

Abstract
Nucleoside reverse transcriptase inhibitors (NRTIs) are an important class of antiviral drugs used to manage infections by human immunodeficiency virus, which causes AIDS. Unfortunately, these drugs cause unwanted side effects, and the molecular basis of NRTI toxicity is not fully understood. Putative routes of NRTI toxicity include the inhibition of human nuclear and mitochondrial DNA polymerases. A strong correlation between mitochondrial toxicity and NRTI incorporation catalyzed by human mitochondrial DNA polymerase has been established both in vitro and in vivo. However, it remains to be determined whether NRTIs are substrates for the recently discovered human X- and Y-family DNA polymerases, which participate in DNA repair and DNA lesion bypass in vivo. Using pre-steady-state kinetic techniques, we measured the substrate specificity constants for human DNA polymerases β, λ, η, ι, κ, and Rev1 incorporating the active, 5′-phosphorylated forms of tenofovir, lamivudine, emtricitabine, and zidovudine. For the six enzymes, all of the drug analogs were incorporated less efficiently (40- to >110,000-fold) than the corresponding natural nucleotides, usually due to a weaker binding affinity and a slower rate of incorporation for the incoming nucleotide analog. In general, the 5′-triphosphate forms of lamivudine and zidovudine were better substrates than emtricitabine and tenofovir for the six human enzymes, although the substrate specificity profile depended on the DNA polymerase. Our kinetic results suggest NRTI insertion catalyzed by human X- and Y-family DNA polymerases is a potential mechanism of NRTI drug toxicity, and we have established a structure-function relationship for designing improved NRTIs.
More than 30 million people worldwide are infected with the human immunodeficiency virus (HIV), which is the causative agent of AIDS. To manage the life-threatening effects of HIV replication, nucleoside reverse transcriptase inhibitors (NRTIs) have been a mainstay in effective combination antiretroviral therapy. NRTIs undergo phosphorylation by host cell kinases to be converted into their active di- or triphosphate (DP or TP) forms, which can serve as nucleotide substrates for HIV reverse transcriptase (RT). Incorporation of the NRTIs into the viral genome by HIV RT terminates the replication process due to the lack of a 3′-hydroxyl in these drugs. Unfortunately, host DNA polymerases (Pols), organized into the A, B, X, and Y families, are susceptible to drug inhibition, because these enzymes catalyze a nucleotidyl transfer reaction similar to that of HIV RT. Incorporation of the drug analog into human DNA will inhibit DNA replication and possibly lead to cell death and drug toxicity. A correlation has been established between the kinetics of nucleotide analog incorporation catalyzed by human DNA Pol γ, an A-family member, and the observed clinical toxicity that presents as mitochondrial dysfunction (12, 13, 23, 26). However, some drug toxicity occurs via Pol γ-independent mechanisms (36, 52), such as the bone marrow toxicity associated with zidovudine (AZT) (2, 49) and the kidney toxicity associated with tenofovir (PMPA) (41, 47). Another possible mechanism is analog incorporation into nuclear DNA (38, 49), since NRTIs induce genomic instability by increasing mutations, structural chromosomal aberrations, abnormal chromatin structure, sister chromatid exchanges, and shortened telomeres (36, 37, 57). Candidates responsible for NRTI insertion into nuclear DNA include three replicative, B-family DNA polymerases (Pols α, δ, and ɛ) and the important base excision repair enzyme, Pol β (an X-family member). Although human replicative DNA polymerases perform the majority of nuclear DNA synthesis, most of these Pols have stringent nucleotide selection mechanisms to exclude antiretroviral nucleotide analogs (3, 31).
In the past 15 years, 10 novel human DNA polymerases, including Pol λ, Pol η, Pol ι, Pol κ, and Rev1, have been identified (18, 58). Pol λ is an X-family member while Pol η, Pol ι, Pol κ, and Rev1 belong to the Y-family of DNA polymerases. The Y-family DNA polymerases have been proposed to function in DNA lesion bypass, somatic hypermutation, base excision repair, nucleotide excision repair, and recombination pathways (18, 29, 55). Pol β, an X-family member, functions in base excision repair, while Pol λ has putative roles in base excision repair, nonhomologous end joining, and V(D)J recombination (58). Incorporation of a chain terminator during these biological processes would result in DNA replication inhibition and single- or double-strand DNA breaks, which can lead to apoptosis, immunosuppression, and genetic diseases (36). These specialized human X- and Y-family DNA polymerases exhibit low fidelity (100 to 10−5) on undamaged DNA, lack 3′-to-5′ exonuclease activity, and possess more flexible active sites that can accommodate altered DNA and nucleotide structures (35). Overall, the X- and Y-family DNA polymerases are up to 100,000 times more error prone than Pol γ in synthesizing undamaged DNA (22, 28, 35).
Due to the relatively recent discovery of these novel DNA polymerases, little is known about whether these noncanonical human Pols can incorporate the active di- or triphosphate forms of the NRTIs. In vitro kinetic studies are a good starting point, because the human genome encodes 16 DNA Pols with overlapping functions; all of these nucleotidyl transferases may be unwanted targets. Currently, cell-based assays and animal models cannot unambiguously identify which DNA polymerases are responsible for inducing cellular toxicity. Thus, the goal of this study was to understand the kinetic basis of incorporation for four widely prescribed anti-HIV nucleoside analogs (emtricitabine [L-FTC], lamivudine [L-3TC], PMPA, and AZT) in their active forms by Pol β, Pol λ, Pol η, Pol ι, Pol κ, and Rev1 by using pre-steady-state kinetics methods (Fig. 1). This study established a structure-function relationship which may be useful in designing more effective and less toxic NRTIs.
FIG. 1.

Chemical structures of NRTIs investigated in this study and their natural counterparts.
MATERIALS AND METHODS
Materials.
The following chemicals used in this study were purchased from the indicated companies: [γ-32P]ATP, MP Biomedicals; deoxyribonucleotide 5′-triphosphates, GE Healthcare; Bio-Spin 6 columns, Bio-Rad Laboratories; OptiKinase, USB Corporation; DNA oligomers (Table 1), Integrated DNA Technologies; 3′-azido-3′-deoxythymidine 5′-triphosphate (AZT-TP), TriLink BioTechnologies. The diphosphate of 9-[2-(phosphonomethoxy)propyl]adenine (PMPA-DP), l-2′,3′-dideoxy-5-fluoro-3′-thiacytidine 5′-triphosphate (L-FTC-TP), and l-2′,3′-dideoxy-3′-thiacytidine 5′-triphosphate (L-3TC-TP) were kind gifts from Gilead Sciences, Inc.
TABLE 1.
Sequences of oligonucleotides
| Oligonucleotide | Sequencea |
|---|---|
| 21-mer | 5′-CGCAGCCGTCCAACCAACTCA-3′ |
| 19-mer C | 5′-CGTCGATCCAATGCCGTCC-3′ |
| 19-mer A | 5′-AGTCGATCCAATGCCGTCC-3′ |
| 41-mer GT | 3′-GCGTCGGCAGGTTGGTTGAGTGTCAGCTAGGTTACGGCAGG-5′ |
| 41-mer TG | 3′-GCGTCGGCAGGTTGGTTGAGTTGCAGCTAGGTTACGGCAGG-5′ |
| 41-mer AG | 3′-GCGTCGGCAGGTTGGTTGAGTAGCAGCTAGGTTACGGCAGG-5′ |
The 21-mer strand was 5′ radiolabeled. For single-nucleotide-gap DNA substrates, the downstream 19-mer strand was 5′ phosphorylated. The identities of important base positions are highlighted in bold.
Preparation of human DNA polymerases and DNA substrates.
The plasmids, expression, and purification of human DNA polymerases β (5, 39), λ (15), η (48), truncated ι (residues 1 to 420) (48), truncated κ (residues 9 to 518) (48), and truncated Rev1 (residues 341 to 829) (32, 33) were described previously. Commercially synthesized oligomers listed in Table 1 were purified using polyacrylamide gel electrophoresis (14, 16). The 21-mer primer was radiolabeled with [γ-32P]ATP and OptiKinase according to the manufacturer's protocol, and the unreacted [γ-32P]ATP was subsequently removed via a Bio-Spin 6 column. The 21-41-mer primer-template DNA substrates (16) and 21-19-41-mer single-nucleotide-gap DNA substrates (14) were annealed as described previously.
Measurements of kp and Kd for single-nucleotide incorporation.
Kinetic assays were completed using buffer B (50 mM Tris-HCl [pH 7.8] at 37°C with 5 mM MgCl2, 50 mM NaCl, 0.1 mM EDTA, 5 mM dithiothreitol [DTT], 10% glycerol, and 0.1 mg/ml of bovine serum albumin [BSA]) for Pol β, buffer L (50 mM Tris-HCl [pH 8.4] at 37°C with 5 mM MgCl2, 100 mM NaCl, 0.1 mM EDTA, 5 mM DTT, 10% glycerol, and 0.1 mg/ml of BSA) for Pol λ (14, 15), and buffer Y (50 mM HEPES [pH 7.5] at 37°C with 5 mM MgCl2, 50 mM NaCl, 0.1 mM EDTA, 5 mM DTT, 10% glycerol, and 0.1 mg/ml of BSA) for Pols η, ι, κ, and Rev1. Pols η, ι, κ, and Rev1 were assayed with the 21-41-mer DNA substrates, and Pols β and λ were assayed with the 21-19-41-mer single-nucleotide-gap DNA substrates. All kinetic experiments described herein were performed at 37°C, and the reported concentrations were final after mixing all the components. Note that AZT-TP was not preincubated with DTT to avoid reduction of the azido group (45). A preincubated solution of the DNA polymerase (120 or 300 nM) and 5′-32P-radiolabeled DNA substrate (30 nM) was mixed with increasing concentrations (0.025 to 2,000 μM) of nucleotide or nucleotide analog in the appropriate buffer at 37°C. The polymerase was present in molar excess over DNA, whereby the enzyme-to-DNA ratio was 4:1 for Pols λ, η, ι, and Rev1 and 10:1 for Pols β and κ. Aliquots of the reaction mixtures were quenched at various times using 0.37 M EDTA. A rapid chemical-quench flow apparatus (KinTek) was utilized for fast nucleotide incorporations. Reaction products were resolved using sequencing gel electrophoresis (17% acrylamide, 8 M urea) and quantitated with a Typhoon TRIO (GE Healthcare). The time course of product formation at each nucleotide concentration was fit to a single-exponential equation, [Product] = A[1 − exp(−kobst)], by using a nonlinear regression program, KaleidaGraph (Synergy Software), to yield an observed rate constant of nucleotide incorporation (kobs). The kobs values were then plotted as a function of nucleotide concentration and fit using the hyperbolic equation, kobs = kp[dNTP]/([dNTP] + Kd), which resolved the maximum rate of incorporation (kp) and the equilibrium dissociation constant (Kd) for nucleotide incorporation catalyzed by each enzyme. Most kinetic parameters in this work were resolved from a plot of seven points (see Fig. 2C), which was a composite of 49 kinetic time points (see Fig. 2B), i.e., kobs values were obtained from seven time points at seven different nucleotide concentrations. These measurements provided sufficient coverage within the appropriate range of the Kd values, and the data-fitting error was low (usually an R value of >0.98). Unlike steady-state kinetic assays, pre-steady-state kinetic assays consume at least 50-fold more enzyme per assay mixure; therefore, the number of time points for the secondary plot was restricted to approximately seven.
FIG. 2.
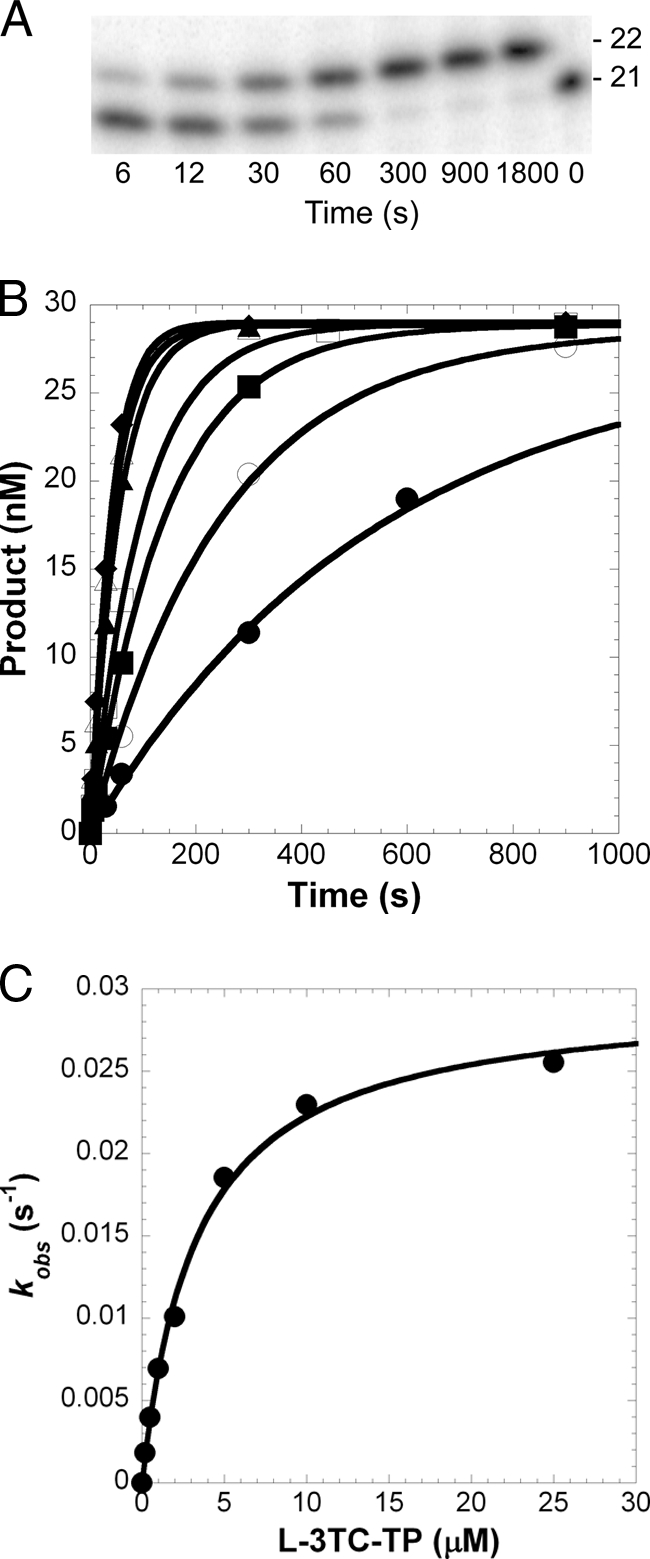
Concentration dependence on the pre-steady-state rate constant of L-3TC-TP incorporation catalyzed by Pol η. A preincubated solution of Pol η (120 nM) and 5′-32P-labeled 21-41-mer GT DNA (30 nM) (Table 1) was rapidly mixed with increasing concentrations of L-3TC-TP·Mg2+ (0.2 μM, •; 0.5 μM, ○; 1 μM, ▪; 2 μM, □; 5 μM, ▴; 10 μM, ▵; 25 μM, ⧫) for various time intervals. (A) A representative gel image is shown for Pol η inserting L-3TC-TP at 25 μM. The lengths of the DNA primer are indicated in the right margin. (B) The concentration of DNA product was plotted as a function of time. The solid lines are the best fits to a single-exponential equation, which determined the observed rate constant, kobs. (C) The kobs values were plotted as a function of L-3TC-TP concentration. The data (•) were then fit to a hyperbolic equation, yielding a kp of 0.0296 ± 0.0009 s−1 and a Kd of 3.3 ± 0.3 μM.
RESULTS
Measurement of selection factors.
Single-turnover kinetic assays were used to determine the kinetic basis of how Pols β, λ, η, ι, κ, and Rev1 discriminate between a correct natural nucleotide versus a nucleotide analog based on the Kd and kp for an incoming nucleotide. To directly observe the conversion of the DNA substrate into the extended DNA product during a single pass through the enzymatic pathway, a preincubated solution of Pol η and 5′-32P-labeled 21-41-mer GT DNA (Table 1) was mixed with increasing concentrations of L-3TC-TP (0.2 to 25 μM) (see Materials and Methods) (24). The data were analyzed to resolve a kp of 0.0296 ± 0.0009 s−1 and a Kd of 3.3 ± 0.3 μM (Fig. 2). Similar single-nucleotide incorporation assays were performed for each enzyme, incorporating dATP, PMPA-DP, dCTP, L-3TC-TP, L-FTC-TP, dTTP, or AZT-TP opposite the complementary template base. Please note, a single-nucleotide-gap DNA substrate was used in the assays for the gap-filling Pols β and λ, since the catalytic efficiencies of these enzymes are enhanced by a 5′-phosphorylated downstream strand (1, 10) and the gap DNA substrate models a more physiologically relevant DNA substrate. Meanwhile, Pols η, ι, κ, and Rev1 were assayed with a primer-template DNA substrate. The kinetic parameters are listed in Tables 2, 3, and 4, and the incorporation efficiency (kp/Kd) and selection factors are defined and calculated. All polymerases preferred the natural deoxynucleotide (dNTP) more than the nucleotide analog.
TABLE 2.
Kinetic parameters for nucleotide incorporation opposite template base dG at 37°C
| DNA polymerase | dNTP | kp (s−1) | Kd (μM) | kp/Kd (μM−1 s−1) | Selection factora |
|---|---|---|---|---|---|
| Pol β | dCTP | 5.02 ± 0.07 | 0.71 ± 0.04 | 7.1 | |
| L-3TC-TP | 0.00390 ± 0.00010 | 0.18 ± 0.02 | 2.2 × 10−2 | 325 | |
| L-FTC-TP | 0.027 ± 0.001 | 11 ± 2 | 2.5 × 10−3 | 2,900 | |
| Pol λ | dCTPb | 1.57 ± 0.04 | 0.9 ± 0.1 | 1.7 | |
| L-3TC-TP | 0.00402 ± 0.00008 | 0.106 ± 0.010 | 3.8 × 10−2 | 46 | |
| L-FTC-TP | 0.0049 ± 0.0001 | 0.36 ± 0.03 | 1.4 × 10−2 | 120 | |
| Pol η | dCTP | 49 ± 2 | 25 ± 4 | 2.0 | |
| L-3TC-TP | 0.0296 ± 0.0009 | 3.3 ± 0.3 | 9.0 × 10−3 | 220 | |
| L-FTC-TP | 0.0244 ± 0.0004 | 2.7 ± 0.1 | 9.0 × 10−3 | 220 | |
| Pol κ | dCTP | 11.8 ± 0.6 | 32 ± 5 | 3.7 × 10−1 | |
| L-3TC-TP | 0.00045 ± 0.00001 | 4.0 ± 0.5 | 1.1 × 10−4 | 3,300 | |
| L-FTC-TP | 0.000225 ± 0.000003 | 4.8 ± 0.2 | 4.7 × 10−5 | 7,900 | |
| Pol ι | dCTP | 0.075 ± 0.002 | 50 ± 5 | 1.5 × 10−3 | |
| L-3TC-TP | 0.00356 ± 0.00009 | 96 ± 8 | 3.7 × 10−5 | 40 | |
| L-FTC-TP | 0.0020 ± 0.0001 | 230 ± 20 | 8.7 × 10−6 | 170 | |
| Rev1 | dCTPc | 22.4 ± 0.9 | 2.2 ± 0.3 | 10 | |
| L-3TC-TP | 0.0236 ± 0.0006 | 1.04 ± 0.10 | 2.3 × 10−2 | 450 | |
| L-FTC-TP | 0.0222 ± 0.0007 | 2.7 ± 0.3 | 8.2 × 10−3 | 1,200 | |
| Pol γ | dCTPd | 44 ± 2 | 1.1 ± 0.1 | 40 | |
| L-3TC-TPd | 0.125 ± 0.005 | 9.2 ± 0.9 | 1.4 × 10−2 | 2,900 | |
| L-FTC-TPe | 0.0086 ± 0.0015 | 62.9 ± 8.4 | 1.4 × 10−4 | 290,000 | |
| HIV-1 RT | dCTPf | 2.9 ± 0.2 | 56 ± 10 | 5.2 × 10−2 | |
| L-3TC-TPg | 0.019 ± 0.001 | 15 ± 3 | 1.3 × 10−3 | 41 | |
| L-FTC-TPe | 0.039 ± 0.003 | 12 ± 3 | 3.3 × 10−3 | 16 |
Calculated as [(kp/Kd)dCTP]/[(kp/Kd)analog].
Kinetic parameters are from reference 15.
Kinetic parameters are from reference 6.
Kinetic parameters are from reference 12.
Kinetic parameters are from reference 13.
Kinetic parameters are from reference 44.
Kinetic parameters are from reference 11.
TABLE 3.
Kinetic parameters for nucleotide incorporation opposite template base dT at 37°C
| DNA polymerase | dNTP | kp (s−1) | Kd (μM) | kp/Kd (μM−1 s−1) | Selection factora |
|---|---|---|---|---|---|
| Pol β | dATP | 32 ± 1 | 9.2 ± 1.0 | 3.5 | |
| PMPA-DP | 4.7 ± 0.5 | 50 ± 10 | 9.4 × 10−2 | 40 | |
| Pol λ | dATPb | 1.5 ± 0.1 | 0.9 ± 0.3 | 1.7 | |
| PMPA-DP | 0.095 ± 0.008 | 3.7 ± 0.9 | 2.6 × 10−2 | 65 | |
| Pol η | dATP | 35 ± 3 | 130 ± 26 | 2.7 × 10−1 | |
| PMPA-DP | 0.0134 ± 0.0007 | 90 ± 10 | 1.5 × 10−4 | 1,800 | |
| Pol κ | dATP | 2.49 ± 0.08 | 7.0 ± 1.0 | 3.6 × 10−1 | |
| PMPA-DP | 0.010 ± 0.005 | 3,000 ± 2,000 | 3.3 × 10−6 | 110,000 | |
| Pol ι | dATP | 0.015 ± 0.001 | 260 ± 40 | 5.8 × 10−5 | |
| PMPA-DP | Could not measure | High | |||
| Rev1 | dATP | 0.00152 ± 0.00005 | 2.0 ± 0.3 | 7.6 × 10−4 | |
| PMPA-DP | Could not measure | High | |||
| Pol γ | dATPc | 45 ± 1 | 0.8 ± 0.1 | 56 | |
| PMPA-DPd | 0.21 ± 0.01 | 40.3 ± 5.7 | 5.2 × 10−3 | 10,800 | |
| T7 exo | dATPe | 156 ± 8 | 8 ± 2 | 19.5 | |
| PMPA-DPe | 0.096 ± 0.009 | 268 ± 39 | 3.6 × 10−4 | 54,400 | |
| HIV-1 RT | dATPe | 41.3 ± 0.6 | 8.1 ± 0.9 | 5.1 | |
| PMPA-DPe | 49 ± 5 | 58 ± 11 | 8.4 × 10−1 | 6 |
TABLE 4.
Kinetic parameters for nucleotide incorporation opposite template base dA at 37°C
| DNA polymerase | dNTP | kp (s−1) | Kd (μM) | kp/Kd (μM−1 s−1) | Selection factora |
|---|---|---|---|---|---|
| Pol β | dTTP | 12.3 ± 0.5 | 3.5 ± 0.5 | 3.5 | |
| AZT-TP | 0.0199 ± 0.0007 | 15 ± 2 | 1.3 × 10−3 | 2,600 | |
| Pol λ | dTTPb | 3.9 ± 0.2 | 2.6 ± 0.4 | 1.5 | |
| AZT-TP | 0.0104 ± 0.0004 | 6 ± 1 | 1.7 × 10−3 | 865 | |
| Pol η | dTTP | 35 ± 1 | 41 ± 5 | 8.5 × 10−1 | |
| AZT-TP | 0.31 ± 0.05 | 500 ± 200 | 6.2 × 10−4 | 1,400 | |
| Pol κ | dTTP | 4.35 ± 0.04 | 11.0 ± 0.5 | 4.0 × 10−1 | |
| AZT-TP | 0.0144 ± 0.0005 | 80 ± 9 | 1.8 × 10−4 | 2,200 | |
| Pol ι | dTTP | 0.75 ± 0.02 | 13 ± 1 | 5.8 × 10−2 | |
| AZT-TP | 0.0037 ± 0.0003 | 90 ± 20 | 4.1 × 10−5 | 1,400 | |
| Rev1 | dTTP | 0.0038 ± 0.0003 | 6 ± 1 | 6.3 × 10−4 | |
| AZT-TP | 0.00119 ± 0.00005 | 200 ± 30 | 5.9 × 10−6 | 110 | |
| Pol γ | dTTPc | 25 ± 2 | 0.6 ± 0.16 | 42 | |
| AZT-TPd | 10,000,000 | ||||
| HIV-1 RT | dTTPe | 16.7 | 19 | 8.8 × 10−1 | |
| AZT-TPe | 0.7 | 2 | 3.5 × 10−1 | 2.5 |
Selection factors for l-cytosine-based analogs.
L-3TC is an NRTI that was approved in 1995, and it has two unique features: l-stereochemistry and an oxathiolane ring (Fig. 1). Despite lacking the natural d-stereochemistry, L-3TC-TP was incorporated by the six Pols examined in this work, with selection factors ranging from 40 to 3,300 (Table 2). In general, the mechanism of L-3TC-TP incorporation was similar for most of the Pols: a lower rate of incorporation (∼6,100-fold on average) but tighter nucleotide ground state binding affinity (1/Kd; ∼6-fold on average) than dCTP. In contrast, a slightly weaker ground state binding affinity for L-3TC-TP to the Pol ι-DNA complex was measured.
L-FTC, an NRTI approved in 2003, consists of L-3TC with a fluorine atom added to the C-5 position (Fig. 1). This subtle modification resulted in selection factors (120 to 7,900) for L-FTC-TP that were equal to or greater than those determined for L-3TC-TP (Table 2). The greatest effect was observed with Pols β and ι, whereby the selection factor for L-FTC-TP increased 9- and 4-fold, respectively, relative to L-3TC-TP. This kinetic effect was due to larger Kd values. In general, the C-5 fluoro group of L-FTC-TP did not affect the rate of incorporation, since the kp values for L-FTC-TP and L-3TC-TP remained similar. Taken together, these results showed that the stereospecificity of an incoming dNTP is important for Pols β, λ, η, ι, κ, and Rev1 during the nucleotide incorporation step.
Selection factors for an acyclic nucleotide analog.
Next, we examined PMPA, which possesses several novel structural characteristics: an acyclic moiety and a phosphonate group that is ∼1 Å closer to the base than the phosphate of dAMP (Fig. 1) (50). The kinetic parameters for Pol ι and Rev1 for inserting PMPA-DP could not be determined, since product formation was barely detectable after 3 h at a relatively high PMPA-DP concentration of 500 μM (data not shown). However, the selection factors of Pols β, λ, η, and κ were calculated to be 40, 65, 1,800, and 110,000, respectively (Table 3). For Pols β and λ, the Kd was weakened by 5-fold on average and the kp was reduced by 11-fold on average compared to dATP. Notably, Pols β and λ have different kinetic parameters, which supports our previous discovery that these two homologs of the X-family possess different enzymatic properties (1, 15). In contrast, Pol η maintained a tight enzyme-DNA-PMPA-DP complex, but the rate of PMPA-DP incorporation opposite template dT dropped by 2,600-fold. Pol κ discriminated PMPA-DP from dATP by decreasing the rate by 250-fold and increasing the Kd value by 430-fold. Thus, the Y-family enzymes use the ribose moiety as a mechanism of discrimination more than the X-family enzymes. Moreover, most of the PMPA-DP selection factors were higher than those determined for L-3TC-TP and L-FTC-TP.
Selection factors for AZT-TP.
The kinetic parameters were measured and the selection factors were calculated for the 5′-triphosphate form of zidovudine, the first NRTI approved by the U.S. Food and Drug Administration (FDA), being incorporated by the six human Pols (Table 4). AZT has a 3′-azido group that would be larger in size than the 3′-hydroxyl (Fig. 1). The selection factors for AZT-TP ranged from 110 to 2,600, and the mechanism of incorporation was altered mostly at the level of incorporation (270-fold reduction on average) and modestly at the ground state binding step (11-fold weaker on average) compared to dTTP. Although Rev1 exhibits the lowest discrimination factor for AZT-TP, the incorporation efficiency was the lowest among the Pols, at 5.9 × 10−6 μM−1 s−1.
Different mechanisms observed for different NRTIs.
As noted above, different mechanisms of analog incorporation were determined for the four NRTIs, indicating that the altered chemical structures differentially affect each polymerase activity, as the preferential order of incorporation varies among the DNA polymerases. Furthermore, it is important to note that the order based on selection factors does not coincide with the order based on substrate specificity constants except for Pol η and Pol κ (i.e., L-FTC-TP ≈ L-3TC-TP > AZT-TP > PMPA-DP for Pol η; AZT-TP > L-3TC-TP > L-FTC-TP > PMPA-DP for Pol κ). Analysis of these orders reveals the following general trends: (i) L-3TC-TP and AZT-TP are usually the most preferred NRTIs, (ii) the fluoro group on L-FTC-TP leads to a higher level of discrimination than L-3TC-TP, and (iii) the Y-family enzymes prefer PMPA-DP the least.
DISCUSSION
Kinetic basis of NRTI selection and inhibition among viral and human DNA polymerases.
Using transient-state kinetic techniques, this work determined the incorporation efficiency values for the active forms of four FDA-approved NRTIs catalyzed by six noncanonical human DNA polymerases: β, λ, η, κ, ι, and Rev1. Most of the NRTIs were inserted into DNA by these DNA polymerases, which suggested that incorporation into nuclear DNA and interference of genomic replication and repair are possible in vivo. The four NRTIs examined herein are associated with a low potential of producing mitochondrial dysfunction (4, 40), thereby suggesting toxicity associated with these NRTIs is induced by other mechanisms (36). The selection factors for an exonuclease-deficient mutant of human DNA polymerase γ, an A-family member, have been measured or estimated using pre-steady-state kinetic techniques similar to those used in this work (12, 13, 19, 23). Based on the selection factors, Pol γ exhibited a greater level of discrimination than most of the X- and Y-family DNA polymerases: up to 70-, 2,400-, 270-, and 90,000-fold for L-3TC-TP, L-FTC-TP, PMPA-DP, and AZT-TP, respectively (Tables 2 to 4). This kinetic finding strengthens the possible role of human X- and Y-family DNA polymerases in the etiology of NRTI toxicity. The kinetics of Pol γ-catalyzed AZT-TP incorporation is complicated by the slow release of the pyrophosphate product; therefore, the lack of a hyperbolic concentration dependence on the rate of incorporation prevented the measurement of single-turnover kinetic parameters, but a selection factor of 1 × 10−7 was calculated (19). The unique mechanism of AZT-TP selection by Pol γ does not apply to the noncanonical polymerases examined in this work, since their observed rate constants were dependent on the concentration of AZT-TP. For the unnatural l-cytosine analogs, Pol γ exhibits a weaker ground state binding affinity than Pols β, λ, η, κ, and Rev1. Perhaps the more spacious active site of low-fidelity enzymes provides greater accessibility for the l-stereoisomer to enter and the oxathiolane ring to bind favorably, leading to a tighter Kd value. Similar to the Y-family enzymes, Pol γ depends on the presence of a ribose moiety for efficient nucleotide incorporation, since PMPA-DP reduced the kp and weakened the Kd. Similar kinetic results were obtained for PMPA-DP incorporation catalyzed by an exonuclease-deficient mutant of T7 DNA polymerase, a model A-family enzyme (50). Unlike Pol γ, the X- and Y-family enzymes lack 3′-to-5′ exonuclease activity; therefore, the incorporated NRTIs can only be removed by pyrophosphorolysis, which requires high cellular pyrophosphate levels due to the weak binding affinity of pyrophosphate, meaning NRTIs will not be excised easily from DNA.
The efficacy of NRTIs depends partially on the selection factor of HIV-1 RT relative to the host DNA polymerases. In general, the selection factors, as calculated from pre-steady-state kinetic parameters, for HIV-1 RT during DNA-dependent DNA synthesis are lower by at least 1-, 8-, 7-, and 22-fold for L-3TC-TP, L-FTC-TP, PMPA-DP, and AZT-TP, respectively, than those measured for human X- and Y-family Pols (Tables 2 to 4). Improving the efficacy of NRTIs may be a challenge, because the kinetic basis of analog incorporation for HIV-1 RT is sometimes similar to the X- and Y-family DNA polymerases. For example, HIV-1 RT incorporates the l-oxathiolane cytosine analogs with an ∼4-fold-tighter binding affinity and ∼100-fold-reduced rate of incorporation, a general trend that is similar to the X- and Y-family polymerases but different than Pol γ (Table 2) (11-13). In contrast, HIV-1 RT prefers L-FTC-TP over L-3TC-TP, whereas Pols β, λ, κ, ι, and Rev1 prefer L-3TC-TP. These results may explain why emtricitabine is a more effective NRTI than lamivudine (46). The smaller and more flexible acyclic moiety of PMPA-DP only affects the Kd during catalysis by HIV-1 RT, unlike the human enzymes. A crystal structure of the HIV-1 RT-DNA-PMPA-DP ternary complex revealed that PMPA-DP forms a noncanonical Watson-Crick base pair (51). Thus, this suboptimal base-pairing scheme for PMPA-DP and dT may contribute to the lower catalytic efficiency for the human enzymes, since hydrogen bonding has been shown to be important for incorporation catalyzed by Pol γ (27), Pol λ (7), Saccharomyces cerevisiae Pol η (54), Pol κ (56), and Rev1 (6). Overall, PMPA-DP appears to be a superior drug analog because it requires only two phosphorylation events to become activated, it is a better substrate for HIV-1 RT than the seven human enzymes examined thus far (Table 3) (23, 50), and it has a favorable resistance profile characterized by activity against most NRTI-resistant viruses and a low propensity for resistance development (30, 34).
Potential cellular implications of NRTIs incorporated into human genomic DNA.
Although the X- and Y-family DNA polymerases perform fewer incorporation events than Pol γ, most of them show a lesser degree of discrimination for the nucleotide analogs versus natural dNTPs than does Pol γ (Tables 2 to 4). The potential incorporation of nucleotide analogs in vivo depends on intracellular concentrations of natural nucleotides relative to the nucleotide analogs. The ratio of dCTP to L-3TC-TP is approximately 10:1 in noninfected phytohemagglutinin (PHA)-activated peripheral blood mononuclear cells (PBMC) (9). Gao et al. determined the dTTP:AZT-TP ratio to be 21:1 and 1.6:1 for resting and PHA-activated PBMC, respectively (17). Unlike the aforementioned works, most studies do not measure the levels of both the natural dNTPs and the nucleotide analogs. Therefore, the dATP:PMPA-DP and dCTP:L-FTC-TP ratios are predicted by us to be 10:1 and 1:1, respectively, based on the values for natural dATP or dCTP (see Table 2 in reference 17) and the average values for PMPA-DP (20, 42) and L-FTC-TP (8, 53), all of which were derived from PBMC. These predicted or measured dNTP:dNTP analog ratios suggest that the relative intracellular concentrations can approach 1:1; therefore, the selection factors (Tables 2 to 4) can be interpreted as the insertion frequencies. For example, the selection factors of AZT-TP range from 110 to 2,600 (Table 4). Thus, the six noncanonical human DNA polymerases are predicted to incorporate 1 AZT-TP molecule for every 110 to 2,600 dTTP incorporations when the cellular concentrations of AZT-TP and dTTP are equal.
The X- and Y-family DNA polymerases are expressed in the tissues affected by drug toxicity, so it is plausible that NRTI incorporation by these enzymes may play a role (18, 58). NRTI incorporation by X- and Y-family DNA polymerases would inhibit critical cellular pathways, such as base excision repair, nonhomologous end joining, translesion DNA synthesis, V(D)J recombination, and somatic hypermutation (18, 29, 43, 55, 58). Therefore, incorporation of a chain terminator at junctions with single-strand or double-strand DNA breaks would lead to genomic instability (21, 37, 57) and eventually trigger apoptosis in noninfected cells, which could lead to unwanted side effects (36). Besides NRTI incorporation by host enzymes, other processes can affect the efficacy and toxicity of the analog, such as drug uptake, transport, catabolism, and metabolism. This interplay of pathways is likely important for a comprehensive understanding of NRTI toxicity.
Concluding remarks.
Although our in vitro kinetic findings do not provide direct evidence that X- and Y-family DNA polymerases are a causative factor of in vivo toxicity, our data did establish the following important points: (i) most antiviral nucleotide analogs are substrates for human X- and Y-family DNA polymerases, (ii) these noncanonical Pols are less selective than Pol γ, (iii) the basis of nucleotide analog selection is sometimes similar for the X- and Y-family Pols and HIV-1 RT, and (iv) NRTI toxicity may involve nucleotide analog incorporation by DNA repair and lesion bypass DNA polymerases.
Acknowledgments
We thank Joy Feng of Gilead Sciences, Inc., for providing us the nucleotide analogs and Michael Miller of Gilead Sciences, Inc., for critical reading of the manuscript.
This work was supported by National Institutes of Health grant GM079403 to Z.S. J.A.B. was supported by a Presidential Fellowship from The Ohio State University and an American Heart Association predoctoral fellowship (grant 0815382D). L.R.P. was supported by an REU supplemental grant from a National Science Foundation Career Award (grant MCB-0447899 to Z.S.). J.D.F. was supported by a postdoctoral fellowship from Pulmonary National Institutes of Health training grant 5T32HL007946 to Mark D. Wewers.
Footnotes
Published ahead of print on 15 November 2010.
REFERENCES
- 1.Ahn, J., V. S. Kraynov, X. Zhong, B. G. Werneburg, and M. D. Tsai. 1998. DNA polymerase beta: effects of gapped DNA substrates on dNTP specificity, fidelity, processivity and conformational changes. Biochem. J. 331:79-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benbrik, E., P. Chariot, S. Bonavaud, M. Ammi-Said, E. Frisdal, C. Rey, R. Gherardi, and G. Barlovatz-Meimon. 1997. Cellular and mitochondrial toxicity of zidovudine (AZT), didanosine (ddI) and zalcitabine (ddC) on cultured human muscle cells. J. Neurol Sci. 149:19-25. [DOI] [PubMed] [Google Scholar]
- 3.Birkus, G., M. Hajek, P. Kramata, I. Votruba, A. Holy, and B. Otova. 2002. Tenofovir diphosphate is a poor substrate and a weak inhibitor of rat DNA polymerases alpha, delta, and epsilon*. Antimicrob. Agents Chemother. 46:1610-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Birkus, G., M. J. Hitchcock, and T. Cihlar. 2002. Assessment of mitochondrial toxicity in human cells treated with tenofovir: comparison with other nucleoside reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 46:716-723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown, J. A., W. W. Duym, J. D. Fowler, and Z. Suo. 2007. Single-turnover kinetic analysis of the mutagenic potential of 8-oxo-7,8-dihydro-2′-deoxyguanosine during gap-filling synthesis catalyzed by human DNA polymerases lambda and beta. J. Mol. Biol. 367:1258-1269. [DOI] [PubMed] [Google Scholar]
- 6.Brown, J. A., J. D. Fowler, and Z. Suo. 2010. Kinetic investigation of nucleotide selection employed by a protein template-dependent DNA polymerase. Biochemistry 49:5504-5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown, J. A., L. R. Pack, S. M. Sherrer, A. K. Kshetry, S. A. Newmister, J. D. Fowler, J. S. Taylor, and Z. Suo. 2010. Identification of critical residues for the tight binding of both correct and incorrect nucleotides to human DNA polymerase lambda. J. Mol. Biol. 403:505-515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Darque, A., G. Valette, F. Rousseau, L. H. Wang, J. P. Sommadossi, and X. J. Zhou. 1999. Quantitation of intracellular triphosphate of emtricitabine in peripheral blood mononuclear cells from human immunodeficiency virus-infected patients. Antimicrob. Agents Chemother. 43:2245-2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dereuddre-Bosquet, N., B. Roy, K. Routledge, P. Clayette, G. Foucault, and M. Lepoivre. 2004. Inhibitors of CTP biosynthesis potentiate the anti-human immunodeficiency virus type 1 activity of 3TC in activated peripheral blood mononuclear cells. Antiviral Res. 61:67-70. [DOI] [PubMed] [Google Scholar]
- 10.Duym, W. W., K. A. Fiala, N. Bhatt, and Z. Suo. 2006. Kinetic effect of a downstream strand and its 5′-terminal moieties on single nucleotide gap-filling synthesis catalyzed by human DNA polymerase lambda. J. Biol. Chem. 281:35649-35655. [DOI] [PubMed] [Google Scholar]
- 11.Feng, J. Y., and K. S. Anderson. 1999. Mechanistic studies comparing the incorporation of (+) and (-) isomers of 3TCTP by HIV-1 reverse transcriptase. Biochemistry 38:55-63. [DOI] [PubMed] [Google Scholar]
- 12.Feng, J. Y., A. A. Johnson, K. A. Johnson, and K. S. Anderson. 2001. Insights into the molecular mechanism of mitochondrial toxicity by AIDS drugs. J. Biol. Chem. 276:23832-23837. [DOI] [PubMed] [Google Scholar]
- 13.Feng, J. Y., E. Murakami, S. M. Zorca, A. A. Johnson, K. A. Johnson, R. F. Schinazi, P. A. Furman, and K. S. Anderson. 2004. Relationship between antiviral activity and host toxicity: comparison of the incorporation efficiencies of 2′,3′-dideoxy-5-fluoro-3′-thiacytidine-triphosphate analogs by human immunodeficiency virus type 1 reverse transcriptase and human mitochondrial DNA polymerase. Antimicrob. Agents Chemother. 48:1300-1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fiala, K. A., W. Abdel-Gawad, and Z. Suo. 2004. Pre-steady-state kinetic studies of the fidelity and mechanism of polymerization catalyzed by truncated human DNA polymerase lambda. Biochemistry 43:6751-6762. [DOI] [PubMed] [Google Scholar]
- 15.Fiala, K. A., W. W. Duym, J. Zhang, and Z. Suo. 2006. Up-regulation of the fidelity of human DNA polymerase lambda by its non-enzymatic proline-rich domain. J. Biol. Chem. 281:19038-19044. [DOI] [PubMed] [Google Scholar]
- 16.Fiala, K. A., and Z. Suo. 2004. Pre-steady-state kinetic studies of the fidelity of Sulfolobus solfataricus P2 DNA polymerase IV. Biochemistry 43:2106-2115. [DOI] [PubMed] [Google Scholar]
- 17.Gao, W. Y., T. Shirasaka, D. G. Johns, S. Broder, and H. Mitsuya. 1993. Differential phosphorylation of azidothymidine, dideoxycytidine, and dideoxyinosine in resting and activated peripheral blood mononuclear cells. J. Clin. Invest. 91:2326-2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo, C., J. N. Kosarek-Stancel, T. S. Tang, and E. C. Friedberg. 2009. Y-family DNA polymerases in mammalian cells. Cell. Mol. Life Sci. 66:2363-2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hanes, J. W., and K. A. Johnson. 2007. A novel mechanism of selectivity against AZT by the human mitochondrial DNA polymerase. Nucleic Acids Res. 35:6973-6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hawkins, T., W. Veikley, R. L. St. Claire III, B. Guyer, N. Clark, and B. P. Kearney. 2005. Intracellular pharmacokinetics of tenofovir diphosphate, carbovir triphosphate, and lamivudine triphosphate in patients receiving triple-nucleoside regimens. J. Acquir. Immune Defic. Syndr. 39:406-411. [DOI] [PubMed] [Google Scholar]
- 21.Igoudjil, A., K. Begriche, D. Pessayre, and B. Fromenty. 2006. Mitochondrial, metabolic and genotoxic effects of antiretroviral nucleoside reverse-transcriptase inhibitors. Anti Infect. Agents Med. Chem. 5:273-292. [Google Scholar]
- 22.Johnson, A. A., and K. A. Johnson. 2001. Fidelity of nucleotide incorporation by human mitochondrial DNA polymerase. J. Biol. Chem. 276:38090-38096. [DOI] [PubMed] [Google Scholar]
- 23.Johnson, A. A., A. S. Ray, J. Hanes, Z. Suo, J. M. Colacino, K. S. Anderson, and K. A. Johnson. 2001. Toxicity of antiviral nucleoside analogs and the human mitochondrial DNA polymerase. J. Biol. Chem. 276:40847-40857. [DOI] [PubMed] [Google Scholar]
- 24.Johnson, K. A. 1992. Transient-state kinetic analysis of enzyme reaction pathways. Enzymes 20:1-61. [Google Scholar]
- 25.Kerr, S. G., and K. S. Anderson. 1997. Pre-steady-state kinetic characterization of wild type and 3′-azido-3′-deoxythymidine (AZT) resistant human immunodeficiency virus type 1 reverse transcriptase: implication of RNA directed DNA polymerization in the mechanism of AZT resistance. Biochemistry 36:14064-14070. [DOI] [PubMed] [Google Scholar]
- 26.Lee, H., J. Hanes, and K. A. Johnson. 2003. Toxicity of nucleoside analogues used to treat AIDS and the selectivity of the mitochondrial DNA polymerase. Biochemistry 42:14711-14719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee, H. R., S. A. Helquist, E. T. Kool, and K. A. Johnson. 2008. Importance of hydrogen bonding for efficiency and specificity of the human mitochondrial DNA polymerase. J. Biol. Chem. 283:14402-14410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee, H. R., and K. A. Johnson. 2006. Fidelity of the human mitochondrial DNA polymerase. J. Biol. Chem. 281:36236-36240. [DOI] [PubMed] [Google Scholar]
- 29.Lehmann, A. R. 2006. New functions for Y family polymerases. Mol. Cell 24:493-495. [DOI] [PubMed] [Google Scholar]
- 30.Margot, N. A., E. Isaacson, I. McGowan, A. K. Cheng, R. T. Schooley, and M. D. Miller. 2002. Genotypic and phenotypic analyses of HIV-1 in antiretroviral-experienced patients treated with tenofovir DF. AIDS 16:1227-1235. [DOI] [PubMed] [Google Scholar]
- 31.Martin, J. L., C. E. Brown, N. Matthews-Davis, and J. E. Reardon. 1994. Effects of antiviral nucleoside analogs on human DNA polymerases and mitochondrial DNA synthesis. Antimicrob. Agents Chemother. 38:2743-2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Masuda, Y., and K. Kamiya. 2006. Role of single-stranded DNA in targeting REV1 to primer termini. J. Biol. Chem. 281:24314-24321. [DOI] [PubMed] [Google Scholar]
- 33.Masuda, Y., M. Takahashi, N. Tsunekuni, T. Minami, M. Sumii, K. Miyagawa, and K. Kamiya. 2001. Deoxycytidyl transferase activity of the human REV1 protein is closely associated with the conserved polymerase domain. J. Biol. Chem. 276:15051-15058. [DOI] [PubMed] [Google Scholar]
- 34.McColl, D. J., N. A. Margot, M. Wulfsohn, D. F. Coakley, A. K. Cheng, and M. D. Miller. 2004. Patterns of resistance emerging in HIV-1 from antiretroviral-experienced patients undergoing intensification therapy with tenofovir disoproxil fumarate. J. Acquir. Immune Defic. Syndr. 37:1340-1350. [DOI] [PubMed] [Google Scholar]
- 35.McCulloch, S. D., and T. A. Kunkel. 2008. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 18:148-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moyle, G. 2000. Toxicity of antiretroviral nucleoside and nucleotide analogues: is mitochondrial toxicity the only mechanism? Drug Saf. 23:467-481. [DOI] [PubMed] [Google Scholar]
- 37.Olivero, O. A. 2007. Mechanisms of genotoxicity of nucleoside reverse transcriptase inhibitors. Environ. Mol. Mutagen. 48:215-223. [DOI] [PubMed] [Google Scholar]
- 38.Olivero, O. A., G. M. Shearer, C. A. Chougnet, A. A. Kovacs, A. L. Landay, R. Baker, A. M. Stek, M. M. Khoury, L. A. Proia, H. A. Kessler, B. E. Sha, R. E. Tarone, and M. C. Poirier. 1999. Incorporation of zidovudine into leukocyte DNA from HIV-1-positive adults and pregnant women, and cord blood from infants exposed in utero. AIDS 13:919-925. [DOI] [PubMed] [Google Scholar]
- 39.Patterson, T. A., W. Little, X. Cheng, S. G. Widen, A. Kumar, W. A. Beard, and S. H. Wilson. 2000. Molecular cloning and high-level expression of human polymerase beta cDNA and comparison of the purified recombinant human and rat enzymes. Protein Expr. Purif. 18:100-110. [DOI] [PubMed] [Google Scholar]
- 40.Perry, C. M., and D. Faulds. 1997. Lamivudine. A review of its antiviral activity, pharmacokinetic properties and therapeutic efficacy in the management of HIV infection. Drugs 53:657-680. [DOI] [PubMed] [Google Scholar]
- 41.Peyriere, H., J. Reynes, I. Rouanet, N. Daniel, C. M. de Boever, J. M. Mauboussin, H. Leray, L. Moachon, D. Vincent, and D. Salmon-Ceron. 2004. Renal tubular dysfunction associated with tenofovir therapy: report of 7 cases. J. Acquir. Immune Defic. Syndr. 35:269-273. [DOI] [PubMed] [Google Scholar]
- 42.Pruvost, A., E. Negredo, H. Benech, F. Theodoro, J. Puig, E. Grau, E. Garcia, J. Molto, J. Grassi, and B. Clotet. 2005. Measurement of intracellular didanosine and tenofovir phosphorylated metabolites and possible interaction of the two drugs in human immunodeficiency virus-infected patients. Antimicrob. Agents Chemother. 49:1907-1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramadan, K., I. Shevelev, and U. Hubscher. 2004. The DNA polymerase X family: controllers of DNA quality? Nat. Rev. Mol. Cell Biol. 5:1038-1043. [DOI] [PubMed] [Google Scholar]
- 44.Ray, A. S., E. Murakami, C. N. Peterson, J. Shi, R. F. Schinazi, and K. S. Anderson. 2002. Interactions of enantiomers of 2′,3′-didehydro-2′,3′-dideoxy-fluorocytidine with wild type and M184V mutant HIV-1 reverse transcriptase. Antiviral Res. 56:189-205. [DOI] [PubMed] [Google Scholar]
- 45.Reardon, J. E., R. C. Crouch, and L. St. John-Williams. 1994. Reduction of 3′-azido-3′-deoxythymidine (AZT) and AZT nucleotides by thiols. Kinetics and product identification. J. Biol. Chem. 269:15999-16008. [PubMed] [Google Scholar]
- 46.Rousseau, F. S., C. Wakeford, H. Mommeja-Marin, I. Sanne, C. Moxham, J. Harris, L. Hulett, L. H. Wang, J. B. Quinn, and D. W. Barry. 2003. Prospective randomized trial of emtricitabine versus lamivudine short-term monotherapy in human immunodeficiency virus-infected patients. J. Infect. Dis. 188:1652-1658. [DOI] [PubMed] [Google Scholar]
- 47.Schaaf, B., S. P. Aries, E. Kramme, J. Steinhoff, and K. Dalhoff. 2003. Acute renal failure associated with tenofovir treatment in a patient with acquired immunodeficiency syndrome. Clin. Infect. Dis. 37:e41-e43. [DOI] [PubMed] [Google Scholar]
- 48.Sherrer, S. M., K. A. Fiala, J. M. Pryor, S. A. Newmister, J. D. Fowler, and Z. Suo. 2010. Quantitative analysis of the efficiency and mutagenic spectra of abasic lesion bypass catalyzed by human Y-family DNA polymerases. Nucleic Acids Res. [Epub ahead of print.] doi: 10.1093/nar/gkq719. [DOI] [PMC free article] [PubMed]
- 49.Sommadossi, J. P., R. Carlisle, and Z. Zhou. 1989. Cellular pharmacology of 3′-azido-3′-deoxythymidine with evidence of incorporation into DNA of human bone marrow cells. Mol. Pharmacol. 36:9-14. [PubMed] [Google Scholar]
- 50.Suo, Z., and K. A. Johnson. 1998. Selective inhibition of HIV-1 reverse transcriptase by an antiviral inhibitor, (R)-9-(2-phosphonylmethoxypropyl) adenine. J. Biol. Chem. 273:27250-27258. [DOI] [PubMed] [Google Scholar]
- 51.Tuske, S., S. G. Sarafianos, A. D. Clark, Jr., J. Ding, L. K. Naeger, K. L. White, M. D. Miller, C. S. Gibbs, P. L. Boyer, P. Clark, G. Wang, B. L. Gaffney, R. A. Jones, D. M. Jerina, S. H. Hughes, and E. Arnold. 2004. Structures of HIV-1 RT-DNA complexes before and after incorporation of the anti-AIDS drug tenofovir. Nat. Struct. Mol. Biol. 11:469-474. [DOI] [PubMed] [Google Scholar]
- 52.Uwai, Y., H. Ida, Y. Tsuji, T. Katsura, and K. Inui. 2007. Renal transport of adefovir, cidofovir, and tenofovir by SLC22A family members (hOAT1, hOAT3, and hOCT2). Pharm. Res. 24:811-815. [DOI] [PubMed] [Google Scholar]
- 53.Wang, L. H., J. Begley, R. L. St. Claire III, J. Harris, C. Wakeford, and F. S. Rousseau. 2004. Pharmacokinetic and pharmacodynamic characteristics of emtricitabine support its once daily dosing for the treatment of HIV infection. AIDS Res. Hum. Retroviruses 20:1173-1182. [DOI] [PubMed] [Google Scholar]
- 54.Washington, M. T., S. A. Helquist, E. T. Kool, L. Prakash, and S. Prakash. 2003. Requirement of Watson-Crick hydrogen bonding for DNA synthesis by yeast DNA polymerase eta. Mol. Cell. Biol. 23:5107-5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Waters, L. S., B. K. Minesinger, M. E. Wiltrout, S. D'Souza, R. V. Woodruff, and G. C. Walker. 2009. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev. 73:134-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wolfle, W. T., M. T. Washington, E. T. Kool, T. E. Spratt, S. A. Helquist, L. Prakash, and S. Prakash. 2005. Evidence for a Watson-Crick hydrogen bonding requirement in DNA synthesis by human DNA polymerase kappa. Mol. Cell. Biol. 25:7137-7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wutzler, P., and R. Thust. 2001. Genetic risks of antiviral nucleoside analogues: a survey. Antiviral Res. 49:55-74. [DOI] [PubMed] [Google Scholar]
- 58.Yamtich, J., and J. B. Sweasy. 2010. DNA polymerase family X: function, structure, and cellular roles. Biochim. Biophys. Acta 1804:1136-1150. [DOI] [PMC free article] [PubMed] [Google Scholar]