remain above the IC50 limit (dashed line).
Concentration in newborns before administration of sdNVP.
Figure 5 represents the simulated neonatal concentrations for 72 h in children if delivery occurred (i) 30 min, (ii) 2 h, (iii) 5 h, and (iv) 8 h after the administration of the sdNVP to the mother. Figure 5 suggests that the shorter that the delay between the administration to the mother and delivery is, the earlier the dose should be given to the neonates. Neonates had a very long half-life, 110 h, compared to adults. If the neonate was born at least 30 min after maternal drug administration, the neonatal nevirapine concentration remained above the IC50 for at least 72 h for all children. If the neonate was born at least 2 h after maternal drug administration, the neonatal nevirapine concentration remained above 10 times the IC50 for at least 72 h for all children.
FIG. 5.

Simulated concentration-time courses of NVP in neonates before dosing for different delays between the time of maternal intake and delivery: (i) 30 min, (ii) 2 h, (iii) 5 h, and (iv) 8 h. Horizontal solid lines, IC50; horizontal dashed line, 10 times the IC50.
DISCUSSION
In the present study, NVP mother and child pharmacokinetics were satisfactorily described by the proposed compartmental model. The following observations support the validity of this model: the population predicted maternal, cord blood, and neonatal concentrations were well correlated with the observed concentrations. The population model was validated by the visual predictive check method and the fact that the values of the typical pharmacokinetic parameters were consistent with those from prior pharmacokinetic studies: for CL/F, 1.42 liters/h in the present study versus 1.40 liters/h in the study of Kunz et al. (17), and for the volume of distribution, 87.3 liters in the present study versus 104.3 liters in the study of Kunz et al. (17).
The elimination of the sdNVP in parturient women is close to the elimination of sdNVP in nonpregnant women. Because CYP 3A4 is upregulated during pregnancy (1, 23), we could expect a more rapid elimination in the women in our study. One explanation for no difference could be that upregulation of CYPs is rapidly reversed after delivery, which is in agreement with recently published data on lopinavir (5), where much higher levels are already found a few days after delivery. Therefore, NVP elimination is mainly determined after delivery instead of during pregnancy.
The long elimination half-life of NVP leads to a long period of decreasing plasma levels. This persistence of NVP predisposes the individual to resistance mutation development (9). Although the minimum concentrations needed to select for the development of resistance is not known, concentrations near the IC50 were supposed to be sufficient. According to our simulations, these concentrations remain above the IC50 for almost 3 weeks in some women, which is in agreement with the findings of the studies of Cressey et al. (9) and Kunz et al. (17). These results suggest that longer treatment with tenofovir-emtricitabine postpartum should be evaluated to prevent the appearance of resistance. These suggestions were based on the plasma concentrations. However, due to a long intracellular half-life, its activity may be much longer and the associated inhibition of viral replication still prevents development of NVP resistance. However, WHO recommends that zidovudine plus lamivudine and not tenofovir-emtricitabine be given for 7 days postpartum (http://www.who.int/hiv/pub/mtct/en/arvdrugswomenguidelinesfinal.pdf.). Longer treatment with these drugs should also be considered.
Few data on NVP placental transfer were reported. In this study, from one sample for each mother-cord pair obtained at delivery (and at various times after drug administration), we were able to assess maternal and cord blood concentrations over time. Placental transfer was estimated as the ratio of the fetal exposure-to-maternal exposure to the drug. We found a relatively constant AUC ratio of 75%, while in other studies, the placental transfer is described by a simple cord blood concentration-to-maternal concentration ratio, which is highly variable: from 71.9 to 122% in the study of Mirochnick et al. (20) and from 57.6% to 93.0% in the study of Musoke et al. (21).
Elimination from the neonate was very slow compared to that from their mothers and to that from older children. This low clearance is in agreement with the developmental pattern of the activity of CYP 3A4, the main enzyme responsible for NVP biotransformation. CYP 3A4 activity appears to be low in newborns, increases to adult levels by 6 to 12 months of age, exceeds adult levels during years 1 to 4, and then declines to adult levels by the end of puberty (18). In contrast, CYP 3A7 activity is high in utero and low during postnatal life. Little is known about the development of CYP 2B6 activity.
In order to keep neonatal concentrations above the IC50, according to our simulations and in agreement with the nevirapine (Viramune) labeling (http://www.pmtctdonations.org/ftp/Guidelines%20use%20of%20VIRAMUNE-EN.pdf.), the newborns have to be dosed (i) within 48 to 72 h of life if the child is born more than 2 h after the mother's intake and (ii) immediately after birth if the child is born less than 30 min after the mother's intake. Contrary to the nevirapine labeling (which recommends administration immediately after birth), our simulations suggested that if the child is born between 30 min and 2 h after the mother's intake, the administration to the newborn is still possible within 48 to 72 h of life.
In conclusion, our model accurately describes the NVP pharmacokinetics in mother, cord, and neonatal blood after the intrapartum and neonatal single dose. We estimated the placental transfer of NVP using an exposure ratio of 75%. NVP concentrations remained above the IC50 for 2 to 3 weeks, and administration of tenofovir-emtricitabine (or the recommended zidovudine plus lamivudine) for at least 3 weeks after delivery could improve the prevention of the emergence of resistant viruses. This hypothesis should be tested, and the efficacy of longer postpartum treatments covering the entire NVP tail needs to be determined. To keep NVP concentrations above the IC50, newborns have to be dosed (i) within 48 to 72 h of life if the child is born more than 30 min after the mother's intake and (ii) immediately after birth if the child is born less than 30 min after the mother's intake. These assumptions should be further evaluated.
Acknowledgments
We acknowledge the French Agence Nationale de Recherche contre le VIH/SIDA et les Hepatitis Virales (ANRS) for sponsoring the trial, as well as the European and Developing Clinical Trials Partnership (EDCTP) for additional financial support. We acknowledge Gilead Sciences for providing the study drugs. Didier K. Ekouévi was an EDCTP senior fellow (2005 to 2007).
We greatly thank the local investigators and their staff in the Formations Sanitaires Urbaines de Youpougon and Abobo and the Centre Hospitalier Universitaire de Yopougon in Abidjan, Côte d'Ivoire; in the Calmette Hospital and Pasteur Institute in Phnom Penh, Cambodia; and the Perinatal HIV Research Unit and Lesedi Clinic in Soweto, South Africa. We also thank the women who agreed to participate in the trial and their infants.
The TEmAA trial group is constituted as follows: the principal investigators are Francois Dabis (Bordeaux, France) and Didier K. Ekouevi (Abidjan, Cote d'Ivoire). Coinvestigators are Christine Rouzioux, Stéphane Blanche, Jean-Marc Treluyer, Marie-Laure Chaix, and Elisabeth Rey (Paris, France); N′dri-Yoman (Abidjan, Côte d'Ivoire); Kruy Leang Sim and Eric Nerrienet (Phnom Penh, Cambodia); and Glenda Gray and James McIntyre (Soweto, South Africa). The trial coordinator is Elise Arrivé (Bordeaux, France). Other members of the TEmAA ANRS 12109 study group (by location and in alphabetical order) are as follows: in Paris, France, Déborah Hirt and Saik Urien; in Abidjan, Côte d'Ivoire, Gérard Allou, Clarisse Amani-Bosse, Divine Avit, Gédéon Bédikou, Kouakou Brou, Patrick Coffié, Patrice Fian, Eulalie Kanga, Mamourou Kone, Broulaye Kone, Suzanne Kouadio, Guy César Kouaho, Jeanne Eliam Kouakou, Sidonie Ngatchou, Touré Pety, and Zenica Seoue; in Phnom Penh, Cambodia, Laurence Borand, Kearena Chhim, Pinn Chou, Meng Ly Ek, Viseth Horm Srey, Seng Hout, Sethikar Im, Saroeum Keo, Vannith Lim, Sopheak Ngin, Vara Ouk, Vibol Ung, and the Magna and Maryknoll associations; and in Soweto, South Africa, Gail Ashford, Portia Duma, Promise Duma, Sarita Lalsab, Shini Legote, Tshepiso Mabena, Joseph Makhura, Modise Maphutha, Selvan Naidoo, and Mandisa Nyati. The scientific board consists of Bernard Koffi Ngoran (Abidjan, Côte d'Ivoire), Koum Kanal (Phnom Penh, Cambodia), Lynn Morris (Johannesburg, South Africa), Séverine Blesson (ANRS, Paris, France), Camille Aubron-Olivier (Gilead Sciences, Paris, France), Gilles Peytavin (Paris, France), Koen Van Rompay (Davis, CA), and Valériane Leroy (Bordeaux, France). The independent committee comprises John Sullivan (Worcester, MA), Philippe Lepage (Brussels, Belgium), Laurent Mandelbrot (Paris, France), Marie-Louise Newell (London, United Kingdom), and Anne-Marie Taburet (Paris, France).
Footnotes
Published ahead of print on 18 October 2010.
REFERENCES
- 1.Acosta, E. P., A. Bardeguez, C. D. Zorrilla, R. Van Dyke, M. D. Hugues, S. Huang, L. Pompeo, A. M. Stek, J. Pitt, D. H. Watts, E. Smith, E. Jimenez, L. Mofenson, and the Pediatric AIDS Clinical Trials Group 386 Protocol Team. 2004. Pharmacokinetics of saquinavir plus low-dose ritonavir in human immunodeficiency virus-infected pregnant women. Antimicrob. Agents Chemother. 48:430-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arrivé, E., M. L. Newell, D. K. Ekouevi, M. L. Chaix, R. Thiebaut, B. Masquelier, V. Leroy, P. V. Perre, C. Rouzioux, F. Dabis, and the Ghent Group on HIV in Women and Children. 2007. Prevalence of resistance to nevirapine in mothers and children after single-dose exposure to prevent vertical transmission of HIV-1: a meta-analysis. Int. J. Epidemiol. 36:1009-1021. [DOI] [PubMed] [Google Scholar]
- 3.Beal, S. L. 2001. Ways to fit a PK model with some data below the quantification limit. J. Pharmacokinet. Pharmacodyn. 28:481-504. [DOI] [PubMed] [Google Scholar]
- 4.Beal, S. L., and L. B. Sheiner. 1998. NONMEM user's guide. NONMEM Project Group, University of California, San Francisco, San Francisco, CA.
- 5.Best, B. M., A. M. Stek, M. Mirochnick, C. Hu, H. Li, S. K. Burchett, S. S. Rossi, E. Smith, J. S. Read, E. V. Capparelli, and the International Maternal Pediatric Adolescent AIDS Clinical Trials Group 1026s Study Team. 2010. Lopinavir tablet pharmacokinetics with an increased dose during pregnancy. J. Acquir. Immune Defic. Syndr. 54:381-388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chi, B. H., M. Sinkala, F. Mbewe, R. A. Cantrell, G. Kruse, N. Chintu, G. M. Aldrovandi, E. M. Stringer, C. Kankasa, J. T. Safrit, and J. S. Stringer. 2007. Single-dose tenofovir and emtricitabine for reduction of viral resistance to nonnucleoside reverse transcriptase inhibitor drugs in women given intrapartum nevirapine for perinatal HIV prevention: an open-label randomised trial. Lancet 370:1698-1705. [DOI] [PubMed] [Google Scholar]
- 7.Coffie, P. A., D. K. Ekouevi, M. L. Chaix, B. Tonwe-Gold, A. B. Clarisse, R. Becquet, I. Viho, T. N′dri-Yoman, V. Leroy, E. J. Abrams, C. Rouzioux, and F. Dabis. 2008. Maternal 12-month response to antiretroviral therapy following prevention of mother-to-child transmission of HIV type 1, Ivory Coast, 2003-2006. Clin. Infect. Dis. 46:622-624. [DOI] [PubMed] [Google Scholar]
- 8.Cooper, C. L., and R. P. G. van Heeswijk. 2007. Once-daily nevirapine dosing: a pharmacokinetics, efficacy and safety review. HIV Med. 8:1-7. [DOI] [PubMed] [Google Scholar]
- 9.Cressey, T. R., G. Jourdain, M. J. Lallemant, S. Kunkeaw, J. B. Jackson, P. Musoke, E. Capparelli, and M. Mirochnick. 2005. Persistence of nevirapine exposure during the postpartum period after intrapartum single-dose nevirapine in addition to zidovudine prophylaxis for the prevention of mother-to-child transmission of HIV-1. J. Acquir. Immune Defic. Syndr. 38:283-288. [PubMed] [Google Scholar]
- 10.Eshleman, S. H., D. R. Hoover, S. Chen, S. E. Hudelson, L. A. Guay, A. Mwatha, S. A. Fiscus, F. Mmiro, P. Musoke, J. B. Jackson, N. Kumwenda, and T. Taha. 2005. Nevirapine (NVP) resistance in women with HIV-1 subtype C, compared with subtypes A and D, after the administration of single-dose NVP. J. Infect. Dis. 192:30-36. [DOI] [PubMed] [Google Scholar]
- 11.Eun Ahn, J., M. O. Karlsson, A. Dunne, and T. M. Ludden. 2008. Likelihood based approaches to handling data below the quantification limit using NONMEM VI. J. Pharmacokinet. Pharmacodyn. 35:401-442. [DOI] [PubMed] [Google Scholar]
- 12.Hirt, D., S. Urien, D. K. Ekouévi, E. Rey, E. Arrivé, S. Blanche, C. Amani-Bosse, E. Nerrienet, G. Gray, M. Kone, S. K. Leang, J. McIntyre, F. Dabis, and J. M. Tréluyer. 2009. Population pharmacokinetics of tenofovir in HIV-1-infected pregnant women and their neonates (ANRS 12109). ANRS 12109. Clin. Pharmacol. Ther. 85:182-189. [DOI] [PubMed] [Google Scholar]
- 13.Hirt, D., S. Urien, E. Rey, E. Arrivé, D. K. Ekouévi, P. Coffié, S. K. Leang, S. Lalsab, D. Avit, E. Nerrienet, J. McIntyre, S. Blanche, F. Dabis, and J. M. Tréluyer. 2009. Population pharmacokinetics of emtricitabine in human immunodeficiency virus type 1-infected pregnant women and their neonates. Antimicrob. Agents Chemother. 53:1067-1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jackson, J. B., G. Becker-Pergola, L. A. Guay, P. Musoke, M. Mracna, M. G. Fowler, L. M. Mofenson, M. Mirochnick, F. Mmiro, and S. H. Eshleman. 2000. Identification of the K103N resistance mutation in Ugandan women receiving nevirapine to prevent HIV-1 vertical transmission. AIDS 14:F111-F115. [DOI] [PubMed] [Google Scholar]
- 15.Jourdain, G., N. Ngo-Giang-Huong, S. Le Coeur, C. Bowonwatanuwong, P. Kantipong, P. Leechanachai, S. Ariyadej, P. Leenasirimakul, S. Hammer, M. Lallemant, and the Perinatal HIV Prevention Trial Group. 2004. Intrapartum exposure to nevirapine and subsequent maternal responses to nevirapine-based antiretroviral therapy. N. Engl. J. Med. 351:229-240. [DOI] [PubMed] [Google Scholar]
- 16.Reference deleted.
- 17.Kunz, A., M. Frank, K. Mugenyi, R. Kabasinguzi, A. Weidenhammer, M. Kurowski, C. Kloft, and G. Harms. 2009. Persistence of nevirapine in breast milk and plasma of mothers and their children after single-dose administration. J. Antimicrob. Chemother. 63:170-177. [DOI] [PubMed] [Google Scholar]
- 18.Leeder, J. S., and G. L. Kearns. 1997. Pharmacogenetics in pediatrics: implications for practice. Pediatr. Clin. North Am. 44:55-77. [DOI] [PubMed] [Google Scholar]
- 19.Lockman, S., R. L. Shapiro, L. M. Smeaton, C. Wester, I. Thior, L. Stevens, F. Chand, J. Makhema, C. Moffat, A. Asmelash, P. Ndase, P. Arimi, E. van Widenfelt, L. Mazhani, V. Novitsky, S. Lagakos, and M. Essex. 2007. Response to antiretroviral therapy after a single, peripartum dose of nevirapine. N. Engl. J. Med. 356:135-147. [DOI] [PubMed] [Google Scholar]
- 20.Mirochnick, M., T. Fenton, P. Gagnier, J. Pav, M. Gwynne, S. Siminski, R. S. Sperling, K. Beckerman, E. Jimenez, R. Yogev, S. A. Spector, and J. L. Sullivan for the Pediatric AIDS Clinical Trials Group Protocol 250 Team. 1998. Pharmacokinetics of nevirapine in human immunodeficiency virus type 1-infected pregnant women and their neonates. J. Infect. Dis. 178:368-374. [DOI] [PubMed] [Google Scholar]
- 21.Musoke, P., L. A. Guay, D. Bagenda, M. Mirochnick, C. Nakabiito, T. Fleming, T. Elliott, S. Horton, K. Dransfield, J. W. Pav, A. Murarka, M. Allen, M. G. Fowler, L. Mofenson, D. Hom, F. Mmiro, and J. B. Jackson. 1999. A phase I/II study of safety and pharmacokinetics of nevirapine in pregnant Ugandan women and their neonates (HIVNET 006). AIDS 13:479-486. [DOI] [PubMed] [Google Scholar]
- 22.Riska, P., M. Lamson, T. Macgregort, J. Sabo, S. Hattox, J. Pav, and J. Keirns. 1999. Disposition and biotransformation of the antiretroviral drug nevirapine in humans. Drug Metab. Dispos. 27:895-901. [PubMed] [Google Scholar]
- 23.Stek, A. M., M. Mirochnick, E. Capparelli, B. M. Best, C. Hu, S. K. Burchett, C. Elgie, D. T. Holland, E. Smith, R. Tuomala, A. Cotter, and J. S. Read. 2006. Reduced lopinavir exposure during pregnancy. AIDS 20:1931-1939. [DOI] [PubMed] [Google Scholar]
- 24.Reference deleted.