

Abstract
Streptococcus pneumoniae is a respiratory pathogen, and mucosal immune response plays a significant role in the defense against pneumococcal infections. Thus, intranasal vaccination may be an alternative approach to current immunization strategies, and effective delivery systems to mucosal organism are necessary. In this study, BALB/c mice were immunized intranasally with chitosan-DNA nanoparticles expressing pneumococcal surface antigen A (PsaA). Compared to levels in mice immunized with naked DNA or chitosan-pVAX1, anti-PsaA IgG antibody in serum and anti-IgA antibody in mucosal lavages were elevated significantly in mice immunized with chitosan-psaA. The balanced IgG1/IgG2a antibody ratio in serum, enhanced gamma interferon (IFN-γ) and IL-17A levels in spleen lymphocytes, and mucosal washes of mice immunized with chitosan-psaA suggested that cellular immune responses were induced. Furthermore, significantly fewer pneumococci were recovered from the nasopharynx of mice immunized with chitosan-psaA than for the control group following intranasal challenge with ATCC 6303 (serotype 3). These results demonstrated that mucosal immunization with chitosan-psaA may successfully generate mucosal and systemic immune responses and prevent pneumococcal nasopharyngeal colonization. Hence, a chitosan-DNA nanoparticle vaccine expressing pneumococcal major immunodominant antigens after intranasal administration could be developed to prevent pneumococcal infections.
Streptococcus pneumoniae is the most common cause of invasive pneumococcal diseases, including meningitis, septicemia, and bacteremic pneumonia, and noninvasive pneumococcal diseases such as acute otitis media, sinusitis, and nonbacteremic pneumonia. Antibiotics often are prescribed for the treatment of pneumococcal infections. However, the morbidity and mortality of pneumococcal diseases are still high in both developed and developing countries due to worldwide increasing antibiotic resistance (41). Therefore, the prevention of pneumococcal diseases is of great interest. S. pneumoniae is part of the commensal flora of the upper respiratory tract and colonizes the nasopharyngeal niche. The asymptomatic nasopharyngeal carriage of pneumococci is widely prevalent among young children worldwide (5). The carriage rate of S. pneumoniae in Chinese children ranges from 5.1 to 40.5% (51). Although colonization with pneumococci is mostly symptomless, it can progress to local or even systemic pneumococcal diseases (6). Therefore, vaccines to prevent pneumococcal diseases should focus on the prevention of nasopharyngeal colonization. However, the current 23-valent capsular polysaccharide vaccine is not able to elicit protective antibodies in infants who suffer the highest rates of pneumococcal carriages and infections (8). Although the current 7-valent pneumococcal conjugate vaccine is effective against invasive pneumococcal diseases and nasopharyngeal colonization (36, 47), the protective effect is restricted to seven serotypes, which has been shown to increase the carriage of the nonvaccine serotype (38). Moreover, the high cost limits their extensive application in developing countries (26).
Recently, a new vaccine strategy has focused on the pneumococcal proteins that contribute to virulence and are common to all serotypes (19). Among these proteins, one of the most promising candidates is pneumococcal surface antigen A (PsaA) (42). PsaA is a member of the family of metal binding lipoproteins and is highly conserved across all pneumococcal serotypes (33). PsaA is required for the full expression of competence and virulence (11), and the virulence of psaA mutants was completely attenuated in systemic, respiratory tract, and otitis media infection models (29). Previous studies have shown that intranasal (i.n.) immunization with PsaA using cholera toxin (CT) or cholera toxin subunit (CTB) as the adjuvant elicits significant protection against colonization in mice (37). On this basis, PsaA has been considered a suitable candidate for pneumococcal vaccine development.
As S. pneumoniae is an extracellular bacterial pathogen, the immunity to pneumococcal diseases has long been assumed to depend on humoral immune responses. However, a recent study by Malley et al. has shown that CD4+ T cells mediate antibody-independent acquired immunity in a pneumococcal colonization model (28). In humans, activated T lymphocytes with TH-1 cytokine profiles are highly engaged in the immune response to S. pneumoniae in vivo (20). These studies indicated that cellular immune responses play an important role in protection against pneumococcal infections. Studies have demonstrated that immunization with DNA vaccines expressing pneumococcal antigens elicited humoral and cellular immune responses (13, 31, 32). Furthermore, DNA vaccine is easy to manufacture, has a low cost, and is more stable for transportation, which make it an ideal vaccine in developing countries.
The upper airway and the mucosal epithelium of the nasopharynx are the primary sites of the colonization of S. pneumoniae. The nasopharyngeal carriage of pneumococci is also a prerequisite of infection and invasive diseases and is the source of transmission (53). Therefore, local mucosal immunity within the nasopharynx may play a crucial role in the reduction of carriage. Mucosal immunization has been proven effective for the induction of systemic and local mucosal immune responses (16). Recent studies from the Lee laboratory demonstrated that nasal immunization with CTB-PsaA fusion protein induced systemic and mucosal immune responses and protected mice against pneumococcal colonization (2, 35). However, nasal immunization with a PsaA DNA vaccine against nasopharyngeal colonization has not been studied.
It is well known that the nasal delivery of naked plasmid DNA induces only weak immune responses due to significant physical and chemical barriers (15). To enhance the immunogenicity of DNA vaccine by mucosal delivery, chitosan was used as a vehicle in this study. Chitosan is a natural biodegradable polysaccharide derived from chitin that possesses biocompatibility and mucoadhesion properties. It has been shown to be nontoxic in both experimental animals and humans (3). In addition, it also is easily obtained, inexpensive, and not restricted by patents. These advantages make it an ideal candidate for the nasal delivery of vaccines (44). Recently, chitosan has been extensively studied as a mucosal gene carrier, and chitosan-DNA nanoparticles have proved effective in inducing mucosal and systemic immune responses in several experiments (46, 49, 52). However, chitosan as a pneumococcal DNA vaccine carrier has not been reported. Therefore, in this study, BALB/c mice were intranasally immunized with the chitosan-encapsulated psaA (chitosan-psaA) nanoparticles, and its efficiency in the generation of mucosal and systemic immune responses and protection against nasopharyngeal colonization was evaluated in this study.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
S. pneumoniae ATCC 6303 (serotype 3) was obtained from the American Type Culture Collection (ATCC) and was grown in Todd-Hewitt broth (Sigma) supplemented with 0.5% yeast extract (THY). Cultures in the exponential phase were frozen and stored at −80°C in THY medium containing 10% glycerol. The bacteria were recovered from stock cultures and were counted prior to challenge. Escherichia coli DH5α was used as the host for routine plasmid cloning and was cultured in Luria broth supplemented with 50-μg/ml kanamycin.
Animals.
Specific-pathogen-free (SPF) female BALB/c mice aged 6 to 8 weeks were purchased from Sino-British SIPPR/BK Lab Animal Ltd. (Shanghai, China) and were maintained under SPF conditions with food and water ad libitum until challenge. Infected mice were raised at a biosafety level 2 (BSL-2) biocontainment animal facility. All animal experiment protocols were approved by the Chinese Science Academy Committee on Care and Use of Laboratory Animals and were performed according to the guidelines of the Laboratory Animal Ethical Board of EENT Hospital, Fudan University.
Construction of pVAX1-psaA and transfection into 293T cells.
According to the methods described by Miyaji (31), with some modifications, the 867-bp psaA gene was amplified from S. pneumoniae genomic DNA (ATCC 6303; serotype 3) without its signal sequence and with the ACC Kozak consensus sequence before the ATG start codon. The primers used were 5′GTCGAAGCTTACCATGGCTAGCGGAAAAAAAGATACAAC 3′ and 5′TAGTAGGGATCCTTATTTTGCCAATCCTTCAGCAATC 3′ containing HindIII and BamHI enzyme restriction sites, respectively. The psaA gene then was cloned into pMD19-T vector (TaKaRa, Dalian, China) and further subcloned into HindIII and BamHI sites of pVAX1 to form pVAX1-psaA. The psaA DNA was purified from Escherichia coli DH5a using a plasmid Mega preparation kit (Qiagen, Germany). The purified plasmid DNA was transfected into 293T cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. After 48 h of transfection, cells were harvested to test the expression level of the psaA gene. The transcription level of the psaA gene was detected by reverse transcription-PCR (RT-PCR) using the psaA primers mentioned above, and the transient expression was analyzed by Western blotting. Briefly, the protein extracts from the transfected 293T cells were separated by SDS-PAGE and transferred onto a polyvinylidene fluoride (PVDF) membrane. The mouse polyclonal anti-PsaA antiserum was raised in BALB/c mice immunized with purified PsaA protein emulsified with Freund's complete adjuvant (1:1) at weeks 0 and 3 and with Freund's incomplete adjuvant (1:1) at week 5. The optimal dilution of polyclonal antiserum, after serial dilutions, was found to be 1:20,000. Horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG at a dilution of 1:10,000 (Santa Cruz Biotechnology Inc., CA) was used as a secondary antibody, and detection was performed using the SuperSignal West Pico chemiluminescent substrate (Pierce, Rockford, IL).
Preparation of chitosan-DNA complex formulations.
Chitosan, about 390 kDa in size and >75% deacetylated, was obtained from Sigma-Aldrich. The chitosan-DNA nanoparticles were prepared as described previously (39). In brief, 200 μl of chitosan (0.02% [wt/vol] in 5 mM sodium acetate-hydrogen acetate buffer, pH 5.5) was added to plasmid DNA (200 μl) (100 μg/ml in 50 mM Na2SO4) at 55°C and promptly vortexed for 30 s.
Particle size, zeta potential, and association efficiency of chitosan-DNA complexes.
Chitosan-DNA nanoparticles were freshly prepared each time. Size and zeta potential were measured using the Malvern Zetasizer (3000HSA; Malvern, United Kingdom) as described previously (52). In the assay of the association efficiency of pVAX1-psaA, the nanoparticles first were centrifuged at 16,000 × g for 1 h. The supernatants for the free pVAX1-psaA were quantified using a Nanodrop ND-1000 spectrophotometer (NanoDrop Technologies, Thermo Fisher Scientific Inc.). The association efficiency (AE) was calculated as follows: %AE = (total amount of DNA − amount of free DNA)/total amount of DNA × 100.
Measurement of chitosan protection against DNase I digestion.
One microgram of naked pVAX1-psaA DNA or chitosan-DNA suspension in a volume of 20 μl was incubated with 2 U DNase I (MBI Fermentas) for 15 min at 37°C. The DNase activity was stopped by adding 0.5 M EDTA to a final concentration of 50 mM, and then the integrity of DNA was analyzed by 1% agarose electrophoresis.
Chitosan-DNA i.n. immunization and sample collection.
Female BALB/c mice at 6 to 8 weeks of age (6 mice per group) were immunized i.n. with 50 μg chitosan-psaA or naked psaA DNA in a volume of 20 μl without general anesthesia. An additional control group received the same dosage of chitosan-pVAX1. The immunization was performed four times at 2-week intervals according to the methods described by Xu et al. (49), with some modifications. For immunization with purified recombinant PsaA (rPsaA) protein, mice were intranasally immunized twice a week for three consecutive weeks with 10 μg rPsaA protein and 2 μg cholera toxin (CT) (Sigma). Recombinant PsaA protein was expressed in Escherichia coli BL21 transformed with pET28a-psaA recombinant plasmid and was purified by one-step Ni2+ affinity chromatography. rPsaA was expressed in E. coli BL21 in a soluble form. Two weeks after the last booster immunization, sera, nasal lavages, bronchoalveolar fluids (BALF), and middle ear lavages (MEL) were collected. Blood samples were collected from mouse tail veins. MEL was collected by injection with 10 5-μl volumes of phosphate-buffered saline (PBS) into the middle ear through the tympanic membrane, and the final volume was adjusted to 100 μl. For the nasal and bronchoalveolar washes, cannula were inserted into the trachea, and then the lungs and nasal cavities were lavaged with 1 ml and 500 μl cold PBS, respectively. All of the samples collected were stored at −80°C until enzyme-linked immunosorbent assay (ELISA) detection.
ELISA measurement of anti-PsaA specific antibody.
PsaA-specific antibody levels in serum, nasal washes, MEL, and BALF were detected by indirect ELISA. Briefly, 96-well plates (Corning Costar Corporation, Cambridge, MA) were coated with rPsaA (1 μg/ml) at 4°C overnight. Plates were washed five times with PBS containing 0.05% Tween 20 (PBS-T) and were blocked with 1% bovine serum albumin (BSA) in PBS for 1 h. The plates then were incubated with serum (2-fold dilution for IgG, IgG1, and IgG2a), nasal washes, MEL, or BALF (no dilution for IgA) at 37°C for 2 h. The plates were incubated with HRP-conjugated goat anti-mouse IgG (1:10,000) (Santa Cruz), IgG1, IgG2a, or IgA (1:2,000) (Southern Biotechnology, Birmingham, AL) at 37°C for 1 h. The plates were developed by adding tetramethylbenzidine substrate (TMB; eBioscience) for IgG or p-nitrophenyl phosphate substrate (pNPP; Sigma) for IgG1, IgG2a, and IgA and were read at an optical density (OD) of 450 nm for IgG and 405 nm for IgG1, IgG2a, and IgA using a 1420 Victor 3 multilabel counter (PerkinElmer Inc., Boston, MA). The reciprocal titer was considered the last dilution of serum that registered an optical density of 0.10.
Detection of cytokines in the supernatants of spleen cell cultures.
Cellular suspensions of splenocytes from immunized mice 2 weeks after the last immunization were obtained by passing spleens through a 70-μm cell strainer, and cells were plated into 12-well tissue culture plates at a concentration of 106 cells/well in 1 ml Dulbecco's modified Eagle's medium (DMEM) (HyClone) containing 10% fetal bovine serum (FBS) (Gibco, Inc.) after being washed and having red blood cells removed by hemolysis. Following 72 h of stimulation with rPsaA (5 μg/ml), splenocyte supernatants were collected and stored at −80°C until analysis by sandwich ELISA for the secretion of gamma interferon (IFN-γ) (PeproTech, Rocky Hill, NJ) or interleukin-17A (IL-17A) (Bender MedSystems, Vienna, Austria). The supernatants were analyzed in duplicate and read against a standard curve according to the instructions provided by the manufacturers.
Pneumococcal nasopharyngeal colonization.
As for the intranasal challenge, a bacterial suspension containing 2 × 105 S. pneumoniae (ATCC 6303, serotype 3) cells was instilled into the nares of each mouse at the second week after the last booster immunization. At day 5 after challenge, all animals were euthanized by intraperitoneal injection with a mixture of xilazine (6 mg/kg of body weight) and ketamine (80 mg/kg), and nasal cavities were washed with 500 μl of sterile PBS as previously described (48). The collected washes were serially diluted in PBS and plated on blood plates containing 4 μg/ml gentamicin. The plates were incubated at 37°C with 5% CO2 for 12 h, and the pneumococci were confirmed by classical colonial morphology and sensitivity to optochin (Becton Dickinson, Franklin Lake, NJ). The recovery of pneumococci from the nasopharynx was counted, and all pneumococcal CFU are reported as log10 values. For graphical representation and statistical analysis, a sterile wash sample was assigned to 10 CFU.
Detection of IL-17A at mucosal washes.
Two weeks after the last booster immunization and 5 days after nasopharyngeal challenge with 2 × 105 S. pneumoniae cells, the BALF of all mice were collected according to the methods mentioned above. The IL-17A levels in BALF were determined according to the instructions provided by the manufacturers.
Statistical analysis.
Statistical calculations were performed with the GraphPad software program (GraphPad Software, San Diego, CA). The levels of antibody and cytokines were compared by one-way analysis of variance (ANOVA). Differences between the pneumococcal log10 CFU were analyzed by the Mann-Whitney U test. P values of <0.05 were considered statistically significant.
RESULTS
Expression of PsaA in vitro.
When the 867-bp psaA gene was amplified and cloned into pVAX1 vector and the generated pVAX1-psaA construct transfected into 293T cells, RT-PCR and Western blotting confirmed PsaA expression in the eukaryotic system. RT-PCR results showed the expected band (867 bp) from the cells transfected with pVAX1-psaA, whereas no similar-sized bands were detected from the cells transfected with pVAX1 as a control (Fig. 1A). The Western blotting also showed specific reaction to anti-PsaA polyclonal serum (∼37 kDa) only from the cells transfected with pVAX1-psaA (Fig. 1B).
FIG. 1.

Analysis of the expression of the pVAX1-psaA construct in 293T cells by RT-PCR (A) and Western blotting (B). Total RNA and proteins were extracted from 293T cells transfected with the pVAX1-psaA construct or pVAX1 vector. Lane 1, DNA marker; lane 2, PCR products from cells transfected with the pVAX1-psaA construct, showing the 867-bp psaA gene and 217-bp glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene; lane 3, PCR products from cells transfected with the pVAX1 vector; lane 4, distilled water as a template serving as a negative control. Lanes 5 and 6, cells transfected with the pVAX1-psaA construct, showing the expression of a 37-kDa PsaA protein; lane 7, cells transfected with the pVAX1 vector only. The migration of standard molecular mass markers is indicated at the left.
Characterization of chitosan-encapsulated DNA particles.
Chitosan-psaA particles were analyzed by the Malvern Zetasizer, showing that the average size of the nanoparticles was 392 nm. The zeta potential was +12.5 mV, suggesting that the chitosan-psaA nanoparticles were positively charged. The association efficiency analyzed by a nanodrop spectrophotometer was 97.5%. As shown in Fig. 2, the encapsulated psaA plasmid was not digested by DNase I (lane 4), while the naked psaA plasmid was completely digested (lane 2).
FIG. 2.
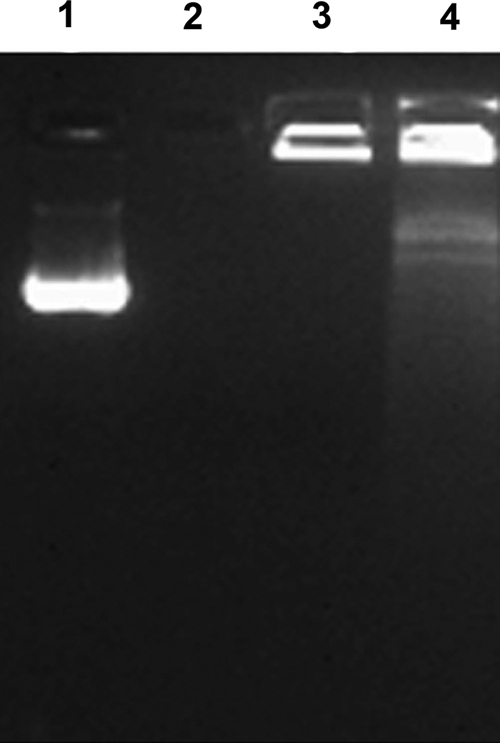
Agarose gel electrophoresis of chitosan-psaA nanoparticles following DNase I digestion. Lane 1, pVAX1-psaA without DNase I; lane 2, pVAX1-psaA with DNase I; lane 3, chitosan-psaA without DNase I; lane 4, chitosan-psaA with DNase I.
Induction of serum and mucosal anti-PsaA antibodies.
To evaluate the ability of chitosan-psaA nanoparticles to induce mucosal and systemic humoral immune responses, groups of mice were intranasally immunized with chitosan-psaA, chitosan-pVAX1, or naked psaA containing 50 μg DNA four times at 2-week intervals. Anti-psaA-specific IgG, IgG1, and IgG2a levels in serum were determined by indirect ELISA, and the results are shown in Fig. 3A. Total IgG levels were significantly higher in mice immunized with chitosan-psaA than in those with naked psaA (P < 0.01) or chitosan-pVAX1 (P < 0.01). As shown in Fig. 3B, IgG1 and IgG2a levels were higher in mice immunized with chitosan-psaA than in either control group (P < 0.01). Balanced IgG1/IgG2a ratios in chitosan-psaA (ratio, 1.6) and naked psaA groups suggested that Th1 immune responses were induced by a DNA vaccine, while mice immunized with rPsaA showed a higher IgG1/IgG2a ratio (5.1) (Fig. 3B). To assess the mucosal immune responses, the nasal washes, BALF, and MEL were collected. As shown in Fig. 3C, detectable levels of IgA were induced in mice immunized with both chitosan-psaA and naked psaA. However, a higher IgA level was induced in the chitosan-psaA mice (P < 0.01), suggesting that chitosan enhances the efficacy of the DNA vaccine for mucosal immune responses.
FIG. 3.

ELISA analysis of total anti-PsaA IgG in serum (A), anti-PsaA IgG isotypes in serum (B), and IgA antibody in nasal washes, BALF, and MEL (C). Mice were intranasally immunized with four doses of chitosan-psaA, naked psaA, or chitosan-pVAX1 at 2-week intervals, and one group was immunized with recombinant PsaA proteins with CT adjuvants as a control. Numbers above columns are mean IgG1/IgG2a reciprocal titer ratios. Statistical analysis was performed using one-way ANOVA, and each column represents means ± standard deviations. Statistical difference between the group immunized with chitosan-psaA and that immunized with naked psaA (P < 0.05) is marked with an asterisk. All samples were obtained from individual mice. These results are representative of three experiments.
Cytokine secretion by splenocytes.
The supernatants of spleen cell cultures stimulated with rPsaA were collected, and IFN-γ and IL-17A levels then were assessed by ELISA. The results showed that splenocytes from mice immunized with chitosan-psaA secreted significantly more IL-17A in response to rPsaA than those from the naked psaA group (P < 0.01) (Fig. 4A) or the chitosan-pVAX1 group (P < 0.01). Compared to levels in the chitosan-pVAX1 group, the expression levels of IFN-γ in the chitosan-psaA group also were significantly increased (P < 0.01), whereas the difference from the naked psaA group was not significant (P = 0.076) (Fig. 4B).
FIG. 4.

ELISA detection of cytokine levels in spleen cells induced by chitosan-psaA. Splenocytes were isolated from immunized BALB/c 2 weeks after the last immunization and then were incubated for 72 h with rPsaA (5 μg/ml). IL-17A and IFN-γ in the supernatants were detected through sandwich ELISA. Each column represents mean concentrations ± standard deviations, and statistical analysis was performed using one-way ANOVA. Statistical difference between the group immunized with chitosan-psaA and that immunized with naked psaA (P < 0.05) is marked with an asterisk. No significant difference was found between naked psaA and chitosan-pVAX1 groups in IL-17A (P = 0.851) and IFN-γ (P = 0.076). All data were the measurements of individual mouse samples. These results are representative of three experiments.
Increased IL-17A secretion in BALF following intranasal challenge.
To investigate the IL-17A levels at the mucosal surface, the BALF were collected before and after pneumococcal challenge. The results showed that the secretion of IL-17A was low in all of the groups before challenge. However, after challenge, the levels of IL-17A in mice immunized with chitosan-psaA were considerably increased compared to those of mice immunized with naked psaA or chitosan-pVAX1 (Fig. 5).
FIG. 5.

IL-17A responses in bronchoalveolar fluids (BALF) during pneumococcal challenge. The BALF were collected before and after pneumococcal challenge, and IL-17A levels were measured through sandwich ELISA. Each column represents mean concentrations ± standard deviations. Before challenge, the secretion of IL-17A was low in each group. Statistical difference after challenge between the group immunized with chitosan-psaA and that immunized with naked psaA (P < 0.05) is marked with an asterisk. These results are representative of three experiments.
Enhanced pneumococcal clearance from the nasopharynx.
To investigate whether i.n. immunization with chitosan-psaA was able to protect mice against pneumococcal nasopharyngeal colonization, BALB/c mice were challenged with 2 × 105 CFU/mouse of type 3 pneumococci intranasally at the second week after the last immunization. Nasopharyngeal CFU counts are shown in Fig. 6. The significant reduction of S. pneumoniae colonization was seen in the chitosan-psaA groups compared to the groups immunized with naked psaA (P < 0.01) or chitosan-pVAX1 (P < 0.01). However, no significant differences were observed between mice inoculated with naked psaA and the chitosan-pVAX1 group (P = 0.0931).
FIG. 6.

Bacterial recovery from mice immunized with chitosan-psaA, naked psaA, or chitosan-pVAX1 after intranasal challenge with S. pneumoniae. Log10 values of total CFU recovered from nasal washes after intranasal challenge with pneumococcal strain ATCC 6303 are shown. The median for each group is displayed as a line, and the absence of colonies in individual nasal washes is represented as 1.0. Statistical analysis was performed using the Mann-Whitney U test. Statistical difference between the groups immunized with chitosan-psaA and naked psaA (P < 0.05) is marked with an asterisk.
DISCUSSION
S. pneumoniae causes serious infections, including pneumonia, bacteremia, and meningitis, in high-risk populations or mucosal infection, such as otitis media and sinusitis, in young children. Since infections occur mainly through respiratory mucosa, the nasal mucosa is considered the first line of defense against S. pneumoniae. Studies have emphasized the fundamental role of mucosal immunity in the control of pneumococcal infections (22); thus, an effective vaccine and immune strategy should be capable of stimulating effective mucosal immunity. Compared to parenteral immunization, intranasal immunization is very effective in eliciting mucosal and systemic immune responses (9). The nasal mucosa is a highly vascularized epithelium mucosa and has a relatively large surface area. More importantly, nasal mucosa is rich in lymphoid tissue, named nasal-associated lymphoid tissue (NALT). Thus, NALT is the inductive site for mucosal immunity, where antigen is encountered and the initial stimulation of naive T and B lymphocytes occurs. These sensitized B and T lymphocytes migrate from the NALT to the draining cervical lymph nodes, enter the blood circulation through the thoracic duct, and subsequently induce the systemic immune response, such as the production of serum IgG (7). Meanwhile, they may migrate back into effector sites, such as nasal, middle ear, and the upper respiratory tract mucosae, where B cells differentiate into IgA plasma cells and produce serum IgA antibody (34). In our study, the results demonstrated that intranasal immunization with chitosan-psaA nanoparticles successfully elicited systemic immune responses in serum and spleen and mucosal immune responses in nasal cavity, middle ear, and bronchoalveolar spaces as well. In addition, intranasal immunization offers a convenient and painless method for vaccine delivery that has been successfully exploited in various vaccination programs (16).
Although mucosal vaccines are thought to be ideal to induce mucosal immunity, the development of a mucosal vaccine has been impeded by poor mucosal absorption due to the low epithelial cell membrane permeability, the mucociliary clearance mechanism, and the possibility of enzymatic degradation (17). Naked DNA is negatively charged and may be degraded by DNase at mucosal surfaces, so it is difficult for naked DNA to permeate across nasal epithelium. Therefore, an appropriate DNA delivery system or adjuvant was demanded. Two major mucosal delivery systems have been evaluated, including viral and nonviral vectors. Although viral vectors have the merit of high transfectability, their potential safety risks (23) as well as immunogenicity hinder the extensive use of viral vectors in gene therapy and vaccination. CT and CTB are the most potent mucosal adjuvants and are widely used in laboratory experiments. However, a potential neurotoxicity of both CT and CTB has been indicated in a report showing that both CT and CTB accumulated in the olfactory nerves and olfactory bulbs of mice following intranasal application (45). The toxicity of CT and CTB restrict their application in humans. During the past decade, there has been increasing interest in the use of chitosan as a nontoxic vector and adjuvant (21). The positively charged chitosan has great potential for complexation with negatively charged DNA and forming nanoparticulates to improve the delivery of DNA (17). Previous studies have suggested that chitosan enhanced the immunity of the vaccine by providing longer residence times in the nasal cavity, and by opening transiently the tight junctions among the mucosal cells, these allowed the vaccine better access to the lymphoid tissue (1). In the present study, chitosan-psaA nanoparticles were prepared by coacervation methods, and the mean size of nanoparticles was 392 nm with +12.5 mV zeta potential. These results suggested that chitosan enhanced the immunogenicity of the psaA DNA vaccine.
Antibody long has been considered to play a key role in the prevention of pneumococcal diseases. However, recent studies from Richard Malley's laboratory confirmed CD4+ T-cell-mediated antibody-independent acquired immunity to pneumococcal colonization in mice immunized intranasally with the cell wall polysaccharide, whole-cell vaccine, or purified pneumococcal proteins. The studies further demonstrated that IL-17A mediated acquired immunity to pneumococcal colonization (4, 24, 25, 27, 28). IL-17A mainly produced by T helper 17 (Th17) cells enhances host defense by coordinating neutrophil recruitment into mucosal sites and increasing neutrophil bactericidal activity (10, 54). In this study, the antigen-specific IL-17A response in splenocytes was higher in the chitosan-psaA group than in naked psaA and chitosan-pVAX1 control groups. In addition, IL-17A levels at the mucosal surface after challenge were enhanced. The higher IL-17A response may be relevant to enhanced protection against nasopharyngeal colonization, but it merits further investigation.
Immunization with DNA vaccine expressing pneumococcal surface protein A (PspA) by the intramuscular route previously has been shown to be protective against pneumococcal challenge, although the antibody levels were lower than those with protein vaccines (12, 30, 31). Previous studies from several laboratories have shown that PsaA immunization is most effective against nasopharyngeal colonization (18, 35, 37). However, the effectiveness of PsaA immunization against systemic pneumococcal challenges varies in different studies (14, 43). The authors explained that bacterial and host factors that affect the synthesis, thickness, and stability of bacterial capsular polysaccharides may determine the exposure of PsaA, thus the efficacy of PsaA immunization (14). However, the specific mechanism still is not clear, and the protective role against systemic infections with DNA vaccine expressing PsaA has not been evaluated until now. DNA vaccines could elicit a more balanced IgG1/IgG2a ratio (13). Antibody-mediated complement-dependent phagocytosis plays an important protective role against systemic pneumococcal challenge, and IgG2a is the isotype with the greatest capacity to mediate complement deposition onto the pneumococcal surface. Therefore, a balanced IgG1/IgG2a response might be beneficial in mediating complement deposition (13). In this study, intranasal immunization with the chitosan-psaA vaccine gave a more balanced IgG1/IgG2a level and induced IFN-γ secretion. The role of IFN-γ in the resolution of pneumococcal infection is well described in different mouse models (40, 50). Our future studies will focus on the evaluation of protective roles of these different responses against systemic challenge following intranasal immunization with chitosan-psaA nanoparticles.
Taken together, mucosal, systemic, and cellular immune responses were induced with an enhanced level by intranasal immunization with chitosan-psaA vaccines, and these findings suggested that chitosan was efficient as a mucosal delivery vector. Nasopharyngeal carriage also was markedly decreased in mice immunized with chitosan-psaA nanoparticles, indicating that nasal vaccination could be a convenient and noninvasive route for the delivery of DNA vaccines against pneumococcal infections.
Acknowledgments
This work was supported by grants from the Shanghai Hospital Cooperation Foundation (no. SHDC12010119) and the National Natural Science Foundation (no. 81000406) and also was supported partially by the Shanghai Hospital Clinical Research Platform for Clinical Research Source (no. SHDC12007703).
We thank Douglas B. Lowrie from the Shanghai Public Health Clinical Center Affiliated with Fudan University for the critical reading and polishing of the manuscript and Sheng Guo from Shanghai Sixth Hospital for animal experiments.
Footnotes
Published ahead of print on 3 November 2010.
REFERENCES
- 1.Alpar, H. O., S. Somavarapu, K. N. Atuah, and V. W. Bramwell. 2005. Biodegradable mucoadhesive particulates for nasal and pulmonary antigen and DNA delivery. Adv. Drug Deliv. Rev. 57:411-430. [DOI] [PubMed] [Google Scholar]
- 2.Arêas, A. P., M. L. Oliveira, E. N. Miyaji, L. C. Leite, K. A. Aires, W. O. Dias, and P. L. Ho. 2004. Expression and characterization of cholera toxin B-pneumococcal surface adhesin A fusion protein in Escherichia coli: ability of CTB-PsaA to induce humoral immune response in mice. Biochem. Biophys. Res. Commun. 321:192-196. [DOI] [PubMed] [Google Scholar]
- 3.Aspden, T. J., J. D. Mason, N. S. Jones, J. Lowe, O. Skaugrud, and L. Illum. 1997. Chitosan as a nasal delivery system: the effect of chitosan solutions on in vitro and in vivo mucociliary transport rates in human turbinates and volunteers. J. Pharm. Sci. 86:509-513. [DOI] [PubMed] [Google Scholar]
- 4.Basset, A., C. M. Thompson, S. K. Hollingshead, D. E. Briles, E. W. Ades, M. Lipsitch, and R. Malley. 2007. Antibody-independent, CD4+ T-cell-dependent protection against pneumococcal colonization elicited by intranasal immunization with purified pneumococcal proteins. Infect. Immun. 75:5460-5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernatoniene, J., and A. Finn. 2005. Advances in pneumococcal vaccines: advantages for infants and children. Drugs 65:229-255. [DOI] [PubMed] [Google Scholar]
- 6.Bogaert, D., R. De Groot, and P. W. Hermans. 2004. Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet Infect. Dis. 4:144-154. [DOI] [PubMed] [Google Scholar]
- 7.Brandtzaeg, P. 2007. Induction of secretory immunity and memory at mucosal surfaces. Vaccine 25:5467-5484. [DOI] [PubMed] [Google Scholar]
- 8.Briles, D. E., S. Hollingshead, A. Brooks-Walter, G. S. Nabors, L. Ferguson, M. Schilling, S. Gravenstein, P. Braun, J. King, and A. Swift. 2000. The potential to use PspA and other pneumococcal proteins to elicit protection against pneumococcal infection. Vaccine 18:1707-1711. [DOI] [PubMed] [Google Scholar]
- 9.Cripps, A. W., and J. M. Kyd. 2007. Comparison of mucosal and parenteral immunisation in two animal models of pneumococcal infection: otitis media and acute pneumonia. Vaccine 25:2471-2477. [DOI] [PubMed] [Google Scholar]
- 10.Curtis, M. M., and S. S. Way. 2009. Interleukin-17 in host defence against bacterial, mycobacterial and fungal pathogens. Immunology 126:177-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dintilhac, A., G. Alloing, C. Granadel, and J. P. Claverys. 1997. Competence and virulence of Streptococcus pneumoniae: Adc and PsaA mutants exhibit a requirement for Zn and Mn resulting from inactivation of putative ABC metal permeases. Mol. Microbiol. 25:727-739. [DOI] [PubMed] [Google Scholar]
- 12.Ferreira, D. M., E. N. Miyaji, M. L. Oliveira, M. Darrieux, A. P. Areas, P. L. Ho, and L. C. Leite. 2006. DNA vaccines expressing pneumococcal surface protein A (PspA) elicit protection levels comparable to recombinant protein. J. Med. Microbiol. 55:375-378. [DOI] [PubMed] [Google Scholar]
- 13.Ferreira, D. M., M. Darrieux, M. L. Oliveira, L. C. Leite, and E. N. Miyaji. 2008. Optimized immune response elicited by a DNA vaccine expressing pneumococcal surface protein a is characterized by a balanced immunoglobulin G1 (IgG1)/IgG2a ratio and proinflammatory cytokine production. Clin. Vaccine Immunol. 15:499-505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gor, D. O., X. Ding, Q. Li, J. R. Schreiber, M. Dubinsky, and N. S. Greenspan. 2002. Enhanced immunogenicity of pneumococcal surface adhesin A by genetic fusion to cytokines and evaluation of protective immunity in mice. Infect. Immun. 70:5589-5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hobson, P., C. Barnfield, A. Barnes, and L. S. Klavinskis. 2003. Mucosal immunization with DNA vaccines. Methods 31:217-224. [DOI] [PubMed] [Google Scholar]
- 16.Holmgren, J., and C. Czerkinsky. 2005. Mucosal immunity and vaccines. Nat. Med. 11:S45-S53. [DOI] [PubMed] [Google Scholar]
- 17.Illum, L. 2003. Nasal drug delivery-possibilities, problems and solutions. J. Control Release 87:187-198. [DOI] [PubMed] [Google Scholar]
- 18.Johnson, S. E., J. K. Dykes, D. L. Jue, J. S. Sampson, G. M. Carlone, and E. W. Ades. 2002. Inhibition of pneumococcal carriage in mice by subcutaneous immunization with peptides from the common surface protein pneumococcal surface adhesin a. J. Infect. Dis. 185:489-496. [DOI] [PubMed] [Google Scholar]
- 19.Kadioglu, A., J. N. Weiser, J. C. Paton, and P. W. Andrew. 2008. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat. Rev. Microbiol. 6:288-301. [DOI] [PubMed] [Google Scholar]
- 20.Kemp, K., H. Bruunsgaard, P. Skinhoj, and P. B. Klarlund. 2002. Pneumococcal infections in humans are associated with increased apoptosis and trafficking of type 1 cytokine-producing T cells. Infect. Immun. 70:5019-5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lai, W., and M. C. Lin. 2009. Nucleic acid delivery with chitosan and its derivatives. J. Control Release 134:158-168. [DOI] [PubMed] [Google Scholar]
- 22.Lee, C. J., L. H. Lee, and X. X. Gu. 2005. Mucosal immunity induced by pneumococcal glycoconjugate. Crit. Rev. Microbiol. 31:137-144. [DOI] [PubMed] [Google Scholar]
- 23.Lehrman, S. 1999. Virus treatment questioned after gene therapy death. Nature 401:517-518. [DOI] [PubMed] [Google Scholar]
- 24.Lu, Y. J., J. Gross, D. Bogaert, A. Finn, L. Bagrade, Q. Zhang, J. K. Kolls, A. Srivastava, A. Lundgren, S. Forte, C. M. Thompson, K. F. Harney, P. W. Anderson, M. Lipsitch, and R. Malley. 2008. Interleukin-17A mediates acquired immunity to pneumococcal colonization. PLoS Pathog. 4:e1000159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu, Y. J., S. Forte, C. M. Thompson, P. W. Anderson, and R. Malley. 2009. Protection against pneumococcal colonization and fatal pneumonia by a trivalent conjugate of a fusion protein with the cell wall polysaccharide. Infect. Immun. 77:2076-2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mahoney, R. T., A. Krattiger, J. D. Clemens, and R. R. Curtiss. 2007. The introduction of new vaccines into developing countries. IV: Global access strategies. Vaccine 25:4003-4011. [DOI] [PubMed] [Google Scholar]
- 27.Malley, R., A. Srivastava, M. Lipsitch, C. M. Thompson, C. Watkins, A. Tzianabos, and P. W. Anderson. 2006. Antibody-independent, interleukin-17A-mediated, cross-serotype immunity to pneumococci in mice immunized intranasally with the cell wall polysaccharide. Infect. Immun. 74:2187-2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malley, R., K. Trzcinski, A. Srivastava, C. M. Thompson, P. W. Anderson, and M. Lipsitch. 2005. CD4+ T cells mediate antibody-independent acquired immunity to pneumococcal colonization. Proc. Natl. Acad. Sci. U. S. A. 102:4848-4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marra, A., S. Lawson, J. S. Asundi, D. Brigham, and A. E. Hromockyj. 2002. In vivo characterization of the psa genes from Streptococcus pneumoniae in multiple models of infection. Microbiology 148:1483-1491. [DOI] [PubMed] [Google Scholar]
- 30.McDaniel, L. S., F. Loechel, C. Benedict, T. Greenway, D. E. Briles, R. M. Conry, and D. T. Curiel. 1997. Immunization with a plasmid expressing pneumococcal surface protein A (PspA) can elicit protection against fatal infection with Streptococcus pneumoniae. Gene Ther. 4:375-377. [DOI] [PubMed] [Google Scholar]
- 31.Miyaji, E. N., W. O. Dias, M. Gamberini, V. C. Gebara, R. P. Schenkman, J. Wild, P. Riedl, J. Reimann, R. Schirmbeck, and L. C. Leite. 2001. PsaA (pneumococcal surface adhesin A) and PspA (pneumococcal surface protein A) DNA vaccines induce humoral and cellular immune responses against Streptococcus pneumoniae. Vaccine 20:805-812. [DOI] [PubMed] [Google Scholar]
- 32.Moore, Q. C., J. R. Bosarge, L. R. Quin, and L. S. McDaniel. 2006. Enhanced protective immunity against pneumococcal infection with PspA DNA and protein. Vaccine 24:5755-5761. [DOI] [PubMed] [Google Scholar]
- 33.Morrison, K. E., D. Lake, J. Crook, G. M. Carlone, E. Ades, R. Facklam, and J. S. Sampson. 2000. Confirmation of psaA in all 90 serotypes of Streptococcus pneumoniae by PCR and potential of this assay for identification and diagnosis. J. Clin. Microbiol. 38:434-437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neutra, M. R., and P. A. Kozlowski. 2006. Mucosal vaccines: the promise and the challenge. Nat. Rev. Immunol. 6:148-158. [DOI] [PubMed] [Google Scholar]
- 35.Oliveira, M. L., A. P. Areas, I. B. Campos, V. Monedero, G. Perez-Martinez, E. N. Miyaji, L. C. Leite, K. A. Aires, and H. P. Lee. 2006. Induction of systemic and mucosal immune response and decrease in Streptococcus pneumoniae colonization by nasal inoculation of mice with recombinant lactic acid bacteria expressing pneumococcal surface antigen A. Microbes Infect. 8:1016-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oosterhuis-Kafeja, F., P. Beutels, and P. Van Damme. 2007. Immunogenicity, efficacy, safety and effectiveness of pneumococcal conjugate vaccines (1998-2006). Vaccine 25:2194-2212. [DOI] [PubMed] [Google Scholar]
- 37.Pimenta, F. C., E. N. Miyaji, A. P. Areas, M. L. Oliveira, A. L. de Andrade, P. L. Ho, S. K. Hollingshead, and L. C. Leite. 2006. Intranasal immunization with the cholera toxin B subunit-pneumococcal surface antigen A fusion protein induces protection against colonization with Streptococcus pneumoniae and has negligible impact on the nasopharyngeal and oral microbiota of mice. Infect. Immun. 74:4939-4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodgers, G. L., A. Arguedas, R. Cohen, and R. Dagan. 2009. Global serotype distribution among Streptococcus pneumoniae isolates causing otitis media in children: potential implications for pneumococcal conjugate vaccines. Vaccine 27:3802-3810. [DOI] [PubMed] [Google Scholar]
- 39.Roy, K., H. Q. Mao, S. K. Huang, and K. W. Leong. 1999. Oral gene delivery with chitosan-DNA nanoparticles generates immunologic protection in a murine model of peanut allergy. Nat. Med. 5:387-391. [DOI] [PubMed] [Google Scholar]
- 40.Rubins, J. B., and C. Pomeroy. 1997. Role of gamma interferon in the pathogenesis of bacteremic pneumococcal pneumonia. Infect. Immun. 65:2975-2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schuchat, A., T. Hilger, E. Zell, M. M. Farley, A. Reingold, L. Harrison, L. Lefkowitz, R. Danila, K. Stefonek, N. Barrett, D. Morse, and R. Pinner. 2001. Active bacterial core surveillance of the emerging infections program network. Emerg. Infect. Dis. 7:92-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tai, S. S. 2006. Streptococcus pneumoniae protein vaccine candidates: properties, activities and animal studies. Crit. Rev. Microbiol. 32:139-153. [DOI] [PubMed] [Google Scholar]
- 43.Talkington, D. F., B. G. Brown, J. A. Tharpe, A. Koenig, and H. Russell. 1996. Protection of mice against fatal pneumococcal challenge by immunization with pneumococcal surface adhesin A (PsaA). Microb. Pathog. 21:17-22. [DOI] [PubMed] [Google Scholar]
- 44.van der Lubben, I. M., J. C. Verhoef, G. Borchard, and H. E. Junginger. 2001. Chitosan for mucosal vaccination. Adv. Drug Deliv. Rev. 52:139-144. [DOI] [PubMed] [Google Scholar]
- 45.van Ginkel, F. W., R. J. Jackson, Y. Yuki, and J. R. McGhee. 2000. Cutting edge: the mucosal adjuvant cholera toxin redirects vaccine proteins into olfactory tissues. J. Immunol. 165:4778-4782. [DOI] [PubMed] [Google Scholar]
- 46.Wang, X., X. Zhang, Y. Kang, H. Jin, X. Du, G. Zhao, Y. Yu, J. Li, B. Su, C. Huang, and B. Wang. 2008. Interleukin-15 enhance DNA vaccine elicited mucosal and systemic immunity against foot and mouth disease virus. Vaccine 26:5135-5144. [DOI] [PubMed] [Google Scholar]
- 47.Whitney, C. G., M. M. Farley, J. Hadler, L. H. Harrison, N. M. Bennett, R. Lynfield, A. Reingold, P. R. Cieslak, T. Pilishvili, D. Jackson, R. R. Facklam, J. H. Jorgensen, and A. Schuchat. 2003. Decline in invasive pneumococcal disease after the introduction of protein-polysaccharide conjugate vaccine. N. Engl. J. Med. 348:1737-1746. [DOI] [PubMed] [Google Scholar]
- 48.Wu, H. Y., A. Virolainen, B. Mathews, J. King, M. W. Russell, and D. E. Briles. 1997. Establishment of a Streptococcus pneumoniae nasopharyngeal colonization model in adult mice. Microb. Pathog. 23:127-137. [DOI] [PubMed] [Google Scholar]
- 49.Xu, W., Y. Shen, Z. Jiang, Y. Wang, Y. Chu, and S. Xiong. 2004. Intranasal delivery of chitosan-DNA vaccine generates mucosal SIgA and anti-CVB3 protection. Vaccine 22:3603-3612. [DOI] [PubMed] [Google Scholar]
- 50.Yamamoto, N., K. Kawakami, Y. Kinjo, K. Miyagi, T. Kinjo, K. Uezu, C. Nakasone, M. Nakamatsu, and A. Saito. 2004. Essential role for the p40 subunit of interleukin-12 in neutrophil-mediated early host defense against pulmonary infection with Streptococcus pneumoniae: involvement of interferon-γ. Microbes Infect. 6:1241-1249. [DOI] [PubMed] [Google Scholar]
- 51.Yao, K. H., and Y. H. Yang. 2008. Streptococcus pneumoniae diseases in Chinese children: past, present and future. Vaccine 26:4425-4433. [DOI] [PubMed] [Google Scholar]
- 52.Yuan, X., X. Yang, D. Cai, D. Mao, J. Wu, L. Zong, and J. Liu. 2008. Intranasal immunization with chitosan/pCETP nanoparticles inhibits atherosclerosis in a rabbit model of atherosclerosis. Vaccine 26:3727-3734. [DOI] [PubMed] [Google Scholar]
- 53.Zhang, Q., and A. Finn. 2004. Mucosal immunology of vaccines against pathogenic nasopharyngeal bacteria. J. Clin. Pathol. 57:1015-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang, Z., T. B. Clarke, and J. N. Weiser. 2009. Cellular effectors mediating Th17-dependent clearance of pneumococcal colonization in mice. J. Clin. Invest. 119:1899-1909. [DOI] [PMC free article] [PubMed] [Google Scholar]