

Abstract
Screening of various bisquaternary bisnaphthalimides against a variety of human pathogens revealed one compound, designated MT02, with strong inhibitory effects against Gram-positive bacteria. The MICs ranged from 0.31 μg/ml against community-acquired methicillin-resistant Staphylococcus aureus (MRSA) lineage USA300 to 20 μg/ml against Streptococcus pneumoniae. Radioactive whole-cell labeling experiments indicated a strong impact of MT02 on bacterial DNA replication. DNA microarray studies generated a transcriptional signature characterized by stronger expression of genes involved in DNA metabolism, DNA replication, SOS response, and transport of positively charged compounds. Furthermore, surface plasmon resonance and gel retardation experiments demonstrated direct binding of MT02 to DNA in a concentration-dependent, reversible, and non-sequence-specific manner. The data presented suggest that the bisquaternary bisnaphthalimide MT02 exerts anti-Gram-positive activity by binding to DNA and thereby preventing appropriate DNA replication.
The ongoing spread of multidrug-resistant bacteria demands an intensive search for new antibacterial substances. Although various novel drugs, especially against Gram-positive pathogens, have been introduced into the market during the past decade, the prevalence of multidrug-resistant staphylococci is still high, and the recent emergence of methicillin-resistant Staphylococcus aureus (MRSA) in the community has raised severe concerns around the globe (10). Promising introductions of new antibiotics on the market, such as the oxazolidinone linezolid and the cyclic lipopeptide daptomycin, could not significantly improve the outcomes of infections associated with multidrug-resistant Gram-positive pathogens. In fact, the emergence of resistant clones during therapy may limit the use of these drugs in the future (20, 24, 39).
The discovery of completely new substance classes with antibiotic potency is a challenging process characterized by a very poor success rate (29). Thus, the screening of already known and slightly derivatized compounds for potential antimicrobial activity seems to be a promising alternative approach applied by pharmaceutical industry and academic research initiatives. Within the framework of a broad screening program for substances active against pathogenic microorganisms, bisquaternary bisnaphthalimides have recently been identified as inhibitory to Plasmodium falciparum (36). Originally, bisnaphthalimides were described as putative anticancer drugs in 1993 by Braña et al. (4). The bistertiary derivatives of this compound are known to have high antitumoral activity against both murine and human tumor cells (5). By providing a planar aromatic moiety, these substances intercalate within the DNA via the major groove (33). Whereas the monomeric 3-nitro- and 3-amino-substituted compounds mitonafide and amonafide did not show sufficient activity in clinical trials (35), the bissecondary analogues elinafide and bisnafide, consisting of a basic linker with a C2-Nsec-C2,3-Nsec-C2 motif (Fig. 1A), exhibited high in vitro antitumoral activity, intercalating twice. Elinafide was transferred to clinical trials against solid tumors but did not succeed (5).
FIG. 1.

Chemical structures of the bissecondary bisnaphthalimides elinafide and bisnafide (A) and the bisquaternary bisnaphthalimide MT02 (B).
By analyzing a series of bisquaternary bisnaphthalimides, Tischer et al. found that members of this compound class are active against the causative agent of malaria, P. falciparum, likely by interfering with phosphatidylcholine biosynthesis (36). Structure-activity relationship (SAR) analysis revealed that a long methylene middle chain of at least eight methylene groups between the two bisquaternary naphthalimides or a quaternary naphthalimide consisting of a long alkyl chain attached to the positively charged nitrogen atom is important for anti-Plasmodium activity (36). Importantly, the substances did not show cytotoxic activity in cell culture systems (27). Further screening against Gram-positive and Gram-negative pathogens elicited high activity of one of these bisquaternary bisnaphthalimides, designated MT02 (Fig. 1B), against Gram-positive bacteria. The aim of the present work was to characterize the antimicrobial activity of MT02 and to decipher the mode of action of the substance against S. aureus. It could be shown that MT02 interacts with bacterial DNA, thereby inhibiting the growth of S. aureus. Moreover, DNA microarray studies revealed strong induction of genes that are involved in DNA metabolism, SOS response, and transport of charged compounds.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
All strains used in this study are listed in Table 1. Except for Streptococcus pneumoniae isolates (see below), all of them were grown in Mueller-Hinton (MH) broth (Fluka, Roth) at 37°C and 220 rpm. MT02 was synthesized as reported previously (36). For all studies described here, compounds were taken from a stock solution of 20 mg/ml in 100% dimethyl sulfoxide (DMSO).
TABLE 1.
MICs of tested Gram-positive bacteria against MT02
| Strain | MIC (μg/ml) |
|---|---|
| S. aureus | |
| 325 | 0.63 |
| 8325 | 2.5 |
| Xen29 | 1.25 |
| MA12 | 1.25 |
| COL | 2.5 |
| RN1 | 1.25 |
| HG001 | 0.63 |
| Newman | 0.63 |
| USA300 | 0.31 |
| SH1000 | 1.25 |
| 113 | 1.25 |
| 8 clinical isolates | 0.15-5 |
| Staphylococcus epidermidis | |
| RP62A | 5 |
| 567 | 2.5 |
| Streptococcus suis S2 | 2.5 |
| Streptococcus canis S23 | 5 |
| Streptococcus equi S31 | 10 |
| Streptococcus agalactiae | |
| S44 | 10 |
| S45 | 2.5 |
| S. pneumoniae | |
| 37wt | 20 |
| 111 | 5 |
| 165 | 20 |
| B. subtilis strain 168 | 5 |
| L. monocytogenes | 5 |
Determination of MIC values.
The MICs of the compounds were determined as follows, except for S. pneumoniae isolates. Serial 2-fold dilutions of the compounds were prepared in MH broth in sterile glass tubes. Bacteria in mid-exponential growth phase at an optical density at 600 nm of 0.5 to 0.6 were added to a final inoculum of 5 × 105 CFU/ml and a total volume of 1 ml. The glass tubes were incubated with shaking at 220 rpm and 37°C for 18 to 24 h. The MIC was defined as the lowest concentration of a compound that completely inhibited bacterial growth. As to different physiological requirements, S. pneumoniae isolates were grown in Todd-Hewitt-yeast broth and incubated under microaerophilic, static conditions. Staphylococcus strains tested under S. pneumoniae growth conditions did not show aberrations in MIC values. Also, MICs for S. aureus did not change when MH broth was supplemented with 10% fetal calf serum (FCS).
Cytotoxicity test.
Cytotoxicity tests were done as described previously (31). J774.1 murine macrophage cells, human embryonic kidney 293 cells, A549 human adenocarcinoma alveolar basal epithelial cells, and Caco-2 human epithelial colorectal adenocarcinoma cells were cultured in complete medium (RPMI with NaHCO3, 10% FCS, 2 mM glutamine, 10 mM HEPES, pH 7.2, 100 U/ml penicillin, 50 μg/ml gentamicin, 50 μM 2-mercaptoethanol) without phenol red in the absence or presence of increasing concentrations of MT02 at a cell density of 1 × 105 cells/ml (200 μl) for 24 h at 37°C, 5% CO2, and 95% humidity. Following the addition of 20 μl of Alamar Blue, the plates were incubated, and the optical densities were measured 24, 48, and 72 h later with a Multiskan Ascent enzyme-linked immunosorbent assay (ELISA) reader (Thermo Electron Corporation, Dreieich, Germany) using a test wavelength of 540 nm and a reference wavelength of 630 nm. Absorbance in the absence of MT02 was set as 100% of growth. The final concentration of DMSO in the medium never exceeded 1% (vol/vol) and had no effect on the proliferation of the cells. For all experiments, each drug concentration was assayed in duplicate wells.
Killing curves.
Four flasks containing 25 ml of MH broth each were inoculated with 1 × 106 CFU/ml of S. aureus strain HG001, supplemented with 0, 1×, 2×, and 4× MIC of MT02, respectively, and incubated with shaking at 37°C. Samples from each flask were taken at 0, 2, 4, 8, 12, 16, 20, and 24 h, diluted appropriately, and plated out in duplicate on MH agar. After incubation of the plates for 24 h, colonies were counted and the respective numbers of CFU/ml were calculated.
Radioactive whole-cell labeling.
The labeling of cells with radioactive compounds was performed as previously described (30). Briefly, S. aureus strain HG001 was grown to an optical density at 600 nm of 0.6 to 0.8 and incubated with 1 μCi/ml [methyl-3H]thymidine, 1 μCi/ml [5,6-3H]uracil, and 5 μCi/ml [4,5-3H]leucine for analysis of DNA, RNA, and protein metabolism, respectively. Inhibitors were added to final concentrations of 10× MIC values. After further incubation at 37°C for 30, 60, and 120 min, samples were taken, centrifuged, and washed twice with PBS buffer to remove extracellular radioactive compounds. Resuspended samples were mixed with scintillation fluid (Rotiszint Eco Plus; Roth, Karlsruhe, Germany) and analyzed using a liquid scintillation counter (Tri-Carb Liquid Scintillation Analyzer; Packard, Meriden, CT). Growth control experiments were carried out under the same conditions. The optical density at 600 nm was measured to estimate the impacts of the different antibiotics on the numbers of cells in the respective cultures during the test period.
Isolation of RNA.
For the isolation of total RNA for microarray experiments, S. aureus strain HG001 was grown to mid-log phase at an optical density at 600 nm of 0.6 to 0.8. Seven milliliters of bacterial culture was mixed with 7 ml of RNAprotect Bacteria Reagent (Qiagen, Hilden, Germany) and immediately incubated on ice. After centrifugation for 10 min at 6,000 × g and 4°C, the supernatant was discarded and the pellet was resuspended in 1 ml RLT buffer (Qiagen) supplemented with 1% (vol/vol) β-mercaptoethanol. Cells were disrupted in Lysing Matrix E (MP Biomedicals) using a FastPrep-24 (MP Biomedicals, Solon, OH), followed by cooling on ice for 2 min. After brief centrifugation, the supernatant was purified using an RNeasyMini Kit (Qiagen). To obtain pure RNA, the eluate was treated with DNase (Roche, Grenzach-Wyhlen, Germany) for 1 h at 37°C and again purified with an RNeasyMini Kit. For RNA precipitation, 1/10 sample volume of aqueous sodium acetate solution (3 M; pH 4.8) and 2.5 volumes of cold 100% ethanol were added, and the samples were incubated for 2 h at −80°C. After centrifugation (15 min; 11,000 × g; 4°C) the supernatant was carefully discarded, and the pellet was washed with 70% cold ethanol and dried at room temperature.
Microarray manufacturing and microarray design.
The microarray was manufactured by in situ synthesis of 10,807 oligonucleotide 60-mer probes (Agilent, Palo Alto, CA), selected as previously described (6). It covered >98% of all open reading frames (ORFs) annotated in strains N315 and Mu50 (23), MW2 (1) and COL (14), NCTC8325, USA300 (11), and MRSA252 and MSSA476 (19), including their respective plasmids.
Expression microarrays.
For labeled nucleic acid preparation, S. aureus strains were grown and total RNA was extracted as described above. After additional DNase treatment, the absence of remaining DNA traces was evaluated by quantitative PCR (SDS 7700; Applied Biosystems, Framingham, MA) with assays specific for 16S rRNA (32, 34). Batches of 5 μg total S. aureus HG001 RNA were labeled with Cy3 dCTP (without MT02) or with Cy5 dCTP (with MT02) using SuperScript II (Invitrogen, Basel, Switzerland) following the manufacturer's instructions. The labeled products were then purified on QiaQuick columns (Qiagen). The labeled cDNA mixture was diluted in 50 μl Agilent hybridization buffer and hybridized at a temperature of 60°C for 17 h. The slides were washed with Agilent proprietary buffers, dried under nitrogen flow, and scanned using 100% photomultiplier tube power for both wavelengths.
Microarray analysis.
Fluorescence intensities were extracted using Feature extraction software (Agilent version 8). Local background-subtracted signals were corrected for unequal dye incorporation or unequal loads of the labeled product. The algorithm consisted of a rank consistency filter and a curve fit using the default LOWESS (locally weighted linear regression) method. Data from two independent biological experiments were expressed as log10 ratios and analyzed using GeneSpring 8.0 (Silicon Genetics, Redwood City, CA). The statistical significance of differentially expressed genes was identified by analysis of variance (ANOVA) (7, 34), performed using GeneSpring, including the Benjamini and Hochberg false-discovery rate correction of 5% (P value cutoff, 0.05) and an arbitrary cutoff of 1.5-fold for expression ratios.
Validation of microarray results.
Data from microarrays were validated by semiquantitative reverse transcriptase PCR of six representative genes, sbcD, lexA, uvrB, opuCA, pbpA, and ftsL, with gyrA as a control. To achieve comparable results, cultures were grown, supplemented with MT02, and harvested, and total RNA was isolated as described above. Four micrograms of RNA was supplemented with 3 μg of random-primer hexamers (Invitrogen) and 1 μl of 10 mM dATP, dCTP, dGTP, and dTTP, respectively, and complemented with water to a total volume of 13 μl. After incubation for 5 min at 65°C and 1 min on ice, 4 μl of 5× first-strand buffer, 1 μl of 0.1 M dithiothreitol (DTT), 1 μl of RNase Out, and 1 μl of Superscript III (Invitrogen) were added. Samples were incubated for 5 min at room temperature, followed by 1 h at 65°C and 15 min at 70°C. The same amount of total cDNA was used for conventional PCR (Go-Taq; Promega, Mannheim, Germany) with primers for the genes mentioned above (Table 2), and the products were separated on 1% agarose gels by electrophoresis.
TABLE 2.
Oligonucleotide primers used in this study
| Purpose of primer | Name | Nucleotide sequence (5′ to 3′) |
|---|---|---|
| Surface plasmon resonance | Bio-TGCA | Biotin-ATATATGCATATATTTTTATATATGCATATAT |
| Bio-control | Biotin-ATATAGACTTATATTTTTATATAAGTCTATAT | |
| Gel shift | SACOL0006for | AAGCAATGGTACGTATGGCTC |
| SACOL0006rev | CTAACAAGTTAGGGAATCGAGCAG | |
| SACOL0935for | CATATGGTCCAACTGAAGCTACG | |
| SACOL0935rev | CAAACTTCGCTTTATCACCAGTG | |
| SACOL1374for | AGTTCAACTGTTCATGGTCA | |
| SACOL1374rev | GAATATGTCGCTATTATGTGTCGA | |
| Microarray validation | ftsLfor | GCAACAACCACAAACTAAGCCCGA |
| ftsLrev | TCGTTCTCAAGGCTCATCCCCTG | |
| opuCAfor | AGCACTTGCGGCCGAACAAGA | |
| opuCArev | TGGCGTATCAAATTGCACCACCT | |
| uvrBfor | TTGCGACCACTGGGTTCGAC | |
| uvrBrev | CGTATGGTCCAGGCGTTGCAGA | |
| sbcDfor | ACACATCAACAGGGAATTACGCGCT | |
| sbcDrev | CCCTTTAGCTTGACCCGCTTCCG | |
| pbpAfor | AGAGAAGCAGCCTAAACGTG | |
| pbpArev | ATGTTGATCCAGGCTCGTATG | |
| rDNAfor | ACACCAGTGGCGAAGGCGAC | |
| rDNArev | CTCCACCGCTTGTGCGGGTC |
Biosensor measurements.
The Biacore2000 system (Biacore) was used for all biosensor experiments. Biotinylated oligonucleotides (∼1,000 resonance units [RU]) were immobilized to streptavidin-coated matrices of biosensor CM5 chips as described previously (28). Interaction analyses were performed using HBS150 buffer (10 mM HEPES, pH 7.4, 150 mM NaCl). Sensorgrams were recorded at a flow rate of 10 μl/min at 25°C. The association time was set to 5 min and the dissociation time to 20 min. The set dissociation time was sufficiently long to gain basal signal levels at the end of each cycle. Therefore, the chips were not regenerated.
Evaluation of recorded sensorgrams.
All apparent binding affinities were calculated using Biaevaluation 2.2.4 software. The affinities of the interactions were calculated by fitting the kinetic data (ka [Kinetic constant of association] and kd [Kinetic constant of dissociation]) to a 1:1 Langmuir binding model [→ KD (kin, the thermodynamic constant of dissociation)]. The standard deviation of KD (kin) is <50%. Differences in binding affinities of more than a factor of 2 can therefore be considered significant.
Agarose gel retardation.
To obtain DNA fragments of defined size, PCR products with similar lengths were amplified from randomly selected genes of S. aureus strain HG001 (primer sequences are listed in Table 2). DNA (150 ng) was incubated for 5 min at room temperature with 20 μg of MT02 or with DMSO as a control. After incubation, the DNA was purified with a PCR Purification Kit (Qiagen) according to the manufacturer's instructions and subsequently used for agarose gel electrophoresis.
Microarray data accession numbers.
The complete microarray data set has been posted on the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/) under accession numbers GPL7137 for the platform design and GSE23077 for the original data set.
RESULTS
In vitro susceptibility studies.
The MIC values for the tested organisms against MT02 are summarized in Table 1. The MIC values of staphylococci are the lowest of all tested Gram-positive bacteria, ranging from 0.31 to 5 μg/ml, depending on the strain. Furthermore, MT02 is active against other Gram-positive species, such as Bacillus subtilis, Listeria monocytogenes, and streptococci. MT02 is highly active against community-acquired MRSA lineage USA300 and ciprofloxacin-resistant clinical isolates. The substance had no effect on Gram-negative bacteria, such as Citrobacter koseri, Escherichia coli, Klebsiella pneumoniae, Proteus mirabilis, Pseudomonas aeruginosa, Salmonella enterica serovar Typhimurium, Serratia marcescens, and Shigella dysenteriae. Cytotoxicity was tested against the murine cell line J-774.1 and the three human cell lines A-549, Caco-2, and 293T. Cytotoxic concentrations were 95 μg/ml, 73 μg/ml, 146 μg/ml, and >152 μg/ml, respectively. Hence, MT02 showed no cytotoxic activity against the four cell lines in antibacterial concentrations. The antibacterial activities of closely related bisquaternary bisnaphthalimides with substitutions or different lengths of the CH2 linker region were also determined against S. aureus HG001 (Fig. 2). Among them, MT02 and a compound with a 12-CH2 linker region had the highest activities against S. aureus HG001. However, there was no correlation between the length of the linker region and the antibacterial activities of the compounds.
FIG. 2.

General structure of tested bisquaternary bishaphthalimides and corresponding MIC values of S. aureus strain HG001.
Killing ability of MT02.
Time-dependent killing of S. aureus strain HG001 (18) by MT02 over a period of 24 h was investigated, and killing curves were determined. Samples were taken at the beginning of the experiment immediately after inoculation and after 2, 4, 8, 12, 16, 20, and 24 h. Thus, the impacts of 1×, 2×, and 4× MIC of MT02 on the total number of CFU/ml were studied and compared to the growth of a control culture without the compound. Supplementation of MT02 leads to inhibition of growth (Fig. 3), which results in a decrease of 3 log phases in CFU/ml of the cultures supplemented with MT02 after 12 h compared to the control culture. Whereas the bacteria are able to regrow in the presence of 1× MIC of MT02 after 12 h, supplementation of 2× and 4× MIC of MT02 leads to further reduction of living bacteria. These results suggest a bactericidal activity of MT02, as the number of live S. aureus cells is reduced by 3 log phases after 12 h of exposure to the compound.
FIG. 3.
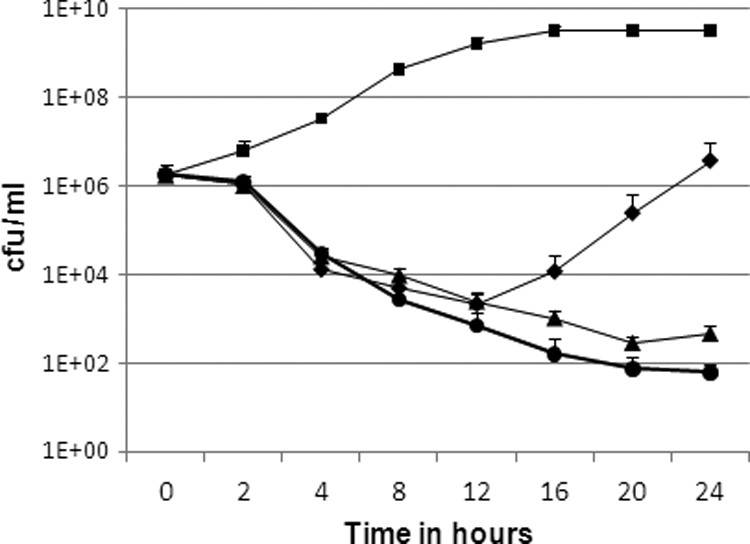
Killing curves of S. aureus HG001 without MT02 (▪), with 1× MIC MT02 (⧫), with 2× MIC MT02 (▴), and with 4× MIC MT02 (•). Mean values from three different experiments are shown. The error bars indicate standard deviations.
Impact of MT02 on major cellular pathways.
Radioactive whole-cell labeling experiments were performed to ascertain the effects of MT02 on three major cellular processes that represent target pathways of many antimicrobials, namely, protein synthesis, RNA synthesis, and DNA replication. Bacterial cultures were incubated with radioactively labeled precursors of these pathways, and the influence of MT02 and control antimicrobials on the incorporation of the radioactive compounds was measured. The labeled precursors were [4,5-3H]leucine for studies on translation, [5,6-3H]uracil for investigations on transcription, and [methyl-3H]thymidine for DNA replication studies. As control antibiotics, gentamicin, which inhibits translation and thus protein biosynthesis by binding to the 30S ribosomal subunit of bacteria; rifampin, which affects transcription by binding DNA-dependent RNA polymerase; and ciprofloxacin, which inhibits DNA replication by binding bacterial DNA gyrase and topoisomerase IV, were used. In addition, a control culture that was supplemented with a radioactive precursor but without an antibiotic substance was included. The radioactive incorporation of this control was set to 100%, and the values of the test samples were referenced accordingly. Additionally, growth controls with antibiotics but without radioactive compounds were performed to estimate possible effects of antibiotics on the overall growth of cells.
The general growth experiments (Fig. 4A) were performed with initial cell numbers of 4.5 × 108 to 6 × 108 per ml and 10× MIC of the respective antibiotic. After 30 min, only rifampin had an inhibitory effect on cell growth. All reference substances diminished growth to a certain extent after 60 min, and this effect was increased after 2 h. In contrast, MT02 did not considerably alter cell growth over the whole period.
FIG. 4.

Results of whole-cell labeling experiments with S. aureus HG001 (mean of two representative experiments). (A) Growth control. (B to D) Labeling with [methyl-3H]thymidine (B), [4,5-3H]leucine (C), and [5,6-3H]uracil (D). White bars, controls with no antibiotics; gray bars, supplementation with 10× MIC of ciprofloxacin (CIP), gentamicin (GEN), or rifampin (RIF); black bars, supplementation with 10× MIC of MT02. x axis, time after supplementation of antibiotics in minutes. OD, optical density.
Importantly, the labeling experiments with MT02 revealed a significant impact of the substance on the incorporation of [methyl-3H]thymidine, as could also be observed for ciprofloxacin (Fig. 4B). After 30 min, the two antibiotics reduced the signal intensity to less than 40% that of the control culture and even to less than 20% after 2 h. The effect of gentamicin on the incorporation of [4,5-3H]leucine was weak after 30 min but increased over the test period, resulting in an 85% reduction of signal intensity compared to the control culture after 2 h (Fig. 4C). MT02 reduced the signal intensity of [4,5-3H]leucine by 50%. Whereas rifampin decreased the incorporation of [5,6-3H]uracil in a time-dependent manner, MT02 had only a weak effect (Fig. 4D).
In summary, MT02 supplementation leads to a drastic decrease of [methyl-3H]thymidine incorporation into S. aureus cells compared to control cultures without MT02, but there is only a marginal effect on the incorporation of [4,5-3H]leucine and [5,6-3H]uracil, respectively. This strongly suggests that MT02 interferes with DNA metabolism and not with protein synthesis or transcription.
Transcriptional analysis.
In order to gain deeper insight into the mode of action of MT02, the changes in global RNA transcription were investigated by DNA microarray analysis. For this, whole-genome arrays covering more than 98% of eight S. aureus genomes were used to compare the influence of 10× MIC of MT02 after 60 min on the transcriptome of S. aureus strain HG001. In total, 112 and 196 transcripts were found to be downregulated and upregulated, respectively. Regulated genes belonging to functional categories of interest are displayed in Table 3 (for complete data, see the supplemental material). Validation of the results was obtained by semiquantitative reverse transcriptase PCR with the upregulated genes sbcD, lexA, and uvrB and the downregulated genes opuCA, pbpA, and ftsL, with gyrA as a control (Fig. 5).
TABLE 3.
Differentially regulated genes belonging to major pathways of S. aureus HG001 following treatment with 10× MIC of MT02 after 60 min
| ORF and functional group | Gene | Annotation | Fold change |
|---|---|---|---|
| DNA metabolism; upregulated ORFs | |||
| SACOL0001 | dnaA | Chromosomal replication initiation protein | 2.52 |
| SACOL0002 | dnaN | DNA polymerase III subunit beta | 2.44 |
| SACOL0004 | recF | Recombination protein RecF | 2.60 |
| SACOL0005 | gyrB | DNA gyrase subunit B | 2.24 |
| SACOL0823 | uvrB | Exinuclease ABC subunit B | 4.36 |
| SACOL0824 | uvrA | Exinuclease ABC subunit A | 3.66 |
| SACOL1304 | recA | Recombinase A | 4.79 |
| SACOL1374 | lexA | LexA repressor | 6.85 |
| SACOL1382 | sbcC | Exonuclease SbcC | 4.96 |
| SACOL1381 | sbcD | SbcD nuclease | 4.51 |
| SACOL1400 | UmuC family polymerase | 18.05 | |
| SACOL1401 | UmuC family polymerase | 25.41 | |
| SACOL2089 | Similar to single-stranded DNA-binding protein | 2.18 | |
| SACOL2727 | Integrase/recombinase, core domain family | 2.06 | |
| Phage related; upregulated ORFs | |||
| SACOL0318 | int | Prophage L54a, integrase | 5.00 |
| SACOL0321 | Prophage L54a, repressor protein, putative | 2.64 | |
| SACOL0325 | Prophage L54a, antirepressor, putative | 48.05 | |
| SACOL0335 | Hypothetical protein | 29.53 | |
| SACOL0336 | Phi PVL ORF 39-like protein | 28.77 | |
| SACOL0343 | Prophage L54a, replicative DNA helicase, putative | 21.63 | |
| SACOL0348 | Conserved hypothetical protein | 12.03 | |
| SACOL0357 | dut | Prophage L54a, deoxyuridine 5′-triphosphate nucleotidohydrolase | 7.31 |
| SACOL0358 | Hypothetical protein | 6.55 | |
| SACOL0361 | Hypothetical protein | 5.33 | |
| SACOL0369 | Prophage L54a, Clp protease, putative | 4.28 | |
| LexA-regulated genes; upregulated ORFs | |||
| SACOL0436 | Hypothetical protein | 18.02 | |
| SACOL0437 | Hypothetical protein | 24.31 | |
| SACOL0823 | uvrB | Exinuclease ABC subunit B | 4.36 |
| SACOL0824 | uvrA | Exinuclease ABC subunit A | 3.66 |
| SACOL1304 | recA | Recombinase A | 4.79 |
| SACOL1374 | lexA | LexA repressor | 6.85 |
| SACOL1375 | Hypothetical protein | 57.47 | |
| SACOL1381 | sbcD | Hypothetical protein | 4.53 |
| SACOL1382 | sbcC | Exonuclease SbcC | 4.96 |
| SACOL1400 | Hypothetical protein | 18.05 | |
| SACOL1401 | Hypothetical protein | 25.41 | |
| SACOL1986 | Hypothetical protein | 16.84 | |
| SACOL1987 | Hypothetical protein | 17.21 | |
| SACOL2162 | Hypothetical protein | 3.88 | |
| Transport- and cell wall-related genes | |||
| Upregulated ORF | |||
| SACOL2525 | ABC transporter, ATP-binding protein | 4.27 | |
| Downregulated ORFs | |||
| SACOL0079 | Hypothetical protein | 0.43 | |
| SACOL0480 | Hypothetical protein | 0.45 | |
| SACOL0682 | Putative monovalent cation/H+ antiporter subunit D | 0.45 | |
| SACOL0684 | Putative monovalent cation/H+ antiporter subunit E | 0.46 | |
| SACOL0991 | oppB | Oligopeptide transport system permease protein | 0.30 |
| SACOL0994 | oppF | Hypothetical protein | 0.32 |
| SACOL1193 | ftsL | Cell division protein | 0.33 |
| SACOL1195 | mraY | Phospho-N-muramic acid-pentapeptide translocase | 0.35 |
| SACOL1196 | murD | UDP-N-acetylmuramoyl-l-alanyl-d-glutamate synthetase | 0.43 |
| SACOL1319 | glpF | Glycerol uptake facilitator | 0.45 |
| SACOL2450 | opuCD | Probable glycine betaine/carnitine/choline ABC transporter OpuCD | 0.43 |
| SACOL2452 | opuCB | Probable glycine betaine/carnitine/choline ABC transporter OpuCB | 0.25 |
| SACOL2453 | opuCA | Glycine betaine/carnitine/choline ABC transporter OpuCA | 0.21 |
| SACOL2475 | Peptide ABC transporter, permease protein, putative | 0.34 | |
| Nucleotide metabolism; upregulated ORFs | |||
| SACOL0791 | nrdI | Ribonucleotide reductase stimulatory protein | 5.89 |
| SACOL0792 | nrdE | Ribonucleotide-diphosphate reductase subunit alpha | 5.68 |
| SACOL0793 | nrdF | Ribonucleotide-diphosphate reductase subunit beta | 4.80 |
| SACOL2606 | pyrD | Dihydroorotate dehydrogenase 2 | 2.50 |
| Toxin production/resistance and pathogenesis | |||
| Upregulated ORFs | |||
| SACOL0096 | sarS | Staphylococcal accessory regulator S | 4.26 |
| SACOL0103 | Hypothetical protein | 2.15 | |
| SACOL2326 | fosB | Fosfomycin resistance protein FosB | 2.38 |
| SACOL2712 | drp35 | Drp35 protein | 2.42 |
| Downregulated ORFs | |||
| SACOL1173 | hla | Alpha-hemolysin precursor | 0.41 |
| SACOL1194 | pbpA | Penicillin-binding protein 1 | 0.31 |
| SACOL2023 | agrB | Accessory gene regulator protein B | 0.36 |
| SACOL2026 | agrA | Accessory gene regulator protein A | 0.33 |
FIG. 5.

Validation of microarray data by semiquantitative RT-PCR. cDNA was synthesized from total RNA isolated from cultures with (+) or without (−) supplementation with MT02 and used as a template for standard-PCR amplification with primers for the respective genes.
The major group of genes regulated under the influence of MT02 represents genes related to DNA metabolism. For example, the upregulation of genes coding for a chromosomal replication initiation protein (dnaA), DNA polymerase III subunit beta (dnaN), DNA gyrase subunit B (gyrB), and a protein similar to single-stranded DNA-binding protein (SACOL2089) reflects the impact of MT02 on DNA replication. Additionally, genes such as sbcC and sbcD, which are involved in DNA repair mechanisms, were upregulated in the presence of inhibitory concentrations of MT02. In line with that, the upregulation of the LexA repressor gene reveals that mechanisms of DNA repair via the SOS response system are induced by MT02. As a consequence, many genes regulated by LexA are found to be upregulated, as well as the exonuclease genes uvrA and uvrB; recA, the gene coding for recombinase A; and recF, the gene coding for the recombinase protein RecF. Finally, a number of phage genes are induced in the presence of MT02, some of which are listed in Table 3. Among them are the genes for a phage integrase (SACOL0318), a putative antirepressor (SACOL0325), and a putative phage helicase (SACOL0343). Overall, 29 phage-related genes and 14 genes related to the SOS response were upregulated after treatment with MT02. Previous studies concluded that DNA-active substances like ciprofloxacin and the DNA cross-linker ELB-21 strongly induce phage proteins in S. aureus (12, 15). These data are in accordance with the results presented here, indicating interference of MT02 with DNA metabolism.
In addition, several membrane proteins, mostly ABC transporters and antiporters (Table 3), were downregulated in the presence of MT02. These classes of proteins are known to be effective transporters of quaternary nitrogen-containing compounds (17). Since MT02 also contains positively charged quaternary nitrogen atoms, those transporters may be involved in the uptake of the compound into the cells, and their downregulation can be regarded as an effort by S. aureus to evade the substance. The genes mraY and murD, which are involved in the transport of cell wall precursors across the membrane and in murein biosynthesis, are also downregulated. In addition, fosB and pbpA, coding for a fosfomycin and a penicillin resistance protein, respectively, are upregulated in the presence of inhibitory concentrations of MT02. Furthermore, drp35, which is known to be induced in S. aureus by cell wall-affecting antibiotics (25), is also upregulated. In contrast, ftsL, which is essential for cell division, was downregulated. Overall, these results suggest an indirect impact of MT02 on cell wall metabolism and cell propagation.
Binding of MT02 to DNA.
Surface plasmon resonance was chosen as a method to investigate direct interactions of MT02 and DNA. For this, binding chips were first coated with streptavidin, and biotinylated double-stranded oligonucleotides (Table 2) were bound to the streptavidin surface. One oligonucleotide contained the sequence 5′-GATC-3′, which was reported to be a specific binding motif of the bisnaphthalimide elinafide (16), and another oligonucleotide contained the sequence 5′-GACT-3′, which was not bound by elinafide. The signal baseline was detected, and after 150 s, solutions with concentrations of 40 to 200 nM MT02 were run through the flowthrough chamber. For both oligonucleotides tested, this was followed by an increase in detected resonance units that was dependent on the concentration of MT02 (Fig. 6). Higher concentrations thus resulted in a faster increase of the signal. After a certain time, the signal reached a maximum value, which was attained earlier when higher concentrations of MT02 were supplemented. These results show that the compound binds DNA in a concentration-dependent manner until saturation occurs. A comparison of the maximum signal levels obtained by surface plasmon resonance studies with the two different oligonucleotides revealed that more molecules of MT02 bound to the oligonucleotide with the 5′-GATC-3′ sequence than to the 5′-GACT-3′ oligonucleotide. After 500 s, the flowthrough chamber was run with buffer again, resulting in a decrease in detected resonance units, implying the withdrawal of the compound from the DNA. This shows that binding of MT02 to DNA is a reversible process. Evaluation of the data obtained revealed thermodynamic dissociation constants of 5.81 × 10−8 mol/liter for the oligonucleotide with the 5′-GATC-3′ sequence and 1.95 × 10−8 mol/liter for the 5′-GACT-3′ oligonucleotide. In summary, the concentration of MT02 required for half saturation of the DNA lies within the nanomolar range for both DNA fragments.
FIG. 6.

Surface plasmon resonance for binding of MT02 to the control oligonucleotide (A) and the oligonucleotide with GATC sequence (B). MT02 concentrations ranged from 40 nM in the bottom curve to 200 nM in the top curve.
In addition to this analysis by surface plasmon resonance, the interaction of MT02 and DNA was investigated by gel retardation experiments. Different fragments of about 250 bp of the three genes SACOL0006, SACOL0935, and SACOL1374 were amplified by PCR to exclude any specific interaction of MT02 with a conserved target DNA sequence. After incubation of the DNA fragments with MT02 and purification, the fragments were analyzed for their gel retardation characteristics in agarose gel electrophoresis in comparison to untreated PCR products. All fragments that were incubated with MT02 were apparently larger than their respective control samples (Fig. 7). It was not possible to define the exact sizes of the fragments, as the DNA appeared as a smear on the gel. Furthermore, the apparent sizes of the treated fragments did not differ among themselves, meaning that the apparently smallest size was that of the untreated fragment. There was a sharp limit at the upper end of the smear that was observed for all PCR products at the same apparent fragment length. This finding is consistent with the results of the surface plasmon resonance experiments, as this sharp limit most probably reflects the saturation of DNA with MT02. Moreover, competitive DNA intercalation experiments using ethidium bromide as the intercalator and Berenil as a positive control suggest an intercalation of MT02 into the DNA (data not shown). Taken together, these data confirm a specific interaction of MT02 with DNA that is concentration dependent, reversible, and not sequence specific.
FIG. 7.
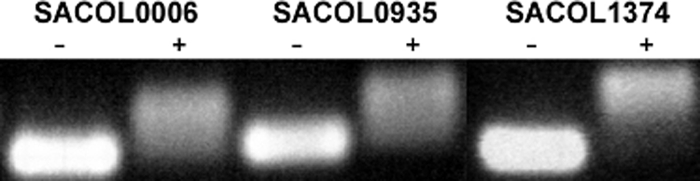
S. aureus DNA fragments amplified by PCR and incubated without (−) or with (+) MT02 prior to purification and gel electrophoresis.
DISCUSSION
In this study, the antibacterial effect of a novel bisquaternary bisnaphthalimide on Gram-positive bacteria was determined, with a focus on S. aureus. Bisnaphthalimides were previously under investigation in antitumor therapy; however, these compounds did not carry quaternary nitrogen atoms within their linker regions (3, 13, 38). The bisquaternary naphthalimides were originally developed as allosteric modulators of muscarinic receptors (2, 26). An initial cytotoxicity screening of a series of compounds with a C3-Nquart-C3-12-Nquart-C3 middle chain (Nquart = quaternary nitrogen) revealed no cytotoxicity against different cell lines (27). The MIC values of the substance MT02 against all tested Gram-positive bacteria were in the low micromolar range, and no cytotoxicity was observed against four tested cell lines. Derivatives of MT02 without the nitro groups and with shorter or longer middle chains also showed activity against S. aureus, but only at higher concentrations of the compounds. In contrast to structure-activity studies of those substances against P. falciparum (36), no relationship between the structures of the compounds and their antibacterial activities could be observed in our study. S. aureus strains with resistance against methicillin or ciprofloxacin did not show any cross-resistance against MT02. The observation that MT02 has no effect on Gram-negative bacteria may be due to the differences in the cell envelope. Most probably, MT02 is not able to pass two membranes in sufficient concentrations and is thus not able to affect Gram-negative bacteria. In contrast to bistertiary bisnaphthalimides, the two permanent positive charges of MT02 could cause its inability to penetrate eukaryotic cells and therefore its low cytotoxicity despite its binding to eukaryotic DNA. Further studies are planned to elucidate the phenomenon of selective toxicity of bisnaphthalimides owing to their individual capabilities to penetrate into different cell types. The radioactive whole-cell labeling experiments revealed that MT02 influences DNA metabolism rather than the synthesis of RNA and proteins. Together with the DNA-binding studies, these results suggest that the antibacterial potential of MT02 is due to its ability to directly bind double-stranded DNA. This process is reversible, concentration dependent, and probably not restricted to a specific base sequence, as reported for the bisnaphthalimide elinafide (16). The binding constants of MT02 to the two DNA fragments investigated here were in the low nanomolar range, comparable to those of other naphthalimides (3). However, the formerly discussed negative effect of methylation of the nitrogen atoms in the linker region of bisnaphthalimides on DNA-binding efficacy could not be confirmed (3). On the other hand, the two quaternary nitrogen atoms in the linker region of MT02 proved to be crucial for its high biological activity.
Microarray experiments revealed that under the influence of 10× MIC of MT02 a large number of genes were differentially regulated after 60 min. This is consistent with earlier studies in which the influence of ciprofloxacin on the transcriptome of S. aureus was investigated (8). It is interesting that antibiotics targeting DNA metabolism obviously have a huge impact on the transcriptomes of bacteria. Some groups of genes regulated after treatment with both ciprofloxacin and MT02 encode virulence factors, such as sarS, agrB, hlY, and genes for nucleotide metabolism, like nrdE and pyrD. Likewise, certain functional groups of genes, namely, DNA metabolism- and phage-related genes, seem to be influenced by substances interacting with DNA, which could also be observed in P. aeruginosa (9). Obviously, phage-related genes, as well as genes for the SOS response of bacteria, are highly activated following inhibition of DNA replication. In line with these results is the reported effect of antibacterial substances on activation of mobile genetic elements and the onset of the SOS response as a consequence of interference of compounds with DNA replication (21, 37).
Other clusters of genes affected by ciprofloxacin were not influenced by MT02, such as those involved in the tricarboxylic acid cycle and lipid biosynthesis, indicating different modes of action of the two substance classes. Interestingly, genes for OpuC-like transporters were found to be downregulated under the influence of MT02. These transporters are involved in the uptake of glycine betaine, carnitine, and choline and play a role in the pathogenicity of S. aureus (22). Remarkably, all of these substrates possess a quaternary nitrogen atom, suggesting that the downregulation is part of the cellular strategy to prevent uptake of MT02.
In summary, this study elucidates the mode of action of the bisquaternary bisnaphthalimide MT02 against Gram-positive bacteria, which includes direct binding of bacterial DNA. Thus, inhibition of DNA synthesis has been identified as the major killing mechanism of the drug. However, further studies will be required to determine if other mechanisms, such as interference of the positively charged compound with cell wall synthesis and the functional integrity of the cell membrane, are also involved.
Supplementary Material
Acknowledgments
We thank Sven Hammerschmidt and Claudia Rennemeier for providing us with the Streptococcus strains and Elena Katzowitsch and Svetlana Sologub for technical assistance.
This work was supported by DFG research grant SFB630.
Footnotes
Published ahead of print on 11 October 2010.
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1.Baba, T., F. Takeuchi, M. Kuroda, H. Yuzawa, K. Aoki, A. Oguchi, Y. Nagai, N. Iwama, K. Asano, T. Naimi, H. Kuroda, L. Cui, K. Yamamoto, and K. Hiramatsu. 2002. Genome and virulence determinants of high virulence community-acquired MRSA. Lancet 359:1819-1827. [DOI] [PubMed] [Google Scholar]
- 2.Bender, W., M. Staudt, C. Trankle, K. Mohr, and U. Holzgrabe. 2000. Probing the size of a hydrophobic binding pocket within the allosteric site of muscarinic acetylcholine M2-receptors. Life Sci. 66:1675-1682. [DOI] [PubMed] [Google Scholar]
- 3.Braña, M. F., M. Cacho, M. A. Garcia, B. de Pascual-Teresa, A. Ramos, M. T. Dominguez, J. M. Pozuelo, C. Abradelo, M. F. Rey-Stolle, M. Yuste, M. Banez-Coronel, and J. C. Lacal. 2004. New analogues of amonafide and elinafide, containing aromatic heterocycles: synthesis, antitumor activity, molecular modeling, and DNA binding properties. J. Med. Chem. 47:1391-1399. [DOI] [PubMed] [Google Scholar]
- 4.Braña, M. F., J. M. Castellano, M. Moran, M. J. Perez de Vega, C. R. Romerdahl, X. D. Qian, P. Bousquet, F. Emling, E. Schlick, and G. Keilhauer. 1993. Bis-naphthalimides: a new class of antitumor agents. Anticancer Drug Des. 8:257-268. [PubMed] [Google Scholar]
- 5.Braña, M. F., and A. Ramos. 2001. Naphthalimides as anti-cancer agents: synthesis and biological activity. Curr. Med. Chem. Anticancer Agents 1:237-255. [DOI] [PubMed] [Google Scholar]
- 6.Charbonnier, Y., B. Gettler, P. Francois, M. Bento, A. Renzoni, P. Vaudaux, W. Schlegel, and J. Schrenzel. 2005. A generic approach for the design of whole-genome oligoarrays, validated for genomotyping, deletion mapping and gene expression analysis on Staphylococcus aureus. BMC Genomics 6:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Churchill, G. A. 2004. Using ANOVA to analyze microarray data. Biotechniques 37:173-175, 177. [DOI] [PubMed] [Google Scholar]
- 8.Cirz, R. T., M. B. Jones, N. A. Gingles, T. D. Minogue, B. Jarrahi, S. N. Peterson, and F. E. Romesberg. 2007. Complete and SOS-mediated response of Staphylococcus aureus to the antibiotic ciprofloxacin. J. Bacteriol. 189:531-539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cirz, R. T., B. M. O'Neill, J. A. Hammond, S. R. Head, and F. E. Romesberg. 2006. Defining the Pseudomonas aeruginosa SOS response and its role in the global response to the antibiotic ciprofloxacin. J. Bacteriol. 188:7101-7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeLeo, F. R., and H. F. Chambers. 2009. Reemergence of antibiotic-resistant Staphylococcus aureus in the genomics era. J. Clin. Invest. 119:2464-2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diep, B. A., S. R. Gill, R. F. Chang, T. H. Phan, J. H. Chen, M. G. Davidson, F. Lin, J. Lin, H. A. Carleton, E. F. Mongodin, G. F. Sensabaugh, and F. Perdreau-Remington. 2006. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 367:731-739. [DOI] [PubMed] [Google Scholar]
- 12.Doyle, M., E. A. Feuerbaum, K. R. Fox, J. Hinds, D. E. Thurston, and P. W. Taylor. 2009. Response of Staphylococcus aureus to subinhibitory concentrations of a sequence-selective, DNA minor groove cross-linking pyrrolobenzodiazepine dimer. J. Antimicrob. Chemother. 64:949-959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gallego, J., and B. R. Reid. 1999. Solution structure and dynamics of a complex between DNA and the antitumor bisnaphthalimide LU-79553: intercalated ring flipping on the millisecond time scale. Biochemistry 38:15104-15115. [DOI] [PubMed] [Google Scholar]
- 14.Gill, S. R., D. E. Fouts, G. L. Archer, E. F. Mongodin, R. T. Deboy, J. Ravel, I. T. Paulsen, J. F. Kolonay, L. Brinkac, M. Beanan, R. J. Dodson, S. C. Daugherty, R. Madupu, S. V. Angiuoli, A. S. Durkin, D. H. Haft, J. Vamathevan, H. Khouri, T. Utterback, C. Lee, G. Dimitrov, L. Jiang, H. Qin, J. Weidman, K. Tran, K. Kang, I. R. Hance, K. E. Nelson, and C. M. Fraser. 2005. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J. Bacteriol. 187:2426-2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goerke, C., J. Koller, and C. Wolz. 2006. Ciprofloxacin and trimethoprim cause phage induction and virulence modulation in Staphylococcus aureus. Antimicrob. Agents Chemother. 50:171-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.González-Bulnes, L., and J. Gallego. 2009. Indirect effects modulating the interaction between DNA and a cytotoxic bisnaphthalimide reveal a two-step binding process. J. Am. Chem. Soc. 131:7781-7791. [DOI] [PubMed] [Google Scholar]
- 17.Hassan, K. A., R. A. Skurray, and M. H. Brown. 2007. Active export proteins mediating drug resistance in staphylococci. J. Mol. Microbiol. Biotechnol. 12:180-196. [DOI] [PubMed] [Google Scholar]
- 18.Herbert, S., A. K. Ziebandt, K. Ohlsen, T. Schäfer, M. Hecker, D. Albrecht, R. Novick, and F. Götz. 2010. Repair of global regulators in Staphylococcus aureus 8325 and comparative analysis with other clinical isolates. Infect. Immun. 78:2877-2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holden, M. T., E. J. Feil, J. A. Lindsay, S. J. Peacock, N. P. Day, M. C. Enright, T. J. Foster, C. E. Moore, L. Hurst, R. Atkin, A. Barron, N. Bason, S. D. Bentley, C. Chillingworth, T. Chillingworth, C. Churcher, L. Clark, C. Corton, A. Cronin, J. Doggett, L. Dowd, T. Feltwell, Z. Hance, B. Harris, H. Hauser, S. Holroyd, K. Jagels, K. D. James, N. Lennard, A. Line, R. Mayes, S. Moule, K. Mungall, D. Ormond, M. A. Quail, E. Rabbinowitsch, K. Rutherford, M. Sanders, S. Sharp, M. Simmonds, K. Stevens, S. Whitehead, B. G. Barrell, B. G. Spratt, and J. Parkhill. 2004. Complete genomes of two clinical Staphylococcus aureus strains: evidence for the rapid evolution of virulence and drug resistance. Proc. Natl. Acad. Sci. U. S. A. 101:9786-9791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones, T., M. R. Yeaman, G. Sakoulas, S. J. Yang, R. A. Proctor, H. G. Sahl, J. Schrenzel, Y. Q. Xiong, and A. S. Bayer. 2008. Failures in clinical treatment of Staphylococcus aureus infection with daptomycin are associated with alterations in surface charge, membrane phospholipid asymmetry, and drug binding. Antimicrob. Agents Chemother. 52:269-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelley, W. L. 2006. Lex marks the spot: the virulent side of SOS and a closer look at the LexA regulon. Mol. Microbiol. 62:1228-1238. [DOI] [PubMed] [Google Scholar]
- 22.Kiran, M. D., D. E. Akiyoshi, A. Giacometti, O. Cirioni, G. Scalise, and N. Balaban. 2009. OpuC—an ABC transporter that is associated with Staphylococcus aureus pathogenesis. Int. J. Artif. Organs 32:600-610. [DOI] [PubMed] [Google Scholar]
- 23.Kuroda, M., T. Ohta, I. Uchiyama, T. Baba, H. Yuzawa, I. Kobayashi, L. Cui, A. Oguchi, K. Aoki, Y. Nagai, J. Lian, T. Ito, M. Kanamori, H. Matsumaru, A. Maruyama, H. Murakami, A. Hosoyama, Y. Mizutani-Ui, N. K. Takahashi, T. Sawano, R. Inoue, C. Kaito, K. Sekimizu, H. Hirakawa, S. Kuhara, S. Goto, J. Yabuzaki, M. Kanehisa, A. Yamashita, K. Oshima, K. Furuya, C. Yoshino, T. Shiba, M. Hattori, N. Ogasawara, H. Hayashi, and K. Hiramatsu. 2001. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 357:1225-1240. [DOI] [PubMed] [Google Scholar]
- 24.Morales, G., J. J. Picazo, E. Baos, F. J. Candel, A. Arribi, B. Peláez, R. Andrade, M. A. de la Torre, J. Fereres, and M. Sánchez-García. 2010. Resistance to linezolid is mediated by the cfr gene in the first report of an outbreak of linezolid-resistant Staphylococcus aureus. Clin. Infect. Dis. 50:821-825. [DOI] [PubMed] [Google Scholar]
- 25.Murakami, H., H. Matsumaru, M. Kanamori, H. Hayashi, and T. Ohta. 1999. Cell wall-affecting antibiotics induce expression of a novel gene, drp35, in Staphylococcus aureus. Biochem. Biophys. Res. Commun. 264:348-351. [DOI] [PubMed] [Google Scholar]
- 26.Muth, M., W. Bender, O. Scharfenstein, U. Holzgrabe, E. Balatkova, C. Trankle, and K. Mohr. 2003. Systematic development of high affinity bis(ammonio)alkane-type allosteric enhancers of muscarinic ligand binding. J. Med. Chem. 46:1031-1040. [DOI] [PubMed] [Google Scholar]
- 27.Muth, M., V. Hoerr, M. Glaser, A. Ponte-Sucre, H. Moll, A. Stich, and U. Holzgrabe. 2007. Antitrypanosomal activity of quaternary naphthalimide derivatives. Bioorg. Med. Chem. Lett. 17:1590-1593. [DOI] [PubMed] [Google Scholar]
- 28.Nickel, J., A. Kotzsch, W. Sebald, and T. D. Mueller. 2005. A single residue of GDF-5 defines binding specificity to BMP receptor IB. J. Mol. Biol. 349:933-947. [DOI] [PubMed] [Google Scholar]
- 29.Ohlsen, K. 2009. Novel antibiotics for the treatment of Staphylococcus aureus. Expert Rev. Clin. Pharmacol. 2:661-672. [DOI] [PubMed] [Google Scholar]
- 30.Onishi, H. R., B. A. Pelak, L. S. Gerckens, L. L. Silver, F. M. Kahan, M. H. Chen, A. A. Patchett, S. M. Galloway, S. A. Hyland, M. S. Anderson, and C. R. Raetz. 1996. Antibacterial agents that inhibit lipid A biosynthesis. Science 274:980-982. [DOI] [PubMed] [Google Scholar]
- 31.Ponte-Sucre, A., R. Vicik, M. Schultheis, T. Schirmeister, and H. Moll. 2006. Aziridine-2,3-dicarboxylates, peptidomimetic cysteine protease inhibitors with antileishmanial activity. Antimicrob. Agents Chemother. 50:2439-2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Renzoni, A., C. Barras, P. Francois, Y. Charbonnier, E. Huggler, C. Garzoni, W. L. Kelley, P. Majcherczyk, J. Schrenzel, D. P. Lew, and P. Vaudaux. 2006. Transcriptomic and functional analysis of an autolysis-deficient, teicoplanin-resistant derivative of methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 50:3048-3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sami, S. M., R. T. Dorr, D. S. Alberts, A. M. Solyom, and W. A. Remers. 2000. Analogues of amonafide and azonafide with novel ring systems. J. Med. Chem. 43:3067-3073. [DOI] [PubMed] [Google Scholar]
- 34.Scherl, A., P. Francois, Y. Charbonnier, J. M. Deshusses, T. Koessler, A. Huyghe, M. Bento, J. Stahl-Zeng, A. Fischer, A. Masselot, A. Vaezzadeh, F. Galle, A. Renzoni, P. Vaudaux, D. Lew, C. G. Zimmermann-Ivol, P. A. Binz, J. C. Sanchez, D. F. Hochstrasser, and J. Schrenzel. 2006. Exploring glycopeptide-resistance in Staphylococcus aureus: a combined proteomics and transcriptomics approach for the identification of resistance-related markers. BMC Genomics 7:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taylor, S. A., C. Rankin, J. J. Townsend, J. B. Craig, R. B. Vance, D. L. Solank, T. D. Brown, and K. Jaeckle. 2002. Phase II trial of amonafide in central nervous system tumors: a Southwest Oncology Group study. Invest. New Drugs 20:113-115. [DOI] [PubMed] [Google Scholar]
- 36.Tischer, M., L. Sologub, G. Pradel, and U. Holzgrabe. 2010. The bisnaphthalimides as new active lead compounds against Plasmodium falciparum. Bioorg. Med. Chem. 18:2998-3003. [DOI] [PubMed] [Google Scholar]
- 37.Ubeda, C., E. Maiques, E. Knecht, I. Lasa, R. P. Novick, and J. R. Penades. 2005. Antibiotic-induced SOS response promotes horizontal dissemination of pathogenicity island-encoded virulence factors in staphylococci. Mol. Microbiol. 56:836-844. [DOI] [PubMed] [Google Scholar]
- 38.Villalona-Calero, M. A., J. P. Eder, D. L. Toppmeyer, L. F. Allen, R. Fram, R. Velagapudi, M. Myers, A. Amato, K. Kagen-Hallet, B. Razvillas, D. W. Kufe, D. D. Von Hoff, and E. K. Rowinsky. 2001. Phase I and pharmacokinetic study of LU79553, a DNA intercalating bisnaphthalimide, in patients with solid malignancies. J. Clin. Oncol. 19:857-869. [DOI] [PubMed] [Google Scholar]
- 39.Yoshida, K., H. Shoji, H. Hanaki, C. Yanagisawa, Y. Ikeda-Dantsuji, K. Fukuchi, M. Adachi, and Y. Niki. 2009. Linezolid-resistant methicillin-resistant Staphylococcus aureus isolated after long-term repeated use of linezolid. J. Infect. Chemother. 15:417-419. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.