

Abstract
In order to develop a completely safe immunogen to replace the traditional inactivated vaccine, a tandem-repeat multiple-epitope recombinant vaccine against foot-and-mouth disease (FMD) virus (FMDV) type O was developed. It contained three copies each of residues 141 to 160 and 200 to 213 of VP1 of the O/China/99 strain of FMDV coupled with a swine immunoglobulin G heavy-chain constant region (scIgG). The data showed that the multiple-epitope recombinant vaccine elicited high titers of anti-FMDV specific antibodies in swine at 30 days postvaccination (dpv) and conferred complete protection against a challenge with 103 50% swine infective doses of the O/China/99 strain. The anti-FMDV specific antibody titers were not significantly different between the multiple-epitope recombinant vaccine and the traditional vaccine (t test, P > 0.05). The number of 50% pig protective doses was 6.47, which is higher than the number recommended by the World Organization for Animal Health. The multiple-epitope recombinant vaccine resulted in a duration of immunity of at least 6 months. We speculate that the multiple-epitope recombinant vaccine is a promising vaccine that may replace the traditional inactivated vaccine for the prevention and control of FMD in swine in the future.
Foot-and-mouth disease (FMD) virus (FMDV) is a member of the genus Aphthovirus of the Picornaviridae family and is classified into seven distinct serotypes (O, A, C, SAT 1 to 3, and Asia 1), as well as numerous subtypes (4, 12). The virus causes highly contagious FMD in cloven-hoofed animals, and its devastating consequences have been demonstrated by the recent outbreaks in Taiwan and the United Kingdom (14, 24). Chemically inactivated whole-virus vaccines play a key role in the control and prevention of FMD (2, 3). However, the traditional vaccines have several disadvantages, such as the requirement for storage under refrigeration, the need for periodic revaccination, and the difficulty in differentiating infected from vaccinated animals (25, 26, 37). Furthermore, the immunogenic diversity of the seven distinct serotypes of FMDV necessitates serologic matching for the formulation of efficacious vaccines. Importantly, there is a potential risk of the escape of live virus from biosafety facilities during vaccine production or from residual live virus inside the vaccines (3, 4, 7). Another problem is that the conventional FMD vaccines do not induce sterile immunity and thus do not prevent a carrier status. For these and other reasons, alternative vaccines that do not require live virus material, such as subunit vaccines, synthetic peptides, DNA vaccines, and recombinant virus vaccines, have been explored extensively (5, 6, 13, 22, 41).
The epitopes located in residues 141 to 160 and 200 to 213 of the VP1 protein are the main immunogenic epitopes of FMDV (5, 11, 29). Previous studies have shown that synthetic peptides or recombinant proteins that contain one or both of the immunogenic epitopes can induce significant titers of neutralizing antibodies against FMDV and confer full protection against a challenge in small animals (36, 39). However, the immunogenicity of these vaccines was substantially lower than that of the traditional inactivated vaccines and afforded limited protection against a challenge in the natural hosts (7, 31, 34, 38, 39). This may be due to the rapid clearance of recombinant proteins or synthetic peptides of small size and the lack of strong and appropriate T-helper cell epitopes (17, 18, 30).
There are several approaches to improving the immunogenicity of antigenic epitopes, such as increasing the number of antigenic epitopes, providing multiple T-helper cell epitopes, and incorporating the antigenic epitopes into a protein “carrier” (8, 27, 29, 40, 42, 44). We have successfully generated a recombinant protein with swine immunoglobulin G (IgG) directed against FMDV as a carrier protein. The results of this study show that vaccinated swine were protected fully against a challenge with 50 50% swine infective doses (ID50) of FMDV.
In this study, to develop a completely safe vaccine that could replace the traditional inactivated vaccines, a recombinant vaccine against FMDV type O was modified further on the basis of the construction developed previously. The potency of this recombinant vaccine in swine was evaluated by a vaccine efficacy test and measurement of the duration of immunity.
MATERIALS AND METHODS
Challenge virus.
The O/China/99 strain of FMDV was obtained from the National FMD Reference Laboratory of the People's Republic of China. The virus was adapted and propagated for five passages in swine, and the titer of the ID50 was determined as described previously (1).
Animals.
Forty-six swine weighing 20 to 30 kg and free of antibodies against the structural proteins and 3ABC nonstructural proteins (NSP) of FMDV were chosen for three experiments. In experiment 1, the potency of the multiple-epitope recombinant vaccine was evaluated by comparison with that of a traditional inactivated vaccine. In experiment 2, the 50% pig protective dose (PD50) was determined according to standard procedures of the World Organization for Animal Health (OIE). In experiment 3, the duration of the immunity induced by the multiple-epitope recombinant vaccine was measured. All experiments were performed in high-containment facilities. All pig pens were separated completely, and each pen had an individual ventilation system. All tests were approved by the Animal Ethics Committee of the Animal Sciences Group of Gansu Province.
Design and synthesis of a tandem-repeat multiple-epitope gene.
Two immunogens corresponding to amino acid (aa) residues 141 to 160 and 200 to 213 of VP1 of the FMDV O/China/99 strain (GenBank accession no. AF506822) were chosen as the antigenic epitopes. A tandem-repeat multiple-epitope gene, 402 bp in length, which contained three copies of each sequence was synthesized by arranging the two epitopes in triplicate in the following order: 141 to 160 and 200 to 213 aa, 141 to 160 and 200 to 213 aa, and 141 to 160 and 200 to 213 aa. Briefly, aa 200 to 213 were linked to the C terminus of aa 141 to 160 and the artificial gene was synthesized by sequential linking of the tandem repeats. To minimize interference between adjacent epitopes and to avoid the development of a new epitope, a linker sequence, GGSSGG, was used to separate adjacent epitopes, and a 402-bp gene was synthesized and cloned into the pUC-18 vector to produce pUC-18-RE (Fig. 1).
FIG. 1.

Construction of a recombinant plasmid containing three copies of two epitopes. E1 represents the antigenic epitope corresponding to aa 141 to 160 of VP1 of FMDV type O. E2 represents the antigenic epitope corresponding to aa 200 to 213 of VP1 of FMDV type O. RE represents the tandem-repeat multiple-epitope gene. MCS, multiple cloning site.
PCR amplification of the swine immunoglobulin G heavy-chain constant region (scIgG) and RE genes.
The genes for RE and scIgG were amplified separately by PCR with the specific primers for recombinant plasmids pUC-18-RE and pET-16b-scIgG, which were constructed previously in our laboratory (Table 1). Briefly, 50.0 μl of reaction mixture contained 5.0 μl of 10× buffer, 5.0 μl of deoxynucleoside triphosphates (5 mM each), 1.0 μl of ExTaq, 1.0 μl of each primer (20 pmol), 0.5 μl of template DNA, and 36.5 μl of distilled water. The PCR was carried out as described below, by using 94°C for 5 min; 30 cycles of 94°C for 1 min, 56°C for 30 s, and 72°C for 30 s; and a final extension for 8 min at 72°C. The amplicon was purified using an agarose DNA purification kit (Takara) and stored at −20°C until use.
TABLE 1.
Sequences of primers used for PCR amplification in this study
| Gene and primera | Sequence (5′-3′)b | Length (bp) |
|---|---|---|
| RE | ||
| 3R5 | AGCTGGATCCGAAACCCAGGTGCAGCGTGT | 30 |
| v3R-IGC3 | TGGGGCCGTCTTGGGGGCAGAGCCTCCCGAACCGCCCAG | 39 |
| scIgG | ||
| 3R-scIgG5 | CTGGGCGGTTCGGGAGGCTCTGCCCCCAAGACGGCCCCA | 39 |
| scIgG3 | AGTCAAGCTTTCATCATTTACCCTGAGT | 28 |
| RE-scIgG | ||
| 3R5 | AGCTGGATCCGAAACCCAGGTGCAGCGTGT | 30 |
| scIgG3 | AGTCAAGCTTTTTACCCTGAGTCTTGGAGATGGA | 34 |
RE represents the tandem-repeat multiple-epitope DNA fragment. scIgG is the swine immunoglobulin heavy-chain constant gene. RE-scIgG is a fusion of the genes for RE and scIgG.
GGATCC, BamHI restriction endonuclease site. AAGCTT, HindIII restriction endonuclease site.
Construction of a recombinant RE-scIgG expression plasmid.
To obtain recombinant expression plasmid pET-22b-RE-scIgG, a chimeric gene for RE-scIgG was amplified by overlapping PCR with a set of specific primers (Table 1). Briefly, 50.0 μl of the reaction mixture contained 25.0 μl of 2× MightyAmp buffer, 1.0 μl of MightyAmp DNA polymerase, 1.0 μl of each primer (20 pmol), 1.0 μl each of purified RE and scIgG, and 20.0 μl of distilled water. The PCR was carried out at 98°C for 2 min; 30 cycles of 94°C for 10 s, 60°C for 10 s, and 68°C for 90 s; and a final extension at 72°C for 8 min. The amplicon was purified and subcloned into an expression vector, pET-22b (+), which resulted in a recombinant expression plasmid, pET-22b-RE-scIgG (Fig. 2). The recombinant expression plasmid was confirmed by digestion with BamHI/HindIII (NEB), and the positive recombinant plasmid was sequenced using Sanger's method to confirm the correct open reading frame (Takara).
FIG. 2.

Construction of a recombinant expression plasmid containing a fusion of the genes for RE and scIgG. MCS, multiple cloning site.
Expression and purification of recombinant protein.
Expression and purification of the recombinant protein in Escherichia coli BL21(DE3) cells (Novagen) were performed as described previously (23), with slight modifications. Briefly, the transformed bacteria were grown in 100 ml of LB medium (Amp+) at 37°C overnight in a shaker. Growth was monitored by measurements of optical density at 600 nm (OD600), and at an OD600 of 0.4 to 0.6, expression was induced with 0.8 mM isopropyl-β-d-thiogalactopyranoside (IPTG). The cells were grown for a further 6 h at 37°C and harvested by centrifugation at 4,000 × g for 20 min. After 1 h, the cells were sonicated on ice (10 × 30 s) and then centrifuged at 20,000 × g for 30 min at 4°C. The RE-scIgG protein was expressed with a six-histidine tag in E. coli BL21 in a formation of inclusion bodies. Cellular pellets that contained insoluble proteins were solubilized with 6 M urea overnight at 4°C. The supernatant was collected, incubated with Ni-nitrilotriacetic acid (NTA) agarose resin (Qiagen) (2.5 ml of resin for 1 liter of induced cells) equilibrated in buffer A (25 mM Tris, pH 8.0, 100 mM NaCl, 6 M urea) containing 5 mM imidazole (Sigma), and then stirred for 1 h to allow binding. The supernatant was poured into a column, and the bound protein and resin were washed with 10 column volumes of buffer A, 10 column volumes of increasing concentrations of imidazole (10, 20, and 40 mM), and 3 column volumes of 90 and 250 mM imidazole. Fractions of the eluate were analyzed by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The concentration of each protein was determined by Bio-Rad Protein Assay according to the manufacturer's procedure (Bio-Rad). Briefly, 100 μl of 2× staining reagent was added to a 96-well microplate, and a series of different volumes (0, 1, 2, 4, 6, 8, and 10 μl) of standard bovine serum albumin (BSA, 1 mg/ml; Sigma) were added in duplicate to the corresponding wells. Meanwhile, appropriate concentrations of the sample (10 μl) were added in duplicate. Subsequently, distilled water was added to a final volume of 200 μl and the plate was placed at room temperature for 5 min. The OD595 values were read on a spectrophotometer (BioTek). A standard curve was drawn, and the concentration of sample protein was determined from the standard curve.
Western blotting.
The immunoreactivity of the recombinant protein was determined by Western blotting. Briefly, the purified protein was subjected to 12% SDS-PAGE and then transferred to a polyvinylidene difluoride membrane, which was blocked by phosphate-buffered saline (PBS) with 10% horse serum for 1 h in a shaker and then washed three times with PBS containing 0.05% Tween 20 (PBST). The membrane was incubated with a 1:500 dilution of positive serum from cattle infected with FMD type O, washed three times with PBST, and incubated with a 1:2,000 dilution of rabbit anti-cattle IgG antibody conjugated with horseradish peroxidase (HRP; Sigma) for 1 h. After five washes with PBST, the signals were developed with 3,3′,5,5′-tetramethylbenzidine.
Preparation of multiple-epitope recombinant vaccine.
The optimal concentration of the recombinant protein (0.25 mg/dose) had been determined previously. The recombinant protein was emulsified with Montanide ISA 206 (Seppic, France) to prepare a double oil emulsion formulation as described previously (2). Briefly, the purified protein was diluted to a final concentration of 0.5 mg/ml and mixed thoroughly with an equal volume of oil adjuvant. The finished vaccine formulation contained 0.25 mg of the purified protein per ml and was stored at 4°C until use.
Statistical analysis.
Statistical comparisons were performed with SPSS software by using the two-tailed Student t test for comparison of two data sets.
Comparison of potency of the multiple-epitope recombinant vaccine with a traditional inactivated vaccine in swine.
Thirteen swine were divided into three groups. Group 1 was inoculated intramuscularly behind the ear with 2 ml of a commercially inactivated vaccine (type O), which had a duration of immunity of at least 6 months (China Agricultural Vet Bio. Science and Technology Co., Ltd.). Group 2 was vaccinated with a full dose of the multiple-epitope recombinant vaccine. Group 3 (three swine) received only 1 ml of PBS in oil adjuvant. At 30 days postvaccination (dpv), serum samples were collected and all animals were challenged intradermally in the bulb of the heel of the left hind foot with 103 ID50 of the O/China/99 strain of FMDV. After the challenge, rectal temperatures and clinical signs were monitored daily for 10 days.
Potency of the multiple-epitope recombinant vaccine in swine.
Eighteen swine were separated into four groups and used to determine the PD50 of the vaccine in swine. Groups 1 to 3 were vaccinated intramuscularly behind the ear with a full dose (1 ml), 1/3 of a dose (0.33 ml), or 1/9 of a dose (0.11 ml) of the multiple-epitope recombinant vaccine, respectively. Group 4 received only 1 ml PBS in oil adjuvant. At 30 dpv, all of the swine were bled and challenged as described above. To prevent the protected animals from receiving an excess challenge from infected animals, animals that showed signs of disease were removed promptly from the group. The PD50 was estimated by Kärber's method (OIE, 2004 version). Antibodies to FMDV were detected by liquid-phase blocking enzyme-linked immunosorbent assay (LPB-ELISA) and microneutralization assay.
Duration of immunity induced by the multiple-epitope recombinant protein in swine.
Three sets of the multiple-epitope recombinant vaccines (A to C) were prepared as described above. Fifteen swine were separated equally into three groups, which were vaccinated intramuscularly behind the ear with a full dose of vaccines A to C, respectively. Serum samples were collected monthly for 7 months (30, 60, 90, 120, 150, 180, and 210 days) postvaccination, and anti-FMDV antibodies were detected by LPB-ELISA. The correlation between the number of dpv and the antibody level was determined. The titers of anti-FMDV antibodies from all vaccinated swine were analyzed by using a statistical method (Student's t test).
LPB-ELISA.
The titers of anti-FMDV antibodies were detected with a commercial LPB-ELISA kit; all procedures were performed by following the manufacturer's instructions. Briefly, 50-μl volumes of a duplicate 2-fold series of each test serum were prepared in U-bottom multiwell plates (Costar). To each well, 50 μl of a constant dose of viral antigen that was homologous to the rabbit antisera used to coat the plates was added, and the mixtures were left overnight at 4°C. Subsequently, 50 μl of the serum-antigen mixture was transferred to an ELISA plate precoated with rabbit anti-FMDV serum and incubated at 37°C for 1 h at a dilution of 1:1,000. After thorough washing with PBST, 50 μl of guinea-pig antiserum was added to each well and the plates were incubated for 1 h at 37°C. The plates were washed five times with PBST, 50 μl of rabbit anti-guinea pig IgG-HRP at a dilution of 1:500 (Sigma) was added, and the plates were incubated at 37°C for 1 h. After five washes with PBST, the enzyme substrate o-phenylenediamine (Sigma) was added to each well for 10 to 15 min of incubation. The reaction was terminated with 2 M H2SO4, and the plate was read at 492 nm on a spectrophotometer (BioTek).
Virus neutralization assay.
Assays to detect neutralizing antibodies were carried out as described previously (20). Endpoint titers were calculated as the reciprocal of the last serum dilution to neutralize 100 50% tissue culture infective doses of homologous FMDV in 50% of the wells.
Lymphocyte proliferation assay.
The proliferation of lymphocytes was analyzed by flow cytometry as described previously (19). Briefly, peripheral blood mononuclear cells (PBMC) were selected by centrifugation over Ficoll-Hypaque (density, 1.007 g/liter). The cells were resuspended at a final concentration of 5 × 107 cells/ml in RPMI 1640 and stained with 2 μM 5(6)-carboxyfluorescein diacetate N-succinimidyl ester (Sigma) for 15 min at 37°C. Five volumes of ice-cold RPMI 1640 with 10% fetal bovine serum was added for 5 to 10 min of incubation at room temperature to quench staining. Finally, the cells were resuspended at 1.5 × 106/ml in complete RPMI 1640 medium (10% fetal bovine serum, 2 mM glutamine, 1 mM sodium pyruvate, 10 mM HEPES, 50 mM β-mercaptoethanol, ampicillin [100 IU/ml], and streptomycin [100 μg/ml]) and dispensed into a 96-well tissue culture plate together with 10 μg of purified FMDV antigen (type O) and 5 μg/ml concanavalin A (Sigma). The negative control received only the culture medium. BSA (10 μg/ml; Sigma) was used as a noncorrelated antigen control in the test. The cultures were incubated for 72 h at 37°C in an incubator with 5% CO2. The proportions of lymphocyte proliferation were determined using a FACScalibur (BD Biosciences).
Virus isolation from heparinized blood and nasal swabs.
Heparinized blood and nasal swabs were obtained daily from a group of five swine vaccinated with the full dose from day 0 to day 9 postchallenge and stored at −70°C until use. Virus isolation from the blood samples and nasal swabs was performed as described previously (28).
Detection of antibodies to NSP of FMDV.
To confirm that antibodies to NSP of FMDV were absent from the swine vaccinated with a full dose of the multiple-epitope recombinant vaccine after challenge, antibodies to NSP of FMDV were detected by a commercially available kit and all procedures were carried out by following the manufacturer's instructions (25).
RESULTS
Expression and characterization of recombinant RE-scIgG protein.
Sequencing results showed that recombinant expression plasmid pET-22b-RE-scIgG was constructed successfully. The recombinant protein was expressed as a formation of inclusion bodies in the E. coli expression system, and a specific band of 52 kDa that was consistent with the expected size of the recombinant protein RE-scIgG could be visualized clearly by 12% SDS-PAGE, whereas no band was found in lysates of E. coli/pET-22b (+) cells (Fig. 3). SDS-PAGE showed that the purity of the recombinant protein was about 95% after purification in the Ni affinity column (Fig. 4). The recombinant protein RE-scIgG could be recognized specifically by anti-FMDV (type O) antibodies (Fig. 5).
FIG. 3.
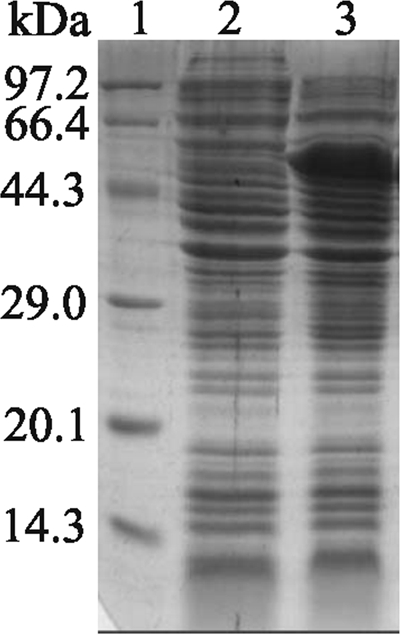
SDS-PAGE analysis of recombinant protein RE-scIgG in E. coli. Lanes: 1, protein molecular size markers; 2, expression of plasmid pET-22b (+) induced with IPTG after 4 h; 3, expression of recombinant plasmid pET-22b-RE-scIgG induced with IPTG after 4 h.
FIG. 4.
Analysis of solubility of expressed RE-scIgG. Lanes: 1, protein molecular size markers; 2, expression of recombinant plasmid pET-22b-RE-scIgG preinduced with IPTG; 3, expression of recombinant plasmid pET-22b-RE-scIgG induced with IPTG after 6 h; 4, deposition of recombinant protein RE-scIgG after sonication; 5, protein RE-scIgG purified with Ni-NTA agarose resin.
FIG. 5.

Western blotting of recombinant protein. Lanes: 1, protein molecular size markers; 2, recombinant protein RE-scIgG reacted with serum negative for FMDV; 3, recombinant protein RE-scIgG reacted with positive serum from cattle infected with FMDV type O.
Comparison of the potency of the multiple-epitope recombinant vaccine with a commercial vaccine in swine.
The development of protective antibody responses in natural hosts is essential to the efficient prevention of viral infection. Therefore, the potency of the multiple-epitope recombinant protein was evaluated according to the titers of anti-FMDV specific antibodies in swine. As shown in Table 2, high titers of anti-FMDV specific antibodies were elicited in swine vaccinated with the recombinant vaccine at 30 dpv. No significant difference in antibody titers was found between the multiple-epitope recombinant vaccine and a traditional inactivated vaccine (t test, P > 0.05). Like the traditional inactivated vaccine, the multiple-epitope recombinant vaccine conferred full protection against a challenge with 103 ID50 of the FMDV O/China/99 strain in swine. The typical signs of FMD, including fever (39 to 42°C), depression, anorexia, lameness, and the formation of vesicles, developed in all four feet and the snout of each animal in the control group.
TABLE 2.
Titers of antibodies against FMDV and swine challenge test results
| Group and no. of dpva | Antibody titers | Ratio of protection (%) |
|---|---|---|
| Recombinant protein | ||
| 0 | <1:4, <1:4, <1:4, <1:4, <1:4 | 5/5 (100) |
| 30 | 1:128, 1:90, 1:90, 1:64, 1:90 | |
| Inactivated vaccine | ||
| 0 | <1:4, <1:4, <1:4, <1:4, <1:4 | 5/5 (100) |
| 30 | 1:128, 1:90, 1:64, 1:128, 1:64 | |
| PBS control | ||
| 0 | <1:4, <1:4, <1:4, <1:4, <1:4 | 0/3 (0) |
| 1 | <1:4, <1:4, <1:4 |
dpv, days postvaccination.
Potency of the multiple-epitope recombinant vaccine in swine.
The potency of the immunity induced by the multiple-epitope recombinant vaccine was evaluated as recommended in the OIE manual (2004 version). As shown in Table 3, high titers of antibodies against FMDV were elicited in the swine. The titers of anti-FMDV antibodies were lower when the dosage of the recombinant vaccine was reduced. One pig in the group given 1/3 of a dose of the vaccine and three pigs in the group given 1/9 of a dose of vaccine developed clinical signs of FMDV, but the time of disease onset was delayed and the severity of disease was reduced compared to that in the control animals. All control swine were completely susceptible and developed clinical signs and vesicles in the feet and snout. The PD50 of 6.47 was higher than that recommended by the OIE (3.0).
TABLE 3.
Titers of antibodies against FMDV, protection ratios, and PD50 in swine
| Group and no. of dpva | LPB-ELISA antibody titers | Log10 virus-neutralizing antibody titers | Protection ratio (%)c |
|---|---|---|---|
| Full dose | |||
| 0 | <1:4, <1:4, <1:4, <1:4, <1:4 | —b | 5/5 (100) |
| 30 | 1:128, 1:90, 1:64, 1:128, 1:64 | 1.98, 1.74, 1.81, 2.11, 1.81 | |
| 1/3 dose | |||
| 0 | <1:4, <1:4, <1:4, <1:4, <1:4 | — | 4/5 (80) |
| 30 | 1:45, 1:64, 1:90, 1:64, 1:22 | 1.81, 1.74, 1.68, 1.68, 1.38 | |
| 1/9 dose | |||
| 0 | <1:4, <1:4, <1:4, <1:4, <1:4 | — | 2/5 (40) |
| 30 | 1:22, 1:11, 1:64, 1:22, 1:45 | 1.20, 1.08, 1.68, 1.38, 1.68 | |
| Negative | |||
| 0 | <1:4, <1:4, <1:4 | — | 0/3 (0) |
| 30 | <1:4, <1:4, <1:4 | — |
dpv, days postvaccination.
—, no detectable neutralizing antibodies to FMDV type O.
The PD50 was 6.47.
Duration of immunity induced by the multiple-epitope recombinant vaccine in swine.
All three sets of the multiple-epitope recombinant vaccine elicited strong immune responses in swine. The titers of anti-FMDV antibodies reached a peak at 30 dpv, a plateau developed from 30 to 90 dpv, and the levels decreased gradually by 210 dpv. The high antibody titers (>1:64) lasted for more than 120 days in swine. However, the titers of anti-FMDV antibodies in 30% of the swine were significantly decreased (<1:45) at 210 dpv (Tables 4). No significant differences between the antibody titers elicited by multiple-epitope recombinant vaccines A, B, and C in swine were observed (t test, P > 0.05).
TABLE 4.
Titers of antibodies to FMDV induced in swine by three batches of vaccine
| No. of dpva and vaccine batch | Antibody titers |
|---|---|
| 0 | |
| A | <1:4, <1:4, <1:4, <1:4, <1:4 |
| B | <1:4, <1:4, <1:4, <1:4, <1:4 |
| C | <1:4, <1:4, <1:4, <1:4, <1:4 |
| 30 | |
| A | 1:360, 1:90, 1:180, 1:360, 1:256 |
| B | 1:360, 1:360, 1:360, 1:360, 1:256 |
| C | 1:90, 1:180, 1:180, 1:360, 1:90 |
| 60 | |
| A | 1:256, 1:90, 1:180, 1:360, 1:180 |
| B | 1:360, 1:360, 1:256, 1:360, 1:180 |
| C | 1:90, 1:180, 1:180, 1:256, 1:90 |
| 90 | |
| A | 1:180, 1:90, 1:180, 1:256, 1:180 |
| B | 1:256, 1:256, 1:256, 1:256, 1:128 |
| C | 1:90, 1:120, 1:90, 1:128, 1:90 |
| 120 | |
| A | 1:90, 1:90, 1:128, 1:256, 1:90 |
| B | 1:90, 1:128, 1:128, 1:128, 1:128 |
| C | 1:45, 1:90, 1:64, 1:90, 1:45 |
| 150 | |
| A | 1:45, 1:90, 1:90, 1:128, 1:90 |
| B | 1:90, 1:128, 1:90, 1:90, 1:90 |
| C | 1:45, 1:64, 1:64, 1:90, 1:45 |
| 180 | |
| A | 1:45, 1:90, 1:90, 1:90, 1:90 |
| B | 1:64, 1:45, 1:90, 1:45, 1:90 |
| C | 1:45, 1:45, 1:45, 1:90, 1:45 |
| 210 | |
| A | 1:22, 1:45, 1:64, 1:90, 1:90 |
| B | 1:45, 1:45, 1:45, 1:64, 1:32 |
| C | 1:22, 1:90, 1:22, 1:90, 1:22 |
dpv, days postvaccination.
Virus neutralization antibodies.
Table 3 shows that a full dose of the multiple-epitope recombinant vaccine produced high titers of FMDV-neutralizing antibodies (≥1.65 log10 [virus neutralization test]), which met or exceeded the value recommended by the OIE. However, one pig in the group given 1/3 of a dose of vaccine and three pigs in the group given 1/9 of a dose of vaccine produced a low titer of neutralizing antibody (<1.65 log10 [virus neutralization test]) that did not meet the value recommended by the OIE.
Lymphocyte proliferation assay.
As shown in Fig. 6, higher percentages of lymphocyte proliferation were obtained with RE-ocIgG, which differed significantly from those of the control group (P < 0.01, t test), but there was no significant difference between the traditional inactivated vaccine and the multiple-epitope recombinant vaccine (P > 0.05, t test).
FIG. 6.

Profile of lymphocyte proliferation from PBMC prior to or after stimulation. N, negative control; BSA, a noncorrelated antigen control. Antigen, purified FMDV (type O) whole-virus antigen. ConA, concanavalin A, a nonspecific stimulator of lymphocyte proliferation used as a positive control.
Virus isolation from heparinized blood and nasal swabs.
All control swine were positive for virus isolation from plasma and nasal swabs from day 2 to day 9 after challenge. Four of five clinically protected animals that were vaccinated with a full dose of vaccine were negative for virus isolation from plasma and nasal swabs. Only one pig in this group was positive for virus isolation from nasal swabs on days 3 to 5 postchallenge, but no virus was recovered from plasma at that time.
Detection of antibodies to NSP of FMDV.
Antibodies to NSP of FMDV could be detected in two of three pigs in the control group for the first time at either 7 or 9 days postinfection. All swine vaccinated with the multiple-epitope recombinant vaccine produced no detectable antibodies to NSP of FMDV throughout the duration of the experiment.
DISCUSSION
Vaccination is still one of the most important policies for the control and prevention of FMD. However, the chemically inactivated whole-virus vaccines are not advocated in most developed countries because of the disadvantages mentioned above. In 2001, an epidemic of FMD in the United Kingdom had a disastrous impact on the livestock industry and led to ardent discussions of the policies for the control and prevention of FMD in the future. Development of a safe, effective, and novel vaccine to replace the traditional vaccines is necessary for the control and eradication of FMD worldwide.
In this study, we improved a multiple-epitope recombinant vaccine against FMDV type O in swine. This vaccine not only elicited high titers of anti-FMDV specific antibodies and strong lymphocyte proliferation responses but offered complete protection against a challenge with 103 ID50 of the FMDV O/China/99 strain in swine. Notably, the recombinant vaccine elicited long-lasting immunity (6 months) in swine after a single vaccination, which is consistent with a vaccine of high potential reported previously (9, 16). In particular, the PD50 of 6.47 (Kärber) was 2-fold higher than the 3.0 recommended in the OIE manual (2004 version). Although FMDV was recovered from the nasal swabs from one clinically protected pig 3 to 5 days after a challenge, no vaccinated pig showed viremia during the trial period. In addition, swine vaccinated with a full dose of the multiple-epitope recombinant vaccine remained negative for antibodies to the NSP of FMDV after a challenge. These results suggest that our vaccine can elicit a strong immune response and offer sterilize immunity in swine. Previous reports have demonstrated that vaccination can induce rapid clinical protection against a challenge in the natural hosts (15, 20). In addition, other reports have indicated that the cellular immune responses and functional cytokines, such as type I and II interferons, can offer clinical protection in swine (10, 21, 28, 32, 33). Therefore, in addition to high titers of neutralizing antibodies, the cellular immune responses and antiviral and/or immunomodulatory molecules play a very important role in conferring clinical protection in the natural hosts. In our study, a vaccine was developed by genetic engineering that showed a high potential, which may be explained as described below. First, the immunogenicity of antigenic epitopes can be enhanced significantly by increasing the number of antigenic epitopes. Second, protein IgG molecules may prolong the half-life of the antigenic epitopes in vivo and show an improved ability to deliver peptides to the major histocompatibility complex molecules in situ (43). Therefore, the recombinant RE-scIgG protein may stimulate the immune system to produce persistent immune responses against the specific epitopes in vivo (8, 42).
The multiple-epitope recombinant vaccine has numerous advantages as an alternative to traditional vaccines. First, a recombinant protein can be developed by genetic engineering and does not involve the use of infectious factors, including RNA and live virus. Second, a recombinant protein, being a denatured protein, can be stored easily. Third, an expressed recombinant protein accounts for more than 50% of the total cellular protein, and the purity of the target protein is greater than 95%. This fusion protein is purified easily with an affinity column, and the production costs can therefore be reduced, which is one of the most important factors in vaccine production. In comparison with expression systems that involve insect or mammalian cells, a large quantity of protein can be obtained easily from the E. coli expression system. Fourth, the development of a multiple-epitope recombinant vaccine is easier than that of a traditional inactivated vaccine because of the use of genetic engineering and bioinformatics. Finally, multiple-epitope recombinant vaccines do not elicit antibodies against NSP of FMDV and therefore NSP tests can be used to distinguish vaccinated from infected animals (25, 26, 35).
In conclusion, we have developed a multiple-epitope recombinant vaccine against FMDV type O in swine. This multiple-epitope recombinant vaccine can elicit high titers of anti-FMDV specific antibodies and confer complete protection against a challenge in swine. The PD50 of 6.47 met or exceeded the value recommended by the OIE. This novel multiple-epitope recombinant vaccine could replace the traditional inactivated vaccines and may be used for the control and eradication of FMD in the future.
Acknowledgments
We thank X. Q. Zhu for critical review of the manuscript, and we also thank J. Chen and W. M. Ma for technical assistance.
This work was supported by the National Science and Technology Pillar Program (2006DAD06A03), the National High Technology Research and Development Program of China (2006AA10A204), and the Gansu Province Key Program of Science and Technology (grant 2009GS00094).
Footnotes
Published ahead of print on 17 November 2010.
REFERENCES
- 1.Alexandersen, S., and A. I. Donaldson. 2002. Further studies to quantify the dose of natural aerosols of foot-and-mouth disease virus for pigs. Epidemiol. Infect. 128:313-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barnett, P. V., L. Pullen, L. Williams, and T. R. Doel. 1996. International bank for foot-and-mouth disease vaccine: assessment of Montanide ISA 25 and ISA 206, two commercially available oil adjuvants. Vaccine 14:1187-1198. [DOI] [PubMed] [Google Scholar]
- 3.Barteling, S. J., and J. Vreeswijk. 1991. Developments in foot-and-mouth disease vaccines. Vaccine 9:75-88. [DOI] [PubMed] [Google Scholar]
- 4.Beck, E., and K. Strohmaier. 1987. Subtyping of European foot-and-mouth disease virus strains by nucleotide sequence determination. J. Virol. 61:1621-1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bittle, J. L., et al. 1982. Protection against foot-and-mouth disease by immunization with a chemically synthesized peptide predicted from the viral nucleotide sequence. Nature 298:30-33. [DOI] [PubMed] [Google Scholar]
- 6.Borrego, B., et al. 2006. DNA vaccines expressing B and T cell epitopes can protect mice from FMDV infection in the absence of specific humoral responses. Vaccine 24:3889-3899. [DOI] [PubMed] [Google Scholar]
- 7.Brown, F. 1992. New approaches to vaccination against foot-and-mouth disease. Vaccine 10:1022-1026. [DOI] [PubMed] [Google Scholar]
- 8.Chan, E. W. C., et al. 2000. An immunoglobulin G based chimeric protein induced foot-and-mouth disease specific immune response in swine. Vaccine 19:538-546. [DOI] [PubMed] [Google Scholar]
- 9.Chung, W. B., et al. 2002. Optimization of foot-and-mouth disease vaccination protocols by surveillance of neutralization antibodies. Vaccine 20:2665-2670. [DOI] [PubMed] [Google Scholar]
- 10.de Avila Botton, S., et al. 2006. Immunopotentiation of a foot-and-mouth disease virus subunit vaccine by interferon alpha. Vaccine 24:3446-3456. [DOI] [PubMed] [Google Scholar]
- 11.DiMarchi, R., et al. 1986. Protection of cattle against foot-and-mouth disease by a synthetic peptide. Science 232:639-641. [DOI] [PubMed] [Google Scholar]
- 12.Doel, T. R. 2003. FMD vaccines. Virus Res. 91:81-99. [DOI] [PubMed] [Google Scholar]
- 13.Doel, T. R., C. Gale, C. M. Do Amaral, G. Mulcahy, and R. Dimarchi. 1990. Heterotypic protection induced by synthetic peptides corresponding to three serotypes of foot-and-mouth disease virus. J. Virol. 64:2260-2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dunn, C. S., and A. I. Donaldson. 1997. Natural adaption to swine of a Taiwanese isolate of foot-and-mouth disease virus. Vet. Rec. 141:174-175. [DOI] [PubMed] [Google Scholar]
- 15.Eblé, P. L., et al. 2004. Vaccination of pigs two weeks before infection significantly reduces transmission of foot-and-mouth disease virus. Vaccine 22:1372-1378. [DOI] [PubMed] [Google Scholar]
- 16.Francis, M. J., and L. Black. 1986. Humoral response of pregnant sows to foot and mouth disease vaccination. J. Hyg. (Lond.) 96:501-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.García-Briones, M. M., et al. 2004. Immunogenicity and T cell recognition in swine of foot-and-mouth disease virus polymerase 3D. Virology 322:264-275. [DOI] [PubMed] [Google Scholar]
- 18.Gerner, W., et al. 2006. Identification of novel foot-and-mouth disease virus specific T-cell epitopes in c/c and d/d haplotype miniature swine. Virus Res. 121:223-228. [DOI] [PubMed] [Google Scholar]
- 19.Gerner, W., S. E. Hammer, K. H. Wiesmüller, and A. Saalmüller. 2009. Identification of major histocompatibility complex restriction and anchor residues of foot-and-mouth disease virus-derived bovine T-cell epitopes. J. Virol. 83:4039-4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Golde, W. T., et al. 2005. Vaccination against foot-and-mouth disease virus confers complete clinical protection in 7 days and partial protection in 4 days: use in emergency outbreak response. Vaccine 23:5775-5782. [DOI] [PubMed] [Google Scholar]
- 21.Guzman, E., G. Taylor, B. Charleston, M. A. Skinner, and S. A. Ellis. 2008. An MHC-restricted CD8+ T-cell response is induced in cattle by foot-and-mouth disease virus (FMDV) infection and also following vaccination with inactivated FMDV. J. Gen. Virol. 89:667-675. [DOI] [PubMed] [Google Scholar]
- 22.He, Y. N., et al. 2008. Construction and immune response characterization of a recombinant pseudorabies virus co-expressing capsid precursor protein (P1) and a multiepitope peptide of foot-and-mouth disease virus in swine. Virus Genes 36:393-400. [DOI] [PubMed] [Google Scholar]
- 23.Kadkhodayan, S., et al. 2000. Cloning, expression, and one-step purification of the minimal essential domain of the light chain of botulinum neurotoxin type A. Protein. Expr. Purif. 19:125-130. [DOI] [PubMed] [Google Scholar]
- 24.Knowles, N. J., A. R. Samuel, P. R. Davies, R. P. Kitching, and A. I. Donaldson. 2001. Outbreak of foot-and-mouth disease virus serotype O in the UK caused by a pandemic strain. Vet. Rec. 148:258-259. [PubMed] [Google Scholar]
- 25.Lu, Z., et al. 2007. Development and validation of a 3ABC indirect ELISA for differentiation of foot-and-mouth disease virus infected from vaccinated animals. Vet. Microbiol. 125:157-169. [DOI] [PubMed] [Google Scholar]
- 26.Mackay, D. K., et al. 1998. Differentiating infection from vaccination in foot-and-mouth disease using a panel of recombinant, non-structural proteins in ELISA. Vaccine 16:446-459. [DOI] [PubMed] [Google Scholar]
- 27.Meloen, R. H., J. I. Casal, K. Dalsgaard, and J. P. Langeveld. 1995. Synthetic peptide vaccines: success at last. Vaccine 13:885-886. [DOI] [PubMed] [Google Scholar]
- 28.Moraes, M. P., et al. 2007. Enhanced antiviral activity against foot-and-mouth disease virus by a combination of type I and II porcine interferons. J. Virol. 81:7124-7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morgan, D. O., and D. M. Moore. 1990. Protection of cattle and swine against foot-and-mouth disease, using biosynthetic peptide vaccines. Am. J. Vet. Res. 51:40-45. [PubMed] [Google Scholar]
- 30.Pfaff, E., M. Mussgay, H. O. Bohm, G. E. Schulz, and H. Schaller. 1982. Antibodies against a preselected peptide recognize and neutralize foot and mouth disease virus. EMBO. J. 1:869-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodriguez, L. L., et al. 2003. A synthetic peptide containing the consensus sequence of the G-H loop region of foot-and-mouth disease virus type O VP1 and a promiscuous T-helper epitope induces peptide-specific antibodies but fails to protect cattle against viral challenge. Vaccine 21:3751-3756. [DOI] [PubMed] [Google Scholar]
- 32.Sanz-Parra, A., et al. 1999. Evidence of partial protection against foot-and-mouth disease in cattle immunized with a recombinant adenovirus vector expressing the precursor polypeptide (P1) of foot-and-mouth disease virus capsid proteins. J. Gen. Virol. 80:671-679. [DOI] [PubMed] [Google Scholar]
- 33.Sanz-Parra, A., et al. 1999. Recombinant viruses expressing the foot-and-mouth disease virus capsid precursor polypeptide (P1) induce cellular but not humoral antiviral immunity and partial protection in pigs. Virology 259:129-134. [DOI] [PubMed] [Google Scholar]
- 34.Sobrino, F., E. Blanco, M. García-Briones, and V. Ley. 1999. Synthetic peptide vaccines: foot-and-mouth disease virus as a model. Dev. Biol. Stand. 101:39-43. [PubMed] [Google Scholar]
- 35.Sørensen, K. J., K. de Stricker, K. C. Dyrting, S. Grazioli, and B. Haas. 2005. Differentiation of foot-and-mouth disease virus infected animals from vaccinated animals using a blocking ELISA based on baculovirus expressed FMDV 3ABC antigen and a 3ABC monoclonal antibody. Arch. Virol. 150:805-814. [DOI] [PubMed] [Google Scholar]
- 36.Su, C. X., et al. 2007. Heterologous expression of FMDV immunodominant epitopes and HSP70 in P. pastoris and the subsequent immune response in mice. Vet. Microbiol. 124:256-263. [DOI] [PubMed] [Google Scholar]
- 37.Sutmoller, P., and R. Casas Olascoaga. 2003. The risks posed by the importation of animals vaccinated against foot and mouth disease and products derived from vaccinated animals: a review. Rev. Sci. Tech. 22:823-835. [DOI] [PubMed] [Google Scholar]
- 38.Taboga, O., et al. 1997. A large-scale evaluation of peptide vaccines against foot-and-mouth disease: lack of solid protection in cattle and isolation of escape mutants. J. Virol. 71:2606-2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Lierop, M. J., J. P. Wagenaar, J. M. van Noort, and E. J. Hensen. 1995. Sequences derived from the highly antigenic VP1 region 140 to 160 of foot-and-mouth disease virus do not prime for a bovine T-cell response against intact virus. J. Virol. 69:4511-4514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Winther, M. D., G. Allen, R. H. Bomford, and F. Brown. 1986. Bacterially expressed antigenic peptide from foot-and-mouth disease virus capsid elicits variable immunologic responses in animals. J. Immunol. 136:1835-1840. [PubMed] [Google Scholar]
- 41.Wu, Q., M. P. Moraes, and M. J. Grubman. 2003. Recombinant adenovirus co-expressing capsid proteins of two serotypes of foot-and-mouth disease virus (FMDV): in vitro characterization and induction of neutralizing antibodies against FMDV in swine. Virus Res. 93:211-219. [DOI] [PubMed] [Google Scholar]
- 42.Yi, J. Z., et al. 2004. Recombinant bivalent vaccine against foot-and-mouth disease virus serotype O/A infection in guinea pig. Acta Biochim. Biophys. Sin. (Shanghai) 36:589-596. [DOI] [PubMed] [Google Scholar]
- 43.Zaghouani, H., et al. 1993. Presentation of a viral T cell epitope expressed in the CDR3 region of a self immunoglobulin molecule. Science 259:224-227. [DOI] [PubMed] [Google Scholar]
- 44.Zamorano, P., et al. 1995. A 10-amino-acid linear sequence of VP1 of foot-and-mouth disease virus containing B- and-T-cell epitopes induces protection in mice. Virology 212:614-621. [DOI] [PubMed] [Google Scholar]