

Abstract
Two-component signal transduction systems are widespread in bacteria and are essential regulatory mechanisms for many biological processes. These systems predominantly rely on a sensor kinase to phosphorylate a response regulator for controlling activity, which is frequently transcriptional regulation. In recent years, an increasing number of atypical response regulators have been discovered in phylogenetically diverse bacteria. These atypical response regulators are not controlled by phosphorylation and exhibit transcriptional activity in their wild-type form. Relatively little is known regarding the mechanisms utilized by these atypical response regulators and the conserved characteristics of these atypical response regulators. Chlamydia spp. are medically important bacteria and encode an atypical OmpR/PhoB subfamily response regulator termed ChxR. In this study, protein expression analysis supports that ChxR is likely exerting its effect during the middle and late stages of the chlamydial developmental cycle, stages that include the formation of infectious elementary bodies. In the absence of detectable phosphorylation, ChxR formed homodimers in vitro and in vivo, similar to a phosphorylated OmpR/PhoB subfamily response regulator. ChxR was demonstrated to bind to its own promoter in vivo, supporting the role of ChxR as an autoactivator. Detailed analysis of the ChxR binding sites within its own promoter revealed a conserved cis-acting motif that includes a tandem repeat sequence. ChxR binds specifically to each of the individual sites and exhibits a relatively large spectrum of differential affinity. Taken together, these observations support the conclusion that ChxR, in the absence of phosphorylation, exhibits many of the characteristics of a phosphorylated (active) OmpR/PhoB subfamily response regulator.
Response regulators are essential regulatory factors of two-component signal transduction systems. They predominantly function as phosphorylation-activated switches to control gene expression at the transcriptional level (16). The largest subfamily of response regulators is the OmpR/PhoB subfamily, in which the vast majority of homologs share a conserved phosphorylation-dependent transcriptional regulation mechanism (16, 17). This subfamily of response regulators is structurally very similar and composed of two domains: a receiver and an effector domain (17, 19, 44, 48). Phosphorylation at an Asp within a highly conserved active site in the receiver domain causes reorientation of two conformational-switch residues and relatively subtle overall changes to the receiver domain (19). These changes promote homodimer formation between receiver domains that is essential for controlling activity of the effector domain.
The effector domain of response regulators binds to either tandem or, more infrequently, inverted repeats of DNA through a subfamily-defining winged helix-turn-helix DNA binding motif to regulate transcription. The DNA recognition site generally ranges from 18 to 23 bp containing a 6- to 10-bp promoter-binding site separated by 2 to 5 bp of intervening sequence (8, 20, 26). The target promoters of OmpR/PhoB subfamily members often contain multiple binding sites that vary in their nucleotide frequency, promoter position, and relative binding affinities (26, 36). As a result, cooperativity and differential binding are commonly incorporated as an important component to transcriptional regulation by OmpR/PhoB response regulators.
Atypical response regulators have recently been discovered and described in phylogenetically diverse organisms, including Chlamydia, Helicobacter, Myxococcus, Streptomyces, and Synechococcus (2, 13, 14, 25, 28, 31, 34, 38). These atypical response regulators do not require phosphorylation to function as transcriptional regulators. In concert with these observations, the receiver domain active site, frequently including the typically phosphorylated Asp, is not conserved. This and other observations support the finding that phosphorylation-dependent activation mechanisms are not utilized by atypical response regulators (2, 25, 31, 35). Highlighting the biological importance of these atypical response regulators to their respective organism, gene disruptions of these transcription factors cause severe phenotypic defects or are requisite for growth (5, 11, 38).
Despite their apparent importance, relatively little information exists regarding the transcriptional regulation mechanisms utilized by atypical OmpR/PhoB subfamily response regulators. Structural analysis of the atypical response regulator homolog HP1043 from H. pylori revealed that the conformational-switch residues were oriented similar to those in a phosphorylated (active) orientation (23). This study also reported that recombinant HP1043 forms stable homodimers and recognizes an inverted repeat of DNA sequences. In contrast, analyses of the atypical response regulator homolog NblR in Synechococcus demonstrated that, unlike phosphorylated (active) response regulators, this essential regulator existed as a monomer both in vitro and in vivo (35). These observations suggest that atypical OmpR/PhoB response regulator mechanisms (e.g., homodimerization) are most likely similar to, but distinct from, the canonical mechanisms.
Chlamydia are phylogenetically distinct from other bacteria and encode an atypical response regulator termed ChxR (42, 43). ChxR is homologous to the OmpR/PhoB subfamily of response regulators; however, none of the active site residues and only one of the conformational switch residues is conserved with other typical OmpR/PhoB subfamily members. Similar to other atypical response regulators, previous studies demonstrated that ChxR activated transcription both in vitro and within a heterologous in vivo system (Escherichia coli) (28). These analyses also revealed that ChxR has a direct autoregulatory role because it recognizes multiple sites within its own promoter region and activates transcription, as do many other atypical response regulators (2, 13).
Chlamydia infections have an immense impact on public health and are associated with diverse disease manifestations including atherosclerosis, blindness, and sterility (37). The pathogenic mechanisms utilized by Chlamydia are still undefined; however, the growth of these obligate intracellular bacteria and their ability to maintain the characteristic biphasic developmental cycle are intrinsically linked with the immune-mediated pathology associated with Chlamydia infections (41). Largely due to the current absence of a system for specific genetic manipulation in Chlamydia, relatively little is known regarding the signals and components that regulate the chlamydial developmental cycle; however, transcriptional regulation has a governing role in the developmental cycle (1, 6, 32).
ChxR is hypothesized to play an important role in regulating the chlamydial developmental cycle and incorporates mechanisms and exhibits properties similar to, but distinct from, the OmpR/PhoB response regulator subfamily. The present study was designed to begin defining the fundamental mechanisms used by and properties of ChxR. Included is the characterization of the cis-acting element recognized by ChxR, which is expected to facilitate the identification of additional ChxR gene targets and eventual assignment of a specific role for ChxR in the developmental cycle.
MATERIALS AND METHODS
Purification of ChxR.
chxR was PCR amplified using Chlamydia trachomatis LGV (L2/484/Bu) genomic DNA and primers specific for chxR (see Table S1 in the supplemental material) (Integrated DNA Technologies, Coralville, IA). The resulting amplicon was digested with NdeI/XhoI, ligated into pET28b (Novagen, San Diego, CA), and transformed into E. coli TOP10 cells (Invitrogen, Carlsbad, CA). After sequence conformation of isogenic clones (DNA Sequencing Laboratory, University of Kansas, Lawrence, KS), the plasmids were transformed into E. coli BL21(DE3) (Invitrogen) and grown to an optical density at 600 nm of 0.7 in Luria broth containing 50 μg of kanamycin/ml. IPTG (isopropyl-β-d-thiogalactopyranoside) at 1 mM was added, and the cells were harvested by centrifugation after overnight incubation at 15°C.
The ChxR-expressing E. coli cells were resuspended in 50 mM Tris (pH 7.0) and 400 mM NaCl, disrupted by sonication, and subjected to centrifugation (30 min at 14,000 × g and 4°C). Residual cell debris was removed by passing the supernatant through a 0.22-μm-pore-size filter before protein purification. ChxR was purified by Co2+-affinity chromatography (Clontech, Mountain View, CA). The hexahistidine-tagged proteins bound to the metal resin were washed with 5 mM imidazole, 50 mM Tris (pH 7.0), and 400 mM NaCl before elution from the resin with the wash buffer that contained 250 mM imidazole. Elution fractions containing ChxR were pooled and further purified by size exclusion chromatography.
The pooled protein mixture was applied to a Sephacryl S-200 16/60 size exclusion column (GE Healthcare, Pittsburgh, PA) equilibrated with 50 mM Tris (pH 7.0) and 400 mM NaCl. Fractions containing ChxR were pooled, and the protein was determined to be >95% pure, as determined by Coomassie staining after sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
Analytical size exclusion chromatography.
After size exclusion (S-200) purification, fractions containing ChxR were concentrated to 100 μM using an Amicon Ultra centrifugal filter (3,000 molecular weight cutoff; Millipore). Equal volumes of protein samples at 100, 10, or 1 μM were applied to a Superdex 75 10/300 GL analytical size exclusion column (GE Healthcare) equilibrated with 50 mM Tris-HCl (pH 7.5), 100 mM NaCl, and 250 mM KCl. In the same buffer, a protein standard containing bovine serum albumin (66 kDa), chicken ovalbumin (44 kDa), and horse myoglobin (17 kDa) (Bio-Rad, Hercules, CA) was used to generate a standard curve.
In vitro chemical cross-linking.
Purified ChxR-His6 was dialyzed in cross-linking buffer (30 mM sodium phosphate [pH 7.0] and 300 mM NaCl). ChxR was exposed to the chemical cross-linker disuccinimidyl suberate (DSS; Pierce, Rockford, IL) at 500 μM. The reactions were incubated at 25°C for 2 min and quenched with 1 M Tris (pH 8.0). The samples were heat denatured in Laemmli buffer, separated by SDS-PAGE, and visualized by Coomassie staining.
Time course of expression of ChxR.
Mouse L929 fibroblast cells (8 × 105 cells/ml) were propagated in RPMI medium (Mediatech, Manassas, VA) supplemented with 5% fetal bovine serum (HyClone, Logan, UT) and 50 μg of vancomycin (MP Biomedicals, Solon, OH)/ml as previously described (39). L929 cells were infected with C. trachomatis LGV (L2/434/Bu) at a dilution that resulted in ca. 80% of the cells infected, as visualized by immunofluorescence microscopy at 24 h postinfection (hpi) (Microtrak, Trinity Biotech, Berkeley Heights, NJ). At 12, 24, and 36 hpi, 1-liter, 500-ml, and 350-ml portions of cells, respectively, were harvested by centrifugation (10 min at 1,400 × g and 15°C). The resulting pellets were washed twice with and resuspended in Hanks balanced salt solution (Mediatech) before being transferred to 40-ml Oakridge tubes. C. trachomatis specimens were liberated from the host cells by gentle sonication. The lysate was layered over 30% Renografin (Bracco Diagnostics, New Jersey) and subjected to ultracentrifugation (10 min at 16,000 × g and 15°C). The resulting chlamydial pellet was resuspended in phosphate-buffered saline (PBS). The samples were subjected to SDS-PAGE, and an immunoblot assay was performed with monospecific-polyclonal antibodies against ChxR (Proteintech, Chicago, IL).
In vivo chemical cross-linking.
Reticulate body (RB)-enriched pellets were resuspended in PBS and exposed to 10 mM DSS (Pierce). After 20 min of incubation at 25°C, the reaction was quenched with the addition of 50 mM Tris (pH 8.0). The samples were subjected to SDS-PAGE, and immunoblot assays were performed with monospecific-polyclonal antibodies against ChxR.
Immunoprecipitation of ChxR.
RB-enriched fractions from 36 hpi cells were obtained as described above. RBs were cross-linked with 1% formaldehyde, and the reaction quenched with 250 μM glycine. After cross-linking, cells were incubated in radioimmunoprecipitation assay (RIPA) buffer (10 mM Tris-Cl [pH 8.0], 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl, 5 mM dithiothreitol) with 15 μl of AEBSF [4-(2-aminoethyl)-benzenesulfonyl fluoride; Thermofisher Scientific] for 45 min on ice. Samples were sonicated to shear DNA and centrifuged (14,000 × g, 15 min, 24°C), and then supernatants were removed. For immunoprecipitation, protein G-Dynabeads (Invitrogen) were washed three times and resuspended in RIPA buffer. Portions (10 μg) of affinity-purified anti-ChxR polyclonal antibodies were added to the beads, followed by incubation at 4°C for 24 h with rotating. Beads were subsequently washed twice with RIPA buffer, and supernatants were added to the beads. Samples were incubated at 4°C for 24 h with rotating. Beads were washed twice with RIPA buffer before resuspension in RIPA buffer and incubated at 25°C for 2 h with rotating. The beads were again washed five times with RIPA buffer prior to 30 μl of TE (10 mM Tris-Cl [pH 7.4], 1 mM EDTA) being added. Samples were boiled 5 min to reverse the cross-links and centrifuged (13,000 × g, 3 min, 25°C), and then the supernatants were collected. PCR was performed on supernatants by using primers to the chxR promoter region or the CT863 promoter region (see Table S1 in the supplemental material). PCR products were separated by agarose gel electrophoresis and detected by ethidium bromide staining.
EMSAs and quantitative binding analysis.
Oligonucleotides were designed to contain each of the putative ChxR binding sites (direct repeats 1 to 6 [DR1 to DR6]) and at least 3 bp of the flanking sequence (see Table S1 in the supplemental material). Three or more base pairs of flanking sequence were shown to result in equal maximal binding (data not shown). IR800-labeled (Eurofins MWG Operon, Huntsville, AL) or unlabeled (Integrated DNA Technologies) oligonucleotides were hybridized prior to use in electrophoretic gel mobility shift assays (EMSAs). Binding reactions (20 μl) contained DNA and ChxR at their respective concentrations, as listed in Results and were performed in triplicate. EMSAs were performed as previously described (28), except the reactions were incubated at 25°C for 20 min. After native PAGE, unlabeled-hybridized DNA fragments were visualized by SYBR green (Invitrogen) staining by using an excitation wavelength of 488 nm and an emission filter of 520 nm on a Typhoon Trio imager (GE Healthcare). IR800-labeled DNA fragments were visualized by using the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE). DNA was quantified by using the software program ImageQuant (GE Healthcare). The percent DNA shifted was determined by the amount of photons emitted for the shifted DNA band relative to the total amount of photons emitted between the shifted and nonshifted DNA bands. To measure the ChxR binding affinity with the six DR sites, EMSAs were performed with 1 nM DR1 to DR6 and increasing concentrations of ChxR (5 nM to 5 μM). Dissociation constants (Kd) were calculated by using nonlinear regression with GraphPad Prism software (GraphPad Software, Inc., San Diego, CA). The dissociation constant is the calculated protein concentration that resulted in 50% ChxR-DNA interaction.
For the analysis of single site mutations in DR2 (see Fig. 7), each individual gel included a ChxR binding reaction and wild-type DR2. Overall, the wild-type DR2 DNA sequence averaged 67% of ChxR shifted DNA through 18 independent reactions. A Student two-tailed t test was used for statistical analysis of triplicate data sets.
For the supershift analysis, binding reactions (20 μl) contained 1 nM DR2 DNA and 500 nM ChxR with or without 1 to 400 nM anti-ChxR antibody in 40 mM Tris-HCl (pH 8.0), 100 mM NaCl, 250 mM KCl, 10% glycerol, and 0.1 mM EDTA. Triplicate reactions were then incubated, subjected to electrophoresis, and visualized as described above.
ChxRE49D site-directed mutagenesis.
PCR was performed using the QuikChange II XL site-directed mutagenesis kit (Stratagene, Cedar Creek, TX) according to the manufacturer's instructions. The wild-type Glu at residue 49 of ChxR (E49) was replaced with an Asp by using chxR plasmid as the reaction template. ChxRE49D was expressed and purified as described above for ChxR.
RESULTS
ChxR forms homodimers in vitro and in vivo.
In response to phosphorylation, homodimer formation has been shown to be critical for OmpR/PhoB subfamily response regulators to bind cognate DNA and activate transcription (17). Only two studies have evaluated the ability of atypical OmpR/PhoB response regulators to form homodimers, and the observations were discordant (23, 35). Prior data indicated that ChxR is an atypical OmpR/PhoB response regulator, although the ability to form homodimers was not evaluated (28). To begin understanding the mechanisms important for ChxR to activate transcription, studies were designed to determine whether ChxR forms homodimers and mimics that of the active conformation of OmpR/PhoB subfamily response regulators.
During purification of recombinant ChxR protein, size exclusion chromatography indicated that ChxR forms stable homodimers. When a relatively high concentration of ChxR (∼100 μM) was applied to the column, a single peak of protein eluted at the size expected (45 kDa) for a ChxR homodimer (Fig. 1). Although the in vivo concentration of ChxR is unknown, OmpR in E. coli has been measured to be present at concentrations of ca. 1 to 3 μM (9). To address the possibility that ChxR forms homodimers only at high concentrations and not at potentially physiologic concentrations, dilutions of ChxR were applied to size exclusion chromatography. As Fig. 1 demonstrates, when the lowest concentration (1 μM) of ChxR was applied to the column, protein was only detected at the expected size of the ChxR homodimer. These data support that ChxR forms stable homodimers and that the molecular interactions between the two protomers form a relatively strong association, albeit within the testing conditions described.
FIG. 1.
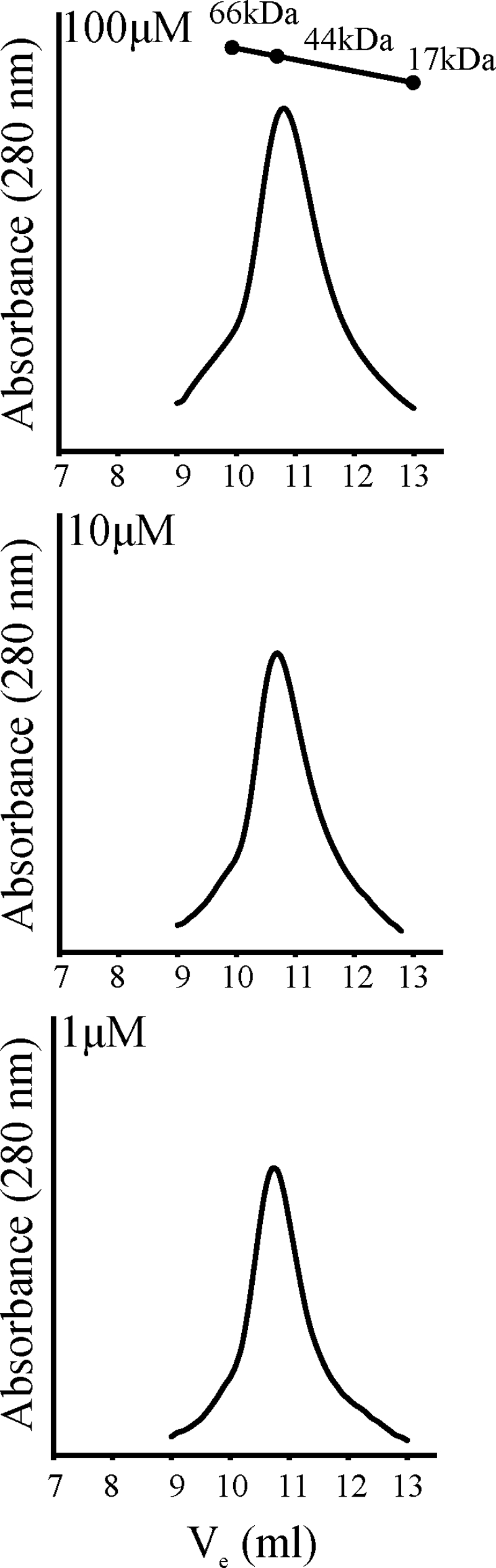
Recombinant ChxR purifies as a stable homodimer. Purified recombinant ChxR at 1, 10, or 100 μM was subjected to analytical size exclusion chromatography to determine the in vitro oligomeric state of the protein. A molecular mass standard curve was generated using bovine serum albumin (66 kDa), chicken ovalbumin (44 kDa), and horse myoglobin (17 kDa).
While the previous data supported that recombinant ChxR forms stable homodimers in the absence of phosphorylation, it was unknown whether ChxR homodimerization occurs in vivo. Given the inability to perform directed genetic studies in Chlamydia, membrane-permeant chemical cross-linkers were used to obtain the most biologically relevant observations. Chemical cross-linkers have previously been utilized to capture homodimer formation by activated OmpR/PhoB subfamily response regulators, even if only within in vitro conditions (13, 30). DSS is a cross-linker that was previously used on the atypical response regulator HP1043 (13). DSS is a membrane-permeant, primary amine homobifunctional cross-linker with a short spacer arm (11.4 Å). ChxR has 17 lysines in addition to the amino terminus (primary amines) that would be expected to serve as targets for DSS and form covalent intermolecular, as well as intramolecular, bonds.
The ability of DSS to capture ChxR homodimers was first tested in vitro. After incubating recombinant ChxR with DSS, two bands of protein were evident following SDS-PAGE analysis (Fig. 2A). One band migrated at the expected size of a ChxR monomer (∼26 kDa), and another migrated at a molecular mass of an expected ChxR homodimer (∼50 kDa). To address the possibility that the detected protein dimerization was due to nonspecific protein interactions, concentrations of ChxR were increased prior to adding DSS. While a small amount of higher-order species were present, after increasing the concentration of ChxR, the predominant species were still homodimers (Fig. 2A). As an additional control for protein-protein interaction specificity, increasing concentrations of a typically monomeric protein, bovine serum albumin, were incubated with DSS. Appreciable formation of higher complexes was not observed (data not shown). These data suggest that the primary amine chemical cross-linker DSS can capture ChxR homodimers in vitro.
FIG. 2.

ChxR forms homodimers in vivo. Recombinant ChxR forms homodimers in vitro; however, the in vivo oligomeric state of ChxR was unknown. (A) To determine whether the primary amine chemical cross-linker DSS could capture ChxR homodimers, increasing concentrations (2.2, 3.5, 7, 14, and 21 μM) of purified, recombinant ChxR were incubated with 500 μM DSS. As a control, 21 μM ChxR was not incubated with DSS. Denatured samples were separated by SDS-PAGE and observed by Coomassie staining. (B) At 12, 24, and 36 hpi, C. trachomatis were enriched from infected L929 cells, and the relative amount of ChxR present was assayed by an immunoblot with polyclonal-monospecific antibodies against ChxR (αChxR). The alpha subunit of RNA polymerase (αRpoA) was used to normalize the amount of chlamydial protein (ChxR) each time point. (C) To test dimer formation in vivo, 10 mM DSS was added to uninfected host cells (Mock) or C. trachomatis-enriched lysates at 30 hpi (Infected). The samples were separated by SDS-PAGE, and an immunoblot was performed with antibodies against ChxR.
Prior to applying DSS to Chlamydia-infected cells to determine whether ChxR forms homodimers in vivo, it was necessary to determine when ChxR is present during the developmental cycle of Chlamydia. Simply described, the biphasic developmental cycle consists of a primarily extracellular, metabolically inactive, and infectious form termed elementary body (EB) converting intracellularly into the metabolically active, replicative, and noninfectious form RB. After numerous rounds of RB replication, asynchronous reciprocal conversion (RB into EB) occurs, and EBs are released to infect new cells (24).
Prior reverse transcription-PCR data indicate that chxR is expressed at 12 hpi and upregulated through 48 hpi (28); however, protein expression has not been determined. To ascertain whether the protein expression pattern complements these findings, the expression profile of ChxR in an RB-enriched fraction of infected cell lysates from 12, 24, and 36 hpi was determined. At 12 hpi, EBs have fully converted to RBs, and the RBs are replicating. At 18 to 24 hpi, some RBs have begun to convert to EBs. At 36 hpi, the inclusion occupies most of the host cell and is composed of RBs and EBs. Immunoblot analysis of these lysates indicated that no ChxR protein was detected at 12 hpi but that ChxR is evident at 24 hpi, and the protein levels dramatically increase by 36 hpi (Fig. 2B). In addition to providing the key times during the developmental cycle to apply DSS, these observations also support that ChxR is most likely exerting its functional activity during the middle (∼24-hpi) and late (>36-hpi) stages of the chlamydial developmental cycle.
Expression data indicated that studies designed to determine the homodimerization capability of ChxR should be performed after 24 hpi. A fraction of C. trachomatis-infected cells enriched for RBs was isolated at 30 hpi and incubated with DSS. Immunoblot analyses of lysates from RB-enriched fractions treated with DSS (Fig. 2C) revealed a protein profile very similar to that in the in vitro experiment (Fig. 2A). Immunoreactive bands were detected near the molecular mass of a ChxR monomer (26 kDa) and homodimer (∼50 kDa). These data support the in vitro observations and indicate that ChxR is forming homodimers in vivo.
ChxR recognizes its own promoter in vivo.
Prior in vitro analyses have demonstrated that ChxR is a transcriptional activator and recognizes its own promoter (28). To provide evidence that ChxR is transcriptionally active in vivo, a commonly utilized immunoprecipitation approach combined with PCR was enlisted (46). RB-enriched fractions were treated with formaldehyde to cross-link ChxR to DNA targets. After immunoprecipitation with anti-ChxR antibodies and extensive washing, DNA was eluted and used in PCRs to determine whether chxR promoter DNA was associated with ChxR. As Fig. 3 indicates, PCR analysis revealed that chxR promoter DNA was specifically associated with ChxR. The specificity of the immunoprecipitation reaction was indicated by the presence of chxR promoter amplicons only when anti-ChxR antibody and cross-linker were applied (Fig. 3).
FIG. 3.

ChxR is associated with its own promoter in vivo. To determine whether ChxR recognizes the chxR promoter during a chlamydial infection, ChxR was cross-linked to DNA, using formaldehyde, at 36 hpi and immunoprecipitated from the lysates using antibodies that recognize ChxR (αChxR). PCR was then performed with primers specific for the chxR promoter. The lack of a PCR product with primers to the CT863 promoter supports that ChxR specifically recognizes the chxR promoter in vivo. The presence (+) or absence (−) of C. trachomatis genomic DNA (DNA) was used as PCR controls for both promoters.
To provide further support for the specificity of ChxR-chxR promoter DNA capture, the association of ChxR with the promoter region of the open reading frame 863 (CT863) was similarly analyzed. CT863 is a gene transcribed at 6 hpi, and transcription levels are constitutively maintained throughout the developmental cycle (6, 22, 32). Based on the expression patterns of ChxR (Fig. 2), it would not be expected that ChxR plays a role in regulating a constitutively expressed CT863. Using primers for this promoter region, a PCR product representative of CT863 promoter was not detected in any of the immunoprecipitated ChxR-DNA samples (Fig. 3). Although these are negative observations (lack of CT863 promoter amplification) from a limited sample size, these observations provide additional support to the specificity of chxR promoter amplification from the immunoprecipitation samples. In combination, these data support that ChxR recognizes its own promoter in vivo and likely plays a key role in regulating its own expression.
Identification of a conserved direct-repeat DNA sequence in each of the ChxR binding sites.
Homodimers of OmpR/PhoB subfamily response regulators generally recognize a region of DNA that ranges from 18 to 23 bp and contains a direct repeat of DNA sequences that are critical for binding (8, 20, 26). Alternatively, examples of DNA binding motifs that consist of inverted repeats have been identified, including the atypical OmpR/PhoB response regulator HP1043 in Helicobacter pylori (23, 47). Observations within the present study support that ChxR exists as a homodimer and would be expected to bind to a similar DNA repeat motif; however, the critical DNA sequences and configuration are undetermined.
Previously, DNase protection assays indicated that ChxR binds to five regions within the chxR promoter, although a consensus recognition sequence was not reported (28). To identify a shared DNA sequence and/or motif, the DNA sequences within these five ChxR binding sites were visually inspected. Within each of the five binding sites, DR sequences (5′-T/A-T/A/C-G-A-T/A-N-T/A/C-3′) separated by 3 to 5 bp were identified (DR1 to DR5; Fig. 4 B), albeit with various degrees of conservation. Using this DNA sequence and arrangement as a guide, an additional sixth site (DR6) was identified upstream of DR5 (Fig. 4A and B). A multiple sequence alignment of all 12 individual binding sites was used to establish a consensus ChxR recognition sequence. The frequency of nucleotides at each position in the DNA recognition sites was calculated, and the computational program Weblogo was utilized to generate a graphic that reflects the nucleotide frequencies (12) (Fig. 4C). Using the nucleotide frequencies at each position, orientation of repeat DNA sequence, and the spacer distance, a ChxR DNA recognition motif was determined (Fig. 4D).
FIG. 4.

chxR promoter region and putative ChxR binding motif. (A) The five putative binding sites (underlined and identified as DR5, DR4, DR3, DR2, and DR1) were derived from a previously reported DNase protection assay (28). A sixth recognition site (DR6) was later identified within the chxR promoter. Transcriptional start site and σ66 holoenzyme promoter element are indicated by +1 and −35/−10, respectively (28) (B) Visual inspection of the six binding sites in the chxR promoter suggested a conserved direct repeat sequence. The two recognition motifs within the six DR sites were aligned. (C) A consensus sequence was generated using Weblogo (12) with each half site from the sequences in Fig. 4B. (D) The recognition sequence and linker length is listed (W = A/T, H = C/A/T, and N = G/C/A/T).
ChxR binds to and exhibits differential affinity for the six individual DR sites in the chxR promoter.
Differential affinity between individual binding sites within a single promoter has been observed for members of the OmpR/PhoB subfamily and is often a central component in the mechanism of regulation (4, 7, 49). Furthermore, it has been reported that binding of a transcription factor to one site can dramatically affect the capability of a neighboring site to become occupied (i.e., cooperativity) (20). To determine the ability of ChxR to bind to each of the DR sites independently and whether a potential binding hierarchy exists, the binding capability of ChxR to each of the six DNA binding sites was analyzed and measured independently. EMSAs were performed with DNA representative to each of the binding sites and ChxR (Fig. 5A and see Table S1 in the supplemental material). ChxR was able to interact with each site independently. In support of the specificity of ChxR to these binding sequences, ChxR did not interact with an oligonucleotide that did not contain a ChxR binding sequence (Fig. 5A; NC). In addition, antibody supershifts by using affinity-purified anti-ChxR demonstrated that the observed DNA shifts are a due to interaction with ChxR (data not shown). The ability of ChxR to interact with DR6 affirms that the visually derived consensus sequence (Fig. 4D) is indeed recognized by ChxR. In addition, by using a static concentration of protein and DNA, it appeared that ChxR has a differential affinity for the individual DR sites.
FIG. 5.

Binding of ChxR to individual DR sites. (A) To determine whether ChxR interacts with the six recognition sites within the chxR promoter individually, EMSAs were performed with 100 nM concentrations of each IR800-labeled binding site (DR1 to DR6) with (+) or without (−) 1 μM ChxR. A DNA sequence corresponding to the −120 to −95 region of the chxR promoter was used as a nonspecific DNA control (NC). The Kd for each binding site is given in Table 1.
To measure the differential affinity for the six DR sites, EMSAs were performed with increasing concentrations of ChxR and each recognition site (data not shown). Kd was then calculated for each site (Table 1). The Kd values listed are for a ChxR/DNA stoichiometric ratio of 2:1 given that recombinant ChxR is a homodimer in vitro. The quantitative analysis revealed that ChxR had the highest affinity (Kd = 43.7 ± 3.9 nM) for DR2 and the lowest affinity (Kd = 1457.0 ± 331.0 nM) for DR6. The affinity for DR2 is more than 33-fold higher than that for DR6. This suggests that there is a hierarchy of binding in the chxR promoter and that the order of binding is as follows: DR2 > DR1 to DR3 > DR4 and DR5 > DR6.
TABLE 1.
Dissociation constants for ChxR and each binding site
| Binding site | Mean Kd (nM) ± SEMa |
|---|---|
| DR1 | 137.6 ± 26.0 |
| DR2 | 43.7 ± 3.9 |
| DR3 | 124.2 ± 27.6 |
| DR4 | 410.5 ± 29.5 |
| DR5 | 479.4 ± 73.8 |
| DR6 | 1457.0 ± 330.6 |
The mean dissociation constants and standard errors of the mean from three replicates are shown.
Three highly conserved bases within the ChxR binding motif are critical to recognition.
As described previously, visual inspection of the six DR sites revealed a conserved recognition sequence (Fig. 4C). Based upon the nucleotide frequency in the deduced recognition site, it is expected that the central GAW nucleotides are critical for DNA binding. To test the hypothesis that ChxR requires the conserved GAW nucleotides for binding, EMSAs were performed with wild-type or mutated DNA constructs from the site (DR2) in the chxR promoter that exhibited the highest affinity for ChxR (Fig. 6A). Increasing concentrations of ChxR were incubated with DR2 DNA containing triple cytosine mutations at either GAW position (DR2-2, −138 to −136; or DR2-1, −126 to −124) or the same mutations at both sites (DR2-1/2). Triple mutations at either GAW site dramatically reduced the ability of ChxR to bind to the DNA fragment relative to the wild-type sequence (Fig. 6B). Mutations in DR2-2 had dramatic negative effects on ChxR binding, although an interaction was still observed at the highest two concentrations of ChxR (50:1 and 100:1). In contrast, mutations in DR2-1 almost completely abolished any detectable interaction with ChxR (Fig. 6B). Mutations at both ChxR monomer-binding sites eliminated any observable ChxR-DNA interaction (Fig. 6B). These data support the hypothesis that the central GAW nucleotides in both sites are important to ChxR binding.
FIG. 6.

Mutations within the DNA recognition motif significantly reduce ChxR-DNA interaction. (A) The DR2 nucleotide sequence from the chxR promoter is shown. The two consensus recognition sequences are underlined, and the boldface nucleotides indicate the sites of triple-cytosine mutations. (B) EMSAs were performed with increasing concentrations of ChxR (39 nM to 3.9 μM) and 39 nM concentrations of each DNA construct: wild-type sequence (WT), DR2-2 mutant, DR2-1 mutant, and DR2-1/2 double mutant.
Single-base-pair contribution to ChxR binding to the DR2 sequence.
The prior analysis supports that the central GAW in the DR2 sequence is critical to ChxR binding. It is expected that additional ChxR-nucleotide interactions are integral to stabilizing the ChxR-DNA complex. To identify these individual bases and measure the effect on ChxR binding, single transversion mutations that would result in a base pair change (A or T↔C or G, respectively) were introduced throughout the DR2 binding site and the intervening sequence. The DR2 site was chosen for single-base-pair contribution analysis because it is the highest affinity binding site in the chxR promoter. The base pair transversions were expected to disrupt both major and minor groove interactions. As described previously in this report, EMSAs were performed with each of the mutated DR2 DNA sequences in triplicate, and the percentage of DR2 DNA relative to wild-type DNA bound by ChxR was determined (Fig. 7).
FIG. 7.

Single-base-pair contributions to ChxR-DNA interactions within the DR2 binding site. EMSAs were performed with 5 μM ChxR and 50 nM DNA containing transversion mutations. The target DNA used in the experiment was the DR2 sequence from the chxR promoter, comprising the DR2 half-sites (underlined). The percentages of DNA shifted with each transversion mutation (n = 3) are shown in the graph relative to the DNA shifted with the wild-type sequence (n = 18). The amount of DNA shifted was quantified using the photon emission of the SYBR green at 520 nm. The mutations that resulted in a significant (P < 0.05) reduction of DNA-interaction are denoted by an asterisk.
Nine of the single-base-pair mutations resulted in a statistically significant decrease of percent DNA shifted relative to wild-type DR2 DNA. Of the nine single-base-pair mutations that had a statistically significant decrease in DNA shifted, four mutations had considerably (>20%) less DNA shifted than wild-type DR2 DNA. The most dramatic decrease in DNA shifting occurred when a transversion was introduced at position −134. Position −134 is located at the 3′ end of the DR2-2 ChxR binding site and resulted in a 70% reduction in DNA shifted relative to the wild-type DR2 sequence. Within the predicted spacer region, at the base immediately 3′ of the DR2-2 site, a transversion caused an ca. 50% reduction of DNA binding by ChxR. In contrast to DR2-2, transversions at four separate locations in DR2-1 (−124, −125, −127, and −128) resulted in a statistically significant decrease in ChxR binding; however, only the mutations in the central GAW positions (−124 and −125) resulted in >20% reduction in the percentage of DNA shifted. Together, these data support that single-base transversions can have a negative effect on ChxR binding to the DR2 region; however, no single mutation eliminated ChxR binding.
ChxRE49D retains dimerization and DNA binding activity.
Typically, members of the OmpR/PhoB response regulator subfamily are phosphorylated through a highly conserved Asp residue in the receiver domain of these proteins (15). This phosphorylation facilitates the reorientation of two switch residues and promotes homodimerization, DNA binding, and transcriptional regulation. ChxR is unique in this respect because the amino acid in the predicted position of the conserved Asp is a Glu residue (E49) (28). Many transcription factors that are activated by a phosphorylated Asp can be converted into a phosphoryl-independent constitutively active state via substitution with Glu (3, 18, 27, 40). Based upon these observations, it was hypothesized that Glu 49 is critical to DNA binding and transcriptional activity of ChxR and that conversion to an Asp could render the molecule inactive. To begin testing this hypothesis, Glu 49 was substituted with an Asp (E49D), and the modified ChxRE49D was assayed for homodimerization and DNA binding capability.
Similar to wild-type ChxR, recombinant ChxRE49D migrated via size exclusion chromatography at a size expected for a homodimer (data not shown), indicating that this modification had a minimal effect on monomer-monomer interactions. To test the capability of ChxRE49D to dimerize, unmodified ChxR or ChxRE49D was exposed to a chemical cross-linker (DSS) to capture homodimers prior to separation via SDS-PAGE. The resulting observations also support that ChxRE49D retained the ability to form homodimers (Fig. 8A). The ability of ChxRE49D to bind to DNA was tested via EMSA by using the DR2 site from the chxR promoter (Fig. 8B). The percentage of DNA shifted with ChxRE49D was quantified and found to be very similar to that with wild-type ChxR. These data indicate the Glu residue (E49) in the position of the conserved Asp in other OmpR family members does not solely account for the constitutive transcriptional activity of ChxR.
FIG. 8.

ChxRE49D retains homodimer formation and DNA binding activity. (A) To test homodimerization, 5 μM ChxR or 6 μM ChxRE49D was incubated with 500 μM DSS. Proteins were separated by SDS-PAGE and visualized by Coomassie staining. (B) To determine the DNA binding activity of ChxRE49D, EMSAs were performed in triplicate with 1.25 μM ChxR or 1.25 μM ChxRE49D and 50 nM DNA from the DR2 site. The percentage of DNA shifted was quantified and normalized as described previously.
DISCUSSION
Predominantly in response to phosphorylation, homodimer formation is a governing step for the regulation of transcription by a large majority of the OmpR/PhoB subfamily of response regulators. Homodimerization orients and stabilizes individual protomers that promote DNA binding and subsequent transcriptional regulation. While prior studies on atypical response regulators support phosphorylation-independent transcriptional activation, the importance of homodimer formation is still uncertain. The data presented here demonstrated that ChxR, in the absence of phosphorylation, forms stable homodimers in vitro, even at concentrations (1 μM) that are likely to be physiologically relevant (Fig. 1And 2). This conclusion is further supported by the utility of the membrane-permeant cross-linker DSS, which showed that ChxR forms homodimers in vivo (Fig. 2). Although it is possible that ChxR has an alternate site of phosphorylation or undetermined modification that promotes homodimerization within Chlamydia, prior analyses showed that unmodified ChxR activated transcription at its own promoter in vitro and within a heterologous E. coli system (28). As such, it is unlikely that this unknown mechanism for phosphorylation at an alternate site or undetermined mechanism is present in E. coli. These observations and those described herein support that wild-type ChxR protein is in a conformation that likely mimics the phosphoryl-activated OmpR/PhoB response regulators, including homodimerization.
While the conformation of ChxR may mimic that of phosphorylated OmpR/PhoB members, ChxR exhibits unique characteristics. Substitution of the phosphorylated Asp with Glu can render response regulators constitutively active (3, 18, 27, 40). However, the ability of ChxR to maintain a homodimer conformation and interact with DNA is not the result of this single substitution (D49E). Supporting this result was the retention of the homodimer formation and DNA binding capability of ChxR after a Glu-to-Asp substitution (ChxRE49D; Fig. 8). These data suggest that the residues (Ser/Thr in β4 and Phe/Tyr in β5) that are typically reoriented in response to Asp phosphorylation might be stabilized in an “active” orientation in unphosphorylated ChxR. Notably, ChxR does not encode a Ser or Thr in the expected β4 strand but does encode a Tyr (Y90) in the anticipated β5 strand. Structural studies on the receiver domain would be useful to identify the molecular orientations, such as that of Tyr90, and interactions that may be critical to forming stable homodimers and eventual transcriptional regulation by this atypical OmpR/PhoB response regulator.
Alternatively, it is possible that phosphorylation of recombinant ChxRE49D in E. coli facilitated the formation of an active ChxR molecule. This appears less likely based upon a number of observations. First, the rate of unaided dephosphorylation in OmpR/PhoB subfamily response regulators typically occurs within seconds to an hour (45), which would suggest that the phosphoryl group would not have been retained throughout the purification process and storage of the protein before the assays were conducted. Second, a cognate sensor kinase that could phosphorylate ChxR is likely not present given the phylogenetic distance between Chlamydia and E. coli. Response regulator phosphorylation by cognate sensor kinases is relatively specific, and cross talk between sensor kinases and response regulators is limited, even within bacteria of the same species. Third, the three-dimensional structure of the ChxR response regulator has been solved (unpublished data), and structural analysis does not indicate the presence of a phosphoryl group.
The affinities for ChxR to each of the six DR sites are lower than those of typical OmpR/PhoB subfamily response regulators. Studies with phosphorylated OmpR have determined that, of the protein-DNA interactions measured, the DNA affinity ranged from ca. 7 to 300 nM (21). Furthermore, DNA interaction is greatly enhanced when multiple repeat sequences are present. In the present study, the affinity for ChxR with the individual six DR sites was measured and ranged from ca. 44 nM to 1.5 μM (Table 1), which is ∼5-fold lower than that of OmpR. It may be possible that cooperativity plays a key role in enhancing affinity to the promoter region. As such, it is currently unclear how the presence of multiple binding sites influences the affinity of ChxR for DNA but is a focus of ongoing studies.
Several individual base pair mutations were demonstrated to have significant effects on ChxR binding to the DR2 DNA binding site (Fig. 7). Interestingly, only one of these mutations (−134) resulted in a >50% reduction in ChxR binding to DNA. The overall tolerance to single-base transversions is likely indicative of relatively strong ChxR affinity to this particular binding site. This is supported by the observation that ChxR has the highest binding to a DR2 DNA fragment compared to the other five sites (Table 1). Furthermore, no single mutation completely eliminated ChxR binding to the DR2 DNA sequence. This is in contrast to the triple-base-pair mutation introduced into either conserved ChxR binding sequence that virtually eliminated all DNA binding by ChxR (Fig. 6). Moreover, no single mutation caused a significant increase in ChxR binding efficiency, suggesting that DR2 is reflective of an optimal ChxR binding site.
Single-base mutations appeared to have more negative influence on DR2-1 ChxR binding site than DR2-2. Four base mutations (−128, −127, −125, and −124) all caused statistically significant reductions in ChxR binding to DR2-1. In contrast, only a mutation at −134 caused a decline in DR2-2 binding, albeit the largest effect by a single mutation. These observations indicate that affinity of ChxR to the DR2-1 wild-type sequence is weaker, since individual base changes are not tolerated well. This is in congruence with the apparent requirement for both ChxR recognition sites for a stable protein-DNA interaction (Fig. 6). Furthermore, the dramatic negative affect at −134 also supports that, in the context of the surrounding sequencing, this residue is critical for ChxR binding. Interestingly, single mutations that resulted in a negative effect were not constrained to the seven bases within a conserved binding site. Three intervening base mutations (−133, −131, and −130) resulted in significant reductions in ChxR binding. This suggests that protein contacts are likely occurring within this region and playing a role in the affinity of ChxR to DNA. Of note, the wing of the OmpR/PhoB subfamily winged-helix motif typically interacts with the DNA minor groove in a non-base-specific fashion. In addition, these mutations could be affecting the topology of the relatively small fragment of DNA and disrupting ChxR interactions in the conserved sequences. A comparison of the intervening sequence between binding sites does not reveal any conserved nucleotide frequencies, which suggests that the latter (DNA topology) is more likely; however, sequence-specific interactions may be involved in only a few of the binding sites, such as DR2. Along these lines, a recent report has emphasized the role of DNA topology on gene regulation in Chlamydia (10).
Based upon the ChxR DNA binding sequence (WHGAWNH; Fig. 4), it may be expected that ChxR has a relatively low level of DNA binding specificity. This may be insightful in regards to the overall role of ChxR in Chlamydia. Bacterial transcription factors that exhibit relatively low levels of specificity are consistently global regulators which are transcription factors that regulate relatively large number of genes and incorporate different response conditions, coregulators, or different sigma factors (29). For example, OmpR has been reported to regulate the transcription of at least 125 genes (33). In contrast, local regulators, transcription factors that regulate a relatively small number of genes associated with a specific stimulus/physiologic response, have high levels of specificity. The ChxR direct repeat binding sequence with a 3- to 5-base spacer occurs at the 3203 and 3303 sites in the C. trachomatis serovar D and serovar L2 genomes, respectively (data not shown). However, when the two genomes are searched with a consensus sequence (WHGAWNW) from the three highest affinity sites (DR1 to DR3), the number of binding sites decreased to 1826 and 1902 for the D and L2 genomes, respectively. Further analyses are clearly needed to correlate the ability of ChxR to recognize any of these sites for transcriptional regulation. It is expected that characterizing other ChxR binding sites will refine the ChxR recognition motif. However, the low nucleotide conservation in the binding sequence supports the hypothesis that ChxR is a global regulator in Chlamydia, in contrast to a local, more restricted transcriptional regulator.
The presence of chxR transcripts during the early stages of the developmental cycle indicates that the previously determined σ66 promoter (28) is active but weakly initiating transcription or that posttranscriptional repression mechanisms are used. Based upon the ability of ChxR to bind to the six sites present in the chxR promoter, we speculate that full expression of chxR may rely on a threshold of ChxR molecules being attained. This threshold would require occupancy of 12 ChxR proteins (6 homodimers) to all six binding sites within the chxR promoter. This potential mechanism is intriguing, since the signal for the asynchronous conversion of RB to EB is unknown. As RBs replicate, it may be expected that the cytosolic contents, including ChxR, are diluted and distributed unequally to the daughter cells. This possibility, combined with differential replication rates of individual organisms, could permit ChxR to accumulate and reach a threshold for full chxR expression in subpopulations of Chlamydia. Many other factors are expected to play a role in the stability of ChxR, and its ability to form homodimers that would also affect this proposed mechanism. While this mechanism is largely speculative, future studies regarding the mechanism of ChxR activation and identifying global ChxR gene targets are expected to address the validity of the proposed model.
Supplementary Material
Acknowledgments
This research and J.M.H., L.W., and P.S.H. were supported by the National Institutes of Health (P20RR17708 and AI079083).
We thank Susan Egan for critical review of the manuscript.
Footnotes
Published ahead of print on 5 November 2010.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Abdelrahman, Y. M., and R. J. Belland. 2005. The chlamydial developmental cycle. FEMS Microbiol. Rev. 29:949-959. [DOI] [PubMed] [Google Scholar]
- 2.Ainsa, J. A., H. D. Parry, and K. F. Chater. 1999. A response regulator-like protein that functions at an intermediate stage of sporulation in Streptomyces coelicolor A3(2). Mol. Microbiol. 34:607-619. [DOI] [PubMed] [Google Scholar]
- 3.Arribas-Bosacoma, R., S. K. Kim, C. Ferrer-Orta, A. G. Blanco, P. J. Pereira, F. X. Gomis-Ruth, B. L. Wanner, M. Coll, and M. Sola. 2007. The X-ray crystal structures of two constitutively active mutants of the Escherichia coli PhoB receiver domain give insights into activation. J. Mol. Biol. 366:626-641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barnard, A., A. Wolfe, and S. Busby. 2004. Regulation at complex bacterial promoters: how bacteria use different promoter organizations to produce different regulatory outcomes. Curr. Opin. Microbiol. 7:102-108. [DOI] [PubMed] [Google Scholar]
- 5.Beier, D., and R. Frank. 2000. Molecular characterization of two-component systems of Helicobacter pylori. J. Bacteriol. 182:2068-2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belland, R. J., G. Zhong, D. D. Crane, D. Hogan, D. Sturdevant, J. Sharma, W. L. Beatty, and H. D. Caldwell. 2003. Genomic transcriptional profiling of the developmental cycle of Chlamydia trachomatis. Proc. Natl. Acad. Sci. U. S. A. 100:8478-8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bergstrom, L. C., L. Qin, S. L. Harlocker, L. A. Egger, and M. Inouye. 1998. Hierarchical and co-operative binding of OmpR to a fusion construct containing the ompC and ompF upstream regulatory sequences of Escherichia coli. Genes Cells 3:777-788. [DOI] [PubMed] [Google Scholar]
- 8.Blanco, A. G., M. Sola, F. X. Gomis-Ruth, and M. Coll. 2002. Tandem DNA recognition by PhoB, a two-component signal transduction transcriptional activator. Structure 10:701-713. [DOI] [PubMed] [Google Scholar]
- 9.Cai, S. J., and M. Inouye. 2002. EnvZ-OmpR interaction and osmoregulation in Escherichia coli. J. Biol. Chem. 277:24155-24161. [DOI] [PubMed] [Google Scholar]
- 10.Case, E. D., E. M. Peterson, and M. Tan. Promoters for Chlamydia type III secretion genes show a differential response to DNA supercoiling that correlates with temporal expression pattern. J. Bacteriol. 192:2569-2574. [DOI] [PMC free article] [PubMed]
- 11.Chater, K. F. 1972. A morphological and genetic mapping study of white colony mutants of Streptomyces coelicolor. J. Gen. Microbiol. 72:9-28. [DOI] [PubMed] [Google Scholar]
- 12.Crooks, G. E., G. Hon, J. M. Chandonia, and S. E. Brenner. 2004. WebLogo: a sequence logo generator. Genome Res. 14:1188-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delany, I., G. Spohn, R. Rappuoli, and V. Scarlato. 2002. Growth phase-dependent regulation of target gene promoters for binding of the essential orphan response regulator HP1043 of Helicobacter pylori. J. Bacteriol. 184:4800-4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fraser, J. S., J. P. Merlie, Jr., N. Echols, S. R. Weisfield, T. Mignot, D. E. Wemmer, D. R. Zusman, and T. Alber. 2007. An atypical receiver domain controls the dynamic polar localization of the Myxococcus xanthus social motility protein FrzS. Mol. Microbiol. 65:319-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedland, N., T. R. Mack, M. Yu, L. W. Hung, T. C. Terwilliger, G. S. Waldo, and A. M. Stock. 2007. Domain orientation in the inactive response regulator Mycobacterium tuberculosis MtrA provides a barrier to activation. Biochemistry 46:6733-6743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galperin, M. Y. 2005. A census of membrane-bound and intracellular signal transduction proteins in bacteria: bacterial IQ, extroverts and introverts. BMC Microbiol. 5:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao, R., T. R. Mack, and A. M. Stock. 2007. Bacterial response regulators: versatile regulatory strategies from common domains. Trends Biochem. Sci. 32:225-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao, R., A. Mukhopadhyay, F. Fang, and D. G. Lynn. 2006. Constitutive activation of two-component response regulators: characterization of VirG activation in Agrobacterium tumefaciens. J. Bacteriol. 188:5204-5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao, R., and A. M. Stock. 2009. Biological insights from structures of two-component proteins. Annu. Rev. Microbiol. 63:133-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harlocker, S. L., L. Bergstrom, and M. Inouye. 1995. Tandem binding of six OmpR proteins to the ompF upstream regulatory sequence of Escherichia coli. J. Biol. Chem. 270:26849-26856. [DOI] [PubMed] [Google Scholar]
- 21.Head, C. G., A. Tardy, and L. J. Kenney. 1998. Relative binding affinities of OmpR and OmpR-phosphate at the ompF and ompC regulatory sites. J. Mol. Biol. 281:857-870. [DOI] [PubMed] [Google Scholar]
- 22.Hefty, P. S., and R. S. Stephens. 2007. Chlamydial type III secretion system is encoded on ten operons preceded by sigma 70-like promoter elements. J. Bacteriol. 189:198-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hong, E., H. M. Lee, H. Ko, D. U. Kim, B. Y. Jeon, J. Jung, J. Shin, S. A. Lee, Y. Kim, Y. H. Jeon, C. Cheong, H. S. Cho, and W. Lee. 2007. Structure of an atypical orphan response regulator protein supports a new phosphorylation-independent regulatory mechanism. J. Biol. Chem. 282:20667-20675. [DOI] [PubMed] [Google Scholar]
- 24.Hybiske, K., and R. S. Stephens. 2007. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc. Natl. Acad. Sci. U. S. A. 104:11430-11435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kato, H., T. Chibazakura, and H. Yoshikawa. 2008. NblR is a novel one-component response regulator in the cyanobacterium Synechococcus elongatus PCC 7942. Biosci. Biotechnol. Biochem. 72:1072-1079. [DOI] [PubMed] [Google Scholar]
- 26.Kenney, L. J. 2002. Structure/function relationships in OmpR and other winged-helix transcription factors. Curr. Opin. Microbiol. 5:135-141. [DOI] [PubMed] [Google Scholar]
- 27.Klose, K. E., D. S. Weiss, and S. Kustu. 1993. Glutamate at the site of phosphorylation of nitrogen-regulatory protein NTRC mimics aspartyl-phosphate and activates the protein. J. Mol. Biol. 232:67-78. [DOI] [PubMed] [Google Scholar]
- 28.Koo, I. C., D. Walthers, P. S. Hefty, L. J. Kenney, and R. S. Stephens. 2006. ChxR is a transcriptional activator in Chlamydia. Proc. Natl. Acad. Sci. U. S. A. 103:750-755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lozada-Chavez, I., V. E. Angarica, J. Collado-Vides, and B. Contreras-Moreira. 2008. The role of DNA-binding specificity in the evolution of bacterial regulatory networks. J. Mol. Biol. 379:627-643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maris, A. E., D. Walthers, K. Mattison, N. Byers, and L. J. Kenney. 2005. The response regulator OmpR oligomerizes via beta-sheets to form head-to-head dimers. J. Mol. Biol. 350:843-856. [DOI] [PubMed] [Google Scholar]
- 31.Mittal, S., and L. Kroos. 2009. A combination of unusual transcription factors binds cooperatively to control Myxococcus xanthus developmental gene expression. Proc. Natl. Acad. Sci. U. S. A. 106:1965-1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nicholson, T. L., L. Olinger, K. Chong, G. Schoolnik, and R. S. Stephens. 2003. Global stage-specific gene regulation during the developmental cycle of Chlamydia trachomatis. J. Bacteriol. 185:3179-3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rhee, J. E., W. Sheng, L. K. Morgan, R. Nolet, X. Liao, and L. J. Kenney. 2008. Amino acids important for DNA recognition by the response regulator OmpR. J. Biol. Chem. 283:8664-8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rotter, C., S. Muhlbacher, D. Salamon, R. Schmitt, and B. Scharf. 2006. Rem, a new transcriptional activator of motility and chemotaxis in Sinorhizobium meliloti. J. Bacteriol. 188:6932-6942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruiz, D., P. Salinas, M. L. Lopez-Redondo, M. L. Cayuela, A. Marina, and A. Contreras. 2008. Phosphorylation-independent activation of the atypical response regulator NblR. Microbiology 154:3002-3015. [DOI] [PubMed] [Google Scholar]
- 36.Schaaf, S., and M. Bott. 2007. Target genes and DNA-binding sites of the response regulator PhoR from Corynebacterium glutamicum. J. Bacteriol. 189:5002-5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schachter, J. 1999. Infection and disease epidemiology, p. 139-169. In R. S. Stephens (ed.), In Chlamydia: intracellular biology, pathogenesis, and immunity. ASM Press, Washington, DC.
- 38.Schar, J., A. Sickmann, and D. Beier. 2005. Phosphorylation-independent activity of atypical response regulators of Helicobacter pylori. J. Bacteriol. 187:3100-3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scidmore, M. A. 2005. Cultivation and Laboratory Maintenance of Chlamydia trachomatis. Curr. Protoc Microbiol. Chapter 11:Unit 11A1. [DOI] [PubMed]
- 40.Siam, R., and G. T. Marczynski. 2003. Glutamate at the phosphorylation site of response regulator CtrA provides essential activities without increasing DNA binding. Nucleic Acids Res. 31:1775-1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stephens, R. S. 2003. The cellular paradigm of chlamydial pathogenesis. Trends Microbiol. 11:44-51. [DOI] [PubMed] [Google Scholar]
- 42.Stephens, R. S. 2002. Chlamydiae and evolution: a billion years and counting, p. 3-12. In C. Schachter, H. Clarke, K. Kaltenboeck, S. Saikku, S. Stephens, P. Timms, and P. Wyrick (ed.),. Proceedings of the Tenth International Symposium on Human Chlamydial Infections. International Chlamydia Symposium, San Francisco, CA.
- 43.Stephens, R. S., S. Kalman, C. Lammel, J. Fan, R. Marathe, L. Aravind, W. Mitchell, L. Olinger, R. L. Tatusov, Q. Zhao, E. V. Koonin, and R. W. Davis. 1998. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 282:754-759. [DOI] [PubMed] [Google Scholar]
- 44.Stock, A. M., V. L. Robinson, and P. N. Goudreau. 2000. Two-component signal transduction. Annu. Rev. Biochem. 69:183-215. [DOI] [PubMed] [Google Scholar]
- 45.Stock, J. B., A. J. Ninfa, and A. M. Stock. 1989. Protein phosphorylation and regulation of adaptive responses in bacteria. Microbiol. Rev. 53:450-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wade, J. T., K. Struhl, S. J. Busby, and D. C. Grainger. 2007. Genomic analysis of protein-DNA interactions in bacteria: insights into transcription and chromosome organization. Mol. Microbiol. 65:21-26. [DOI] [PubMed] [Google Scholar]
- 47.Wang, S., J. Engohang-Ndong, and I. Smith. 2007. Structure of the DNA-binding domain of the response regulator PhoP from Mycobacterium tuberculosis. Biochemistry 46:14751-14761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.West, A. H., and A. M. Stock. 2001. Histidine kinases and response regulator proteins in two-component signaling systems. Trends Biochem. Sci. 26:369-376. [DOI] [PubMed] [Google Scholar]
- 49.Yoshida, T., L. Qin, L. A. Egger, and M. Inouye. 2006. Transcription regulation of ompF and ompC by a single transcription factor, OmpR. J. Biol. Chem. 281:17114-17123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.