

Abstract
The protein scaffold and signaling regulator p62 is important in critical cellular functions, including bone homeostasis, obesity, and cancer, because of its interactions with various signaling intermediaries. p62 is overexpressed in human cancers and is induced during cell transformation. Its genetic ablation inhibits lung tumorigenesis in vivo and cell proliferation in culture by regulating the TRAF6/NF-κB signaling cascade to control reactive oxygen species (ROS) production and apoptosis. Here we show that cdk1 phosphorylates p62 in vitro and in vivo at T269 and S272, which is necessary for the maintenance of appropriate cyclin B1 levels and the levels of cdk1 activity necessary to allow cells to properly enter and exit mitosis. The lack of cdk1-mediated phosphorylation of p62 leads to a faster exit from mitosis, which translates into enhanced cell proliferation and tumorigenesis in response to Ras-induced transformation. Therefore, p62 emerges as a node for the control of not only cell survival but also cell transit through mitosis.
Scaffold and adaptor proteins are required for the efficient and selective transmission of information during cell signal transduction. They function by restraining the nonspecific access of enzymes to substrates, which could produce undesirable cellular effects if not properly controlled. One such adaptor is p62 (also known as sequestosome 1), which was initially isolated as an interacting partner of the atypical protein kinase C isoforms (21). p62 has been implicated in crucial cellular processes through biochemical assays that demonstrated its ability to interact with important signaling intermediaries (15, 16). The phenotypic analysis of genetically modified mice lacking p62 shows that, in fact, p62 regulates several physiological processes (1, 2, 20). These include osteoclastogenesis and bone homeostasis, through the E3 ubiquitin ligase TRAF6 by acting as an important intermediary of the RANK pathway in the activation of the transcription factor NF-κB (2). Another informative feature of p62-deficient mice is that they develop late-onset obesity that leads to impaired glucose tolerance and insulin resistance (20). Recently, we demonstrated, through both in vitro and in vivo assays, that p62 is an important regulator of extracellular signal-regulated kinase 1 (Erk1) in metabolism (10). Taken together, these observations indicate that p62 plays critical roles in bone remodeling and obesity.
In addition to these physiological roles, there is evidence that p62 also contributes to certain pathologies. That is, p62 levels are increased in human tumors and in cells transformed by the Ras oncogene, which is crucial for tumorigenesis (1, 5). Indeed, a lack of p62 markedly inhibits Ras-induced cell transformation in cell cultures and in a mouse model of Ras-induced lung carcinogenesis due to the impaired activation of NF-κB by Ras in the absence of p62 (1). In this paradigm, the reduced NF-κB levels observed in the Ras-transformed p62-deficient mice leads to more apoptosis than in wild-type (WT) cells and mice, without apparent changes in the proportion of cells in the G1, S, or G2/M phases of the cell cycle (1). Therefore, in the context of Ras-induced tumorigenesis, the role of p62 is to induce cell survival through NF-κB and the ensuing control of ROS production by Ras (1). However, cell division is a fundamental process that mediates the growth, development, and maintenance of all organisms. The cell cycle integrates innumerable cellular activities whose execution is rigorously coordinated to maintain chromosome stability and to produce healthy progeny (3, 6). Any misregulation in this coordinated progression through the cell division cycle can lead to genome instability that, in turn, may result in reduced fitness, uncontrolled proliferation, or death of the progeny cells (6, 9). For example, subtle alterations in the timing of a cell's entrance or exit from the mitotic phase of the cell cycle could have consequences in tumorigenesis (9, 24). In this study, we sought to determine whether p62 plays a role in cell cycle regulation that could contribute to its role in cell transformation.
Cell division in mammalian cells is driven by cyclin-dependent kinases (cdk's) that regulate progression through the various phases of the cell cycle (18). cdk's are heterodimeric protein kinases each composed of a catalytic subunit known as cdk and a regulatory subunit known as cyclin (14). The mammalian genome has 12 loci encoding cdk's, although only five of them (i.e., cdk1, cdk2, cdk3, cdk4, and cdk6) have been directly implicated in cell cycle progression (14). Evidence from knockout (KO) mice has shown that cdk1, a mitotic kinase, is the only one that is not redundant and plays an essential role in cell cycle control (22). The other cdk's are believed to play several redundant roles in the early phases of cell division (22). Therefore, cdk1 emerges as a central driver of the cell cycle, specifically during mitosis. The activities of cdk's are regulated by different mechanisms, including phosphorylation/dephosphorylation and transcription, but more importantly by the levels of cyclins (11). The activity of cdk1 is regulated by the availability of cyclin B1, whose expression fluctuates throughout the cell cycle. During S phase, cyclin B1 mRNA and protein begin to accumulate and reach their maximum levels at G2/M phase. As cells pass through mitosis, cyclin B1 is ubiquitinated and degraded by the anaphase-promoting complex (APC) (17). Although the cell cycle regulation of cdk's has been extensively characterized, only a few physiological substrates have been identified, and the detailed mechanisms by which cdk's regulate cell cycle transitions are still unclear (13). Here, we demonstrate that p62 is phosphorylated by cdk1 at early mitosis and that this phosphorylation is necessary for efficient activity of the cdk1/cyclin B1 complex by regulating the levels and stability of cyclin B1 and the ensuing transition of cells through mitosis. We also demonstrate that the phosphorylation of p62 is necessary to restrain tumorigenesis in vivo in response to Ras-induced transformation.
MATERIALS AND METHODS
Cell culture and treatments.
The HEK293-derived virus-packaging cell line Phoenix-GP and the A549, U2OS, and HEK293 cell lines were obtained from the American Type Culture Collection (ATCC). p62 KO immortal embryo fibroblasts (EFs) have been previously described (1). p62-floxed (p62fl/fl) EFs were derived from day 13.5 post coitum (D13.5) embryos, according to standard procedures. Cells were grown in Dulbecco modified Eagle medium (DMEM; Gibco BRL) supplemented with 10% (vol/vol) fetal calf serum (FCS), 1% glutamine, and 1% penicillin-streptomycin (Gibco-Invitrogen) in an atmosphere of 95% air and 5% CO2. For G1/S-phase cell cycle synchronization, a double thymidine block (dTB) was performed, as follows. Cells (30% to 40% confluent) were incubated with 2 mM thymidine for 18 h at 37°C. After incubation, cells were rinsed with phosphate-buffered saline (PBS) and incubated with thymidine-free medium for 8 h. Cells were then reincubated with 2 mM thymidine for 17 h. For M-phase synchronization, cells blocked in G1/S phase were released for 3 h in complete medium and subsequently treated for the indicated times with nocodazole at 100 ng/ml. Mitotic cells were then detached by tapping the culture flasks, washed in PBS, and used for mitotic exit time course experiments, for which they were replated on either polylysine-coated cover slides for immunofluorescence analysis or on tissue culture dishes for subsequent protein extraction. Synchronized and released cells were also fixed in ethanol and analyzed by propidium iodide (PI) staining (50 μg/ml in PBS) and flow cytometry (FACScan; Becton-Dickinson) for cell cycle distribution. Treatments with nocodazole were performed in complete medium for the indicated times. At each time point, cells were collected by trypsinization and divided into two aliquots for protein extraction and flow cytometry analysis. For cyclin B1 stability assays, cells were treated with 100 μg/ml of cycloheximide. Kinase inhibitors (10 μM purvalanol, 50 μM VX-680, 50 μM PD98059, 50 μM SB202190, and 50 μM SB216763, all purchased from Sigma) were added to the blocked cells 1 h before the end of incubation. Extracts from cells that had been released from the thymidine block into nocodazole were treated with alkaline phosphatase (Takara), according to the manufacturer's instructions.
Retroviral, lentiviral, and Cre recombinase adenovirus transduction.
The following retroviral expression vectors were used: pWZL-Hygro-FLAG-p62, pWZL-Hygro-FLAG-p62T269A, pWZL-Hygro-FLAG-p62S272A, pWZL-Hygro-FLAG-p62T269A-S272A, pBabe-Puro, and pBabe-Puro-K-RasV12. Retroviruses were produced in Phoenix-GP cells by transient transfection with Lipofectamine 2000 (Invitrogen). Culture supernatants were collected at 24 h, 48 h, and 72 h posttransfection, filtered (0.45 μm), and supplemented with 4 μg/ml Polybrene. U2OS cells were infected with three rounds of viral supernatants to generate stable clones expressing wild-type p62 (p62WT), p62T269A, p62S272A, p62T269A-S272A (p62AA), and p62T269E-S272E (p62EE). Clones of U2OS cells were selected with hygromycin (75 μg/ml) over a course of 7 days in the indicated experiments. p62 KO EFs were infected with three rounds of viral supernatants to generate stable clones expressing p62WT and p62AA. Clones of EFs were selected with hygromycin (75 μg/ml) over a course of 7 days, and the generated clones were then transformed with three rounds of viral supernatant of pBabe-Puro or pBabe-Puro-K-RasV12 to generate Ras-transformed EFs. Lentivirus Mission short hairpin RNA (shRNA) plasmids (Sigma) were utilized for the knockdown of cdk1, according to the manufacturer's instructions. To infect EFs, we used an empty adenovirus and adeno-Cre (empty Ad5 cytomegalovirus [CMV] and Ad5 CMV Cre, respectively; University of Iowa Gene Transfer Vector Core), with approximate multiplicities of infection of 100. We incubated EFs for 24 h in medium containing adenovirus to allow for efficient infection and lox site excision.
Time-lapse experiments.
Monitoring of cell mitosis was carried out on double thymidine-blocked cells. Five hours after release from the block, cell mitosis was followed with time-lapse cinematography using image analysis software (Discovery-1; Universal Imaging). The experimental apparatus used for time-lapse experiments consisted of an inverted microscope (IX50 Olympus), a charge-coupled-device (CCD) CoolSnap camera (RS Photometrix), an XYZ computer-controlled stage (ProScan; Prior), and a mini-incubator set on the stage. The camera and the computerized stage were synchronized by specific codes to follow several cells in the same experiment. Cells were observed for 12 h, and images were acquired every 5 min. Movies were created from the acquired images and analyzed by MetaMorph Offline software.
In vitro kinase assays.
Recombinant MBP-p62 (Upstate) and FLAG-tagged p62 immunoprecipitated from cell extracts were subjected to phosphorylation with the recombinant cdk1/cyclin B1 complex (Cell Signaling) in phosphorylation buffer (35 mM Tris-HCl, 10 mM MgCl2, 0.1 mM CaCl2, 0.5 mM EGTA, 100 μM ATP, 1 mM dithiothreitol [DTT], 5 μCi of [32P]ATP) for 1 h at 30°C. The phosphorylation was stopped with sample buffer, and proteins were then resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to nitrocellulose membranes, and analyzed by autoradiography for incorporation of 32P. To measure cdk1 activity, hemagglutinin (HA)-tagged cdk1 immunoprecipitated from cell extracts was incubated in phosphorylation buffer with histone H1 (H1) as the in vitro target substrate.
Mice.
All mice were kept under pathogen-free conditions. Animal handling and experimental procedures conformed to institutional guidelines (University of Cincinnati Institutional Animal Care and Use Committee). For the in vivo tumor growth assays, cell suspensions (5 × 105 cells/ml) were injected intradermally into each flank of male athymic 4- to 6-week-old nu/nu mice.
RESULTS
p62 is phosphorylated at early mitosis.
As an initial assessment of the likelihood of a role for p62 in cell cycle regulation, we measured the levels of p62 in extracts from cells of the lung adenocarcinoma cell line A549 that had been synchronized at G1 by release from a double thymidine block (dTB). We also determined the levels of p62 in A549 cells that had been synchronized by a dTB, as described above, but were released into treatment with nocodazole, which allows for the accumulation of synchronized cells at early mitosis. Interestingly, although total p62 protein levels were not changed by any of these treatments (Fig. 1), we observed the appearance of a supershifted band at 10 and 14 h after the exposure to nocodazole, which suggests the phosphorylation of p62 during mitosis (Fig. 2A). To address this possibility, extracts from cells that had been released from a dTB into nocodazole for 14 h were treated with alkaline phosphatase. Figure 2B demonstrates that phosphatase treatment reduced the intensity of the supershifted band, such that p62 exhibited faster mobility, comparable to that of the nontreated cells. These data suggest that p62 undergoes phosphorylation at the beginning of the M phase of the cell cycle.
FIG. 1.

p62 levels do not change in early mitosis. A549 cells were arrested in G1 by a double thymidine block (time zero) and were released for the indicated times in medium with nocodazole in three independent experiments. Cell lysates were immunoblotted for p62, and its levels were measured using ImageJ software. The graph shows the means ± standard deviations.
FIG. 2.

cdk1 phosphorylates p62 during mitosis. (A) A549 cells were arrested in G1 by a double thymidine block (time zero) and were released for the indicated times in fresh medium or medium with nocodazole. Cell lysates were immunoblotted for p62 or for actin as a loading control. (B) A549 cells were arrested in G1 by a double thymidine block and were released into nocodazole for 14 h. Cell lysates were treated with alkaline phosphatase and analyzed by immunoblotting for p62. (C) A549 cells treated as described in the legend to panel B were incubated with purvalanol A (cdk1 inhibitor), VX-680 (aurora kinase inhibitor), PD98059 (MEK inhibitor), SB202190 (p38 inhibitor), or SB216763 (GSK3β inhibitor) for 1 h, and the levels of p62 were determined by immunoblotting. (D) A549 cells were transduced with a cdk1 shRNA lentivirus (sh-cdk1). Cells were then treated as described in the legend to panel B. Cell lysates were immunoblotted to detect p62, cdk1, and actin as a loading control. NT, nontargeted. (E) In vitro phosphorylation of recombinant p62 with [32P]ATP in the presence or absence of recombinant cdk1/cyclin B1.
cdk1 phosphorylates p62 at T269 and S272.
cdk1 is a cyclin-associated kinase that plays critical roles in the control of cell cycle progression at late S/G2 and in early mitosis (14). As cells arrested by nocodazole show maximum cdk1 activity, it is conceivable that p62 could be targeted by cdk1, which could account for its phosphorylation upon nocodazole treatment. To test this possibility and rule out the hypothetical actions of another kinase, cells treated as shown in Fig. 2B were incubated with selective inhibitors of cdk1, aurora kinase, MEK, p38, and glycogen synthase kinase 3β (GSK3β) for 1 h, after which p62 levels were analyzed by immunoblotting. Interestingly, the supershifted band triggered by nocodazole treatment was completely abolished only by cdk1 inhibition (Fig. 2C). We also transduced A549 cells with a cdk1 short hairpin RNA (shRNA) lentivirus, after which cells were treated as shown in Fig. 2B, and cell lysates were immunoblotted with anti-p62 antibody. Interestingly, cdk1 downregulation abolished the supershifted p62 band (Fig. 2D). These results indicate that cdk1 is responsible for p62 phosphorylation at early mitosis. To determine whether p62 is actually a direct cdk1 substrate, we incubated bacterially expressed recombinant MBP-p62 with an active cdk1 complex in an in vitro kinase assay. Under these conditions, cdk1 was able to directly phosphorylate MBP-p62 but not the MBP control (Fig. 2E). All together, these results demonstrate that p62 is directly phosphorylated by cdk1 at early mitosis, when cdk1 activity is maximal.
We next determined whether there is a stable, direct p62-cdk1 interaction by performing in vitro pulldown experiments in which recombinant MBP-p62 protein was incubated with the GST-cdk1/cyclinB1 complex. Samples were immunoprecipitated with anti-GST antibody to precipitate the recombinant cdk1/cyclin B1 complex, and the association with p62 was determined by immunoblotting with an anti-p62 antibody. The results shown in Fig. 3 A demonstrate a direct interaction of p62 with the cdk1/cyclin B1 complex in vitro. In addition, HEK293 cells were cotransfected with FLAG-tagged p62 and HA-tagged cdk1 constructs. Cell lysates were immunoprecipitated with an anti-HA antibody to precipitate the transfected cdk1, and the association with p62 was determined by immunoblotting the precipitates with an anti-FLAG antibody. The results shown in Fig. 3B demonstrate that, in fact, p62 interacts with cdk1. Consistent with this, the results shown in Fig. 4 demonstrate that p62 interacts with cdk1 independently of the presence of cyclin B1. In another experiment, A549 cells that ectopically express FLAG-tagged p62 were synchronized by a dTB and incubated with or without nocodazole, after which endogenous cdk1 was immunoprecipitated, followed by immunoblotting with an anti-FLAG antibody. Interestingly, from this experiment, it is clear that, although p62 is hyperphosphorylated under the nocodazole-treated condition, it constitutively interacts with cdk1 independently of the presence or absence of nocodazole (Fig. 3C). Therefore, we next determined whether this interaction could be detected in a more physiologically relevant setting. For this, endogenous cdk1 was immunoprecipitated from A549 cells and treated as shown in Fig. 3C, followed by immunoblotting with anti-p62. The results shown in Fig. 3D demonstrate that endogenous p62 interacts with cdk1. To map the precise sites on p62 that are phosphorylated by cdk1, we used A549 cells constitutively expressing FLAG-tagged p62. These cells were synchronized by a dTB, followed by nocodazole (or control) treatment. p62 was purified by using affinity chromatography with an anti-FLAG antibody, followed by elution with a FLAG peptide. The purified p62 was subjected to in-gel trypsin digestion, followed by phosphopeptide enrichment using titanium dioxide columns and liquid chromatography coupled to mass spectrometry (LC-MS). Following use of this strategy, we found that p62 was phosphorylated at S272 and T269 upon nocodazole treatment (Fig. 5A). Comparison of p62 sequences around these sites from different species highlights the high degree of conservation in this region (Fig. 5B).
FIG. 3.

cdk1 interacts with p62. (A) Bacterially expressed recombinant MBP and MBP-p62 were incubated with the recombinant GST-cdk1/cyclin B1 complex, and the ability of p62 to bind directly to the cdk1/cyclin B1 complex was determined in a pulldown experiment. WB, Western blot. (B) Human HEK293 cells were cotransfected with expression plasmids encoding FLAG-tagged p62 and HA-tagged cdk1, and cell lysates were immunoprecipitated (IP) with HA epitope-tagged antibody. The immunoprecipitates were analyzed by immunoblotting with an anti-FLAG antibody. Cell lysates were analyzed for HA and FLAG expression. (C) A549 cells that express FLAG-tagged p62 were arrested in G1 by a double thymidine block and released in nocodazole for 14 h. Cell extracts were immunoprecipitated with anti-cdk1 or IgG control antibody. The immunoprecipitates were analyzed by immunoblotting with an anti-FLAG antibody. (D) A549 cells were treated as described in the legend to panel C. Cell extracts were immunoprecipitated with anti-cdk1 or IgG control antibody. The immunoprecipitates were analyzed by immunoblotting with an anti-p62 antibody.
FIG. 4.
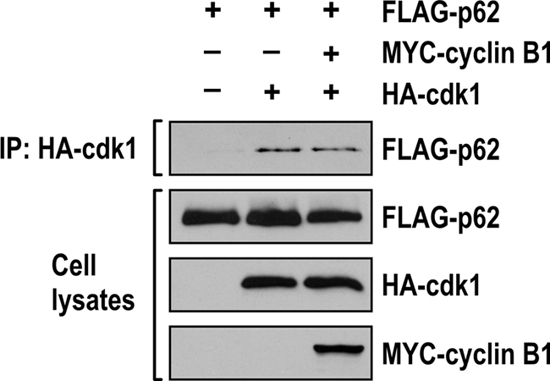
p62 interacts with cdk1 in the absence of cyclin B1. Human HEK293 cells were cotransfected with expression plasmids encoding FLAG-tagged p62, HA-tagged cdk1, and MYC-tagged cyclin B1, and cell lysates were immunoprecipitated with HA epitope-tagged antibody. The immunoprecipitates were analyzed by immunoblotting with an anti-FLAG antibody. Cell lysates were immunoblotted to detect HA, MYC, and FLAG expression.
FIG. 5.

Cdk1 phosphorylates p62 at threonine 269 and serine 272. (A) A549 cells expressing FLAG-tagged p62 were synchronized by a double thymidine block, followed by release into nocodazole for 14 h, and the phosphorylation sites in p62 were determined by LC-MS in the FLAG immunoprecipitates. Tandem mass spectrometry (MS-MS) spectra of the phosphopeptides corresponding to sites T269 and S272 in p62 are shown. (B) Human p62 sequence containing the consensus cdk1 phosphorylation sites aligned with the corresponding sequences from different species. H. sapiens, Homo sapiens; E. caballus, Equus caballus; M. mulatta, Macaca mulatta; B. taurus, Bos taurus; M. musculus, Mus musculus; R. norvegicus, Rattus norvegicus. (C) A549 cells that express FLAG-tagged p62WT, p62T269A, p62S272A, or p62T269AS272A were arrested in G1 by a double thymidine block and were released for 14 h in medium with nocodazole. Cell lysates were immunoblotted using FLAG antibody. (D) Extracts from HEK293 cells cotransfected with FLAG-tagged p62WT or p62AA were immunoprecipitated with the anti-FLAG antibody. The resulting immunoprecipitates were subjected to in vitro phosphorylation with [32P]ATP in the presence of recombinant cdk1/cyclin B1. (E) Extracts of HEK293 cells cotransfected with FLAG-tagged p62WT or p62AA and HA-tagged cdk1 were immunoprecipitated with the anti-HA antibody. The resulting immunoprecipitates were subjected to an in vitro phosphorylation with [32P]ATP, using histone H1 as the target substrate.
To test the functional relevance of these phosphorylations, we used FLAG-p62 with T269 and S272 mutated to alanines, either individually or simultaneously. These constructs were transfected into A549 cells, which were incubated in the presence of nocodazole for 14 h, as described above. Cell extracts were prepared, and the mobility of p62 under these conditions was determined by immunoblotting with an anti-FLAG antibody. Figure 5C clearly shows that the individual nonphosphorylatable mutations of residue T269 or S272 to alanine partially reduced the mobility shift of p62 in nocodazole-treated cells. However, the T269 S272 double mutant completely abolished the p62 supershift in nocodazole-treated cells. To determine whether these residues account for the majority of p62 phosphorylation by cdk1 in mitosis, cell lysates from HEK293 cells transfected with FLAG-tagged p62WT or the p62 T269 S272 double mutant (p62AA) were immunoprecipitated with anti-FLAG antibody to precipitate transfected p62WT or p62AA and then were phosphorylated in vitro by a recombinant cdk1/cyclin B1 complex. Figure 5D demonstrates that cdk1 is unable to phosphorylate the p62AA mutant. We concluded from these studies that residues T269 and S272 account for a large majority of p62 phosphorylation by cdk1 in mitosis.
To further test whether p62 phosphorylation is involved in cdk1 activation, HEK293 cells cotransfected with HA-tagged cdk1 and FLAG-tagged p62WT or p62AA were immunoprecipitated with anti-HA antibody to precipitate transfected cdk1. A kinase assay was then performed using histone protein (H1) as the target substrate. The results shown in Fig. 5E demonstrate that cdk1 activity was markedly reduced in cells transfected with p62AA compared with that in cells expressing p62WT. These results demonstrate that p62 phosphorylation plays an important role in cdk1 activation.
Cells with mutant p62 display altered transition through mitosis.
Once we established that p62 is phosphorylated by cdk1 at residues T269 and S272, we next addressed the functional role of these phosphorylations in the cell cycle. For this, we ectopically expressed FLAG-tagged p62WT or p62AA in U2OS cells to generate the two corresponding cell lines. U2OS is a particularly appropriate cell line for these studies because endogenous p62 levels are nearly undetectable and it has been extensively used in cell cycle studies. We reasoned that, since p62 is phosphorylated by cdk1 in early mitosis, it is possible that the role of p62 phosphorylation is to regulate transition through this phase of the cell cycle. Therefore, cells were synchronized at the G1/S boundary with a dTB and then released into the cell cycle in the presence of nocodazole for different amounts of time. Consistent with the hypothesis, the data shown in Fig. 6 A demonstrate that the levels of phospho-S10-H3 (a well-established marker of mitosis) were induced in the p62WT cells but that this induction was impaired in the p62AA cell line. Similar results were obtained when a different stable clone expressing p62WT or p62AA was examined (Fig. 7). This indicates that a lack of p62 phosphorylation at T269/S272 affects mitosis upon synchronized nocodazole arrest. Interestingly, under these conditions, the synthesis of cyclin A is properly induced in the two cell lines, but that of cyclin B1 is impaired in the p62AA mutant-expressing cells (Fig. 6A). Consistent with this, cdk1 activity, as determined by histone H1 phosphorylation, was also impaired in these mutant cells (Fig. 6A). Binding of cyclin B1 to cdk1 was not affected by p62 phosphorylation (Fig. 8). Likewise, p62 interaction with its previously identified partners (atypical protein kinase C isoforms [aPKCs], TRAF6, and ERK) was not modified in the nonphosphorylatable p62 mutant (Fig. 9). Of note, flow cytometry analysis showed that there was a reproducible reduction in the percentage of p62AA cells accumulating in the G2/M fraction on nocodazole exposure compared to that of the p62WT cell line (Fig. 6B and C). The mutation of p62 phosphorylation sites to phosphomimetic glutamate (p62EE) reverted the defect of p62AA cells on the entry to mitosis (Fig. 10). Interestingly, nocodazole treatment, followed by the shake-off and counting of cells in mitosis, demonstrated that the lack of p62 phosphorylation by cdk1 leads to a significant reduction in the percentage of cells in mitosis in the G2/M fraction (Fig. 6D). On the other hand, because prolonged cell arrest in mitosis due to nocodazole treatment typically results in cell death by apoptosis (25), and because p62 has been shown to be an important survival molecule (1), we tested whether phosphorylation of p62 by cdk1 could regulate the life/death decision point in nocodazole-treated cells. We reasoned that a reduction in phospho-S10-H3 levels and the percentage of p62AA mutant cells in mitosis could conceivably be due to reduced survival when arrested by nocodazole. However, we found that, although nocodazole treatment produced a detectable increase in cell death, there were no significant differences between the WT and p62AA-expressing cells (Fig. 6E). Collectively, these results demonstrate that the inability of p62AA to be phosphorylated by cdk1 leads to a selective reduction in cyclin B1 levels and a subsequent inhibition of cdk1 activity that translates into a reduced mitotic index in the mutant cells.
FIG. 6.

Altered G2/M transition in cells expressing the nonphosphorylatable p62 mutant. (A) U2OS cells expressing either p62WT or p62AA were synchronized by a double thymidine block and released into nocodazole-containing medium for the indicated times. Cell lysates were immunoblotted for phospho-S10-H3 [p-H3 (S10)]; cyclin A; p62; cyclin B1; phospho-histone 1 (p-H1), a marker of cdk1 activity; and actin as a loading control. The experiment shown is representative of two others with similar results. (B) Flow cytometry analysis of U2OS cells expressing either p62WT or p62AA treated as described in the legend to panel A. The experiment shown is representative of two others with similar results. (C) Percentages of cells in each phase treated as described in the legend to panel A. (D) Percentages of cells in mitosis after nocodazole treatment, followed by shake-off detachment and counting. The graph shows the means ± standard deviations of triplicates. *, P < 0.05. (E) Percentages of cells with cleaved caspase-3 in cells treated as described in the legend to panel A.
FIG. 7.

Altered G2/M transition in cells expressing the nonphosphorylatable p62 mutant. A clone of U2OS cells that is different from the one used in Fig. 4A expressing either p62WT or p62AA was synchronized by a double thymidine block and released into nocodazole-containing medium for the indicated times. Cell lysates were immunoblotted for phospho-S10-H3 (p-H3), cyclin A, cyclin B1, phospho-histone 1 (p-H1), p62, and actin as a loading control. The experiment shown is representative of two others with similar results.
FIG. 8.
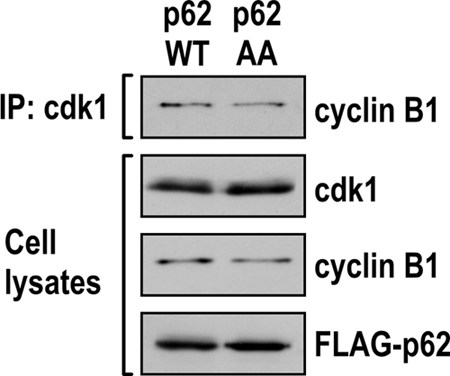
p62 phosphorylation does not affect cdk1-cyclin B1 complex formation. Cell lysates from U2OS cells expressing either p62WT or p62AA were immunoprecipitated with anti-cdk1 antibody. The immunoprecipitates were analyzed by immunoblotting to detect cyclin B1. Cells lysates were immunoblotted to detect cdk1, cyclin B1, and FLAG expression.
FIG. 9.

p62 phosphorylation does not affect complex formation with p62 partners. U2OS cells expressing either p62WT or p62AA were synchronized by a double thymidine block and released into nocodazole-containing medium for 14 h. Cell lysates were immunoprecipitated with an anti-FLAG antibody. The immunoprecipitates were analyzed by immunoblotting to detect PKCλ, PKCζ, Traf6, and Erk (left). Cell lysates were immunoblotted to detect PKCλ, PKCζ, Traf6, Erk, and p62 expression (right).
FIG. 10.

Mutation of the p62 phosphorylation sites to phosphomimetic glutamate reverses the p62AA defect on the entry to mitosis. (A) Flow cytometry analysis from U2OS cells expressing either p62WT, p62AA, or p62EE synchronized by a double thymidine block and released into nocodazole-containing medium for the indicated times. The experiment shown is representative of two others with similar results. (B) Percentages of cells in each phase when treated as described in the legend to panel A.
p62 phosphorylation by cdk1 regulates cdk1 activity and influences mitotic exit.
cdk1 in complex with cyclin B1 (cdk1/cyclin B1) is the key mitotic kinase, whose inactivation is required for cells to exit from mitosis. The activity of the cdk1/cyclin B1 complex is regulated at the exit of mitosis by the availability of cyclin B1, which is ubiquitinated and degraded by the anaphase-promoting complex (APC) (17, 19). To test whether cdk1/cyclin B1 is regulated by the phosphorylation state of p62 during mitosis exit, we synchronized the p62WT and p62AA cell lines by using a dTB, followed by nocodazole treatment for 14 h. Afterward, mitotic cells were shaken off and, when in suspension, were reseeded in nocodazole-free medium, allowing for cell cycle progression from the prometaphase block. Cells were then collected at different time points and analyzed for different mitotic biochemical parameters. Of potential functional relevance, the decrease in the levels of phosphorylated histone H1 and of cyclin B1 during the release phase was reproducibly faster in cells that expressed p62AA than in those expressing p62WT (Fig. 11A). Of note, the levels of securin, another substrate of APC that must be degraded for cells to exit mitosis, were also lower in the p62AA cells when released from the nocodazole block (Fig. 11A). Interestingly, p62 was also quickly degraded upon mitotic block release, consistent with the notion that it is part of a functional mitotic complex (Fig. 11B). Of note, the degradation of mutant p62AA was somehow slower than that of the WT (Fig. 11B), suggesting that p62 phosphorylation, in addition to being a signal to promote enhanced cyclin B1 levels and cdk1 activity, makes p62 more vulnerable to its degradation during mitotic exit.
FIG. 11.

p62 phosphorylation regulates the exit from mitosis. (A) U2OS cells expressing either p62WT or p62AA were isolated by mitotic shake-off and released for 30, 60, and 90 min. Cell lysates were immunoblotted for cyclin B1, securin, and p-H1. The experiment shown is representative of two others with similar results. (B) Cells were treated as described in the legend to panel A, and cell lysates were immunoblotted for p62 and actin as a loading control. (C) Flow cytometry analysis of cells treated as described in the legend to panel A. This experiment is representative of another two with very similar results. (D) Quantification of total mitosis length in the two cell lines, as assessed by video time-lapse microscopy. Results (means ± standard deviations; *, P < 0.05) are representative of 30 mitotic cells (n = 30) followed independently. (E) Immunofluorescence analysis of p62WT- and p62AA-expressing mitotic cells stained for α-tubulin (green) and propidium iodide (PI; red). Representative confocal sections showing normal telophase in a p62WT cell (left) and telophase with missegregated (lagging) chromosomes (white arrow on the right) in a p62AA cell. (F) Quantification of mitosis with missegregated chromosomes in p62WT and p62AA cells. Cells were examined 10 h after the double thymidine block (left) and 60 min after the mitotic shake-off (right). n, total number of mitoses analyzed. (G) Immunofluorescence analysis of p62WT and p62AA cells stained for PI. Representative confocal sections showing normal interphase in a p62WT cell (left) and interphase with the presence of micronuclei (white arrow on the right) in a p62AA cell. (H) Quantification of micronuclei in p62WT and p62AA cells. Cells were examined 10 h after the double thymidine block (left) and 90 min after the mitotic shake-off (right). n, total number of mitoses analyzed.
When these cells were analyzed in parallel by flow cytometry, it was apparent that there was a reproducible increase in the percentage of the p62 mutant cells in G1, compared to that of p62WT cells, as early as 30 and 60 min after reseeding, indicating that the exit from mitosis, and the transition from mitosis to G1, is faster in cells that cannot have p62 phosphorylated by cdk1 (Fig. 11C). Since nocodazole treatment by itself might have an effect on mitosis length, we decided to analyze the mitotic progression of undisturbed WT and mutant cells by using video time-lapse microscopy. To do this, p62WT and p62AA cells were synchronized by a dTB, followed by release into fresh medium, after which they were followed for up to 12 h by video microscopy. The analysis of cells of each type confirmed that, in the absence of p62 phosphorylation, a lower percentage of cells exhibited a slowed prophase-telophase transition (Fig. 11D). These results demonstrate that p62 phosphorylation plays an important role in the regulation of the mitotic transition/exit. One consequence of a faster mitotic exit is the presence of cells containing lagging chromosomes and micronuclei during mitosis. We looked for defects in DNA segregation by immunofluorescence of cells released after a dTB and subsequently released after a mitotic shake-off. Our data clearly showed a significant increase in the percentage of cells containing lagging chromosomes and micronuclei for those expressing the nonphosphorylatable form of p62 (Fig. 11E to H). Collectively, these data indicate that p62 phosphorylation by cdk1 is necessary to maintain adequate cdk1/cyclin B1 kinase activity levels, which prevents a premature exit from mitosis. Failure of p62AA cells to do so would lead to a faster-than-normal exit from mitosis and possibly increased proliferation. Consistent with this model, we observed in growth curve experiments that p62AA cells cultured in complete medium grew faster over a 6-day period than the p62WT cells (Fig. 12A).
FIG. 12.

A lack of p62 phosphorylation by cdk1 leads to enhanced cell proliferation and tumorigenesis in Ras-transformed cells. (A) Growth curve comparing p62AA (black squares) and p62WT (white squares) cells over the course of 6 days in complete medium. Results are shown as means ± standard deviations. (B) p62 KO EFs reconstituted with either p62WT or p62AA were infected with K-RasV12. Cell lysates were immunoblotted to detect p62, Ras, and actin as a loading control. (C) Growth curve comparing p62AA (black squares) and p62WT (white squares) Ras-transformed p62-KO EFs in 0.5% FBS medium. Results are shown as means ± standard deviations. (D) Suspensions of p62 KO EFs reconstituted with either p62WT or p62AA, and infected with K-RasV12, were intradermally injected into each flank of nude mice, and tumors were allowed to develop for 17 days. Tumor size was measured every other day. Results are shown as means ± standard deviations (n = 5). (E) Tumor weights at 17 days (means ± standard deviations; *, P < 0.05).
Lack of p62 phosphorylation by cdk1 leads to enhanced cell proliferation and tumorigenesis in Ras-transformed cells.
We recently showed that p62 was important for Ras-induced transformation in EFs and in lung cancer in vivo (1). That is, the lack of p62 in EFs from KO mice showed reduced proliferation when transformed by the Ras oncogene compared with those from WT controls, due to the inability of Ras to induced NF-κB in the absence of p62 (1). As we have shown here that p62, in addition to its role in cell survival during transformation, also plays a role in the control of mitosis transit, we next determined the impact of p62 phosphorylation by cdk1 in Ras-induced cell proliferation. To do this, we generated immortalized embryo fibroblasts (EFs) in a p62 knockout (KO) background that were reconstituted with either p62WT or p62AA expression constructs and transformed by expression of the K-RasV12 oncogene. The results shown in Fig. 12B demonstrate that both cell lines displayed similar levels of ectopically expressed p62 and oncogenic Ras. Interestingly, and consistent with the data obtained using U2OS cells, Ras-transformed EFs that expressed the p62AA mutant form grew faster than the Ras-transformed EFs expressing p62WT (Fig. 12C).
To determine whether phosphorylation of p62 affects the ability of Ras-transformed EFs to form solid tumors in vivo, we injected cell suspensions of Ras-transformed EFs that expressed p62WT or p62AA, as described above, intradermally into each flank of nude mice, and tumors were allowed to develop for 17 days. Interestingly, from the results of this experiment, it is clear that inoculation with Ras-expressing p62AA cells resulted in larger tumors that grew faster than the tumors resulting from Ras-expressing p62WT cells (Fig. 12D and E). Collectively, these results demonstrate that while p62 is necessary for Ras to induce cell survival and transformation, its phosphorylation by cdk1 is required for the restraint of cell growth and tumor progression in response to Ras.
Lack of p62 produces a slower exit from mitosis.
Once we had established that p62 phosphorylation by cdk1 controls the timely transit of cells through mitosis, we sought to determine whether the absence of p62 affects cell cycle progression. To test this possibility, U2OS cells, transfected with small interfering RNA (siRNA) to deplete p62 levels, were synchronized at the G1/S boundary with a dTB and then released into the cell cycle in the presence of nocodazole for different durations. Surprisingly, flow cytometry analysis showed that the absence of p62 did not affect cell cycle progression and entry into mitosis (Fig. 13A and B).
FIG. 13.

The absence of p62 produces a slower exit from mitosis. (A) Flow cytometry analysis of U2OS cells transfected with p62 siRNA and control siRNA synchronized by a double thymidine block and released into nocodazole-containing medium for the indicated times. The experiment shown is representative of two others with similar results. (B) Percentages of cells in each phase, treated as described in the legend to panel A. (C) Flow cytometry analysis of U2OS cells transfected with p62 siRNA and control siRNA, isolated by mitotic shake-off, and released for 30, 60, and 90 min. The experiment shown is representative of two others with similar results. (D) Percentages of cells in each phase, treated as described in the legend to panel C. (E) Cells were treated as described in the legend to panel C, and cell lysates were immunoblotted for p62, cyclin B1, and actin as a loading control. The experiment shown is representative of another two with very similar results. (F) Flow cytometry analysis of p62fl/fl EFs infected with Cre or GFP control adenoviruses, isolated by mitotic shake-off, and released for 60, 90, and 120 min. The experiment shown is representative of two others with similar results. (G) Percentages of cells in each phase, treated as described in the legend to panel F.
To test whether the absence of p62 affects the exit from mitosis, we synchronized p62 knockdown and control cells by a dTB, followed by nocodazole treatment for 14 h. Afterward, mitotic cells were shaken off and reseeded in nocodazole-free medium, allowing for their cell cycle progression. Cells were then collected at different time points. Interestingly, when these cells were analyzed by flow cytometry, it was apparent that there was a reproducible reduction in the percentage of the p62 knockdown cells in G1 compared with that of control cells as early as 60 and 90 min after reseeding, indicating that the exit from mitosis and the transition from mitosis to G1 are slower in cells that have reduced levels of p62 (Fig. 13C and D). Of potential functional relevance, the decrease in the levels of cyclin B1 during the release phase was reproducibly slower in p62 knockdown cells than in control cells (Fig. 13E). Similar results were obtained using p62-floxed mouse embryo fibroblasts (p62fl/fl EFs) infected with Cre and green fluorescent protein (GFP) control adenoviruses (Fig. 13F and G). Interestingly, these results demonstrated that when p62 is missing, cells have the opposite phenotype to that of p62AA-expressing cells, which supports the idea that p62 is phosphorylated by cdk1 to prevent a premature exit from mitosis.
p62 phosphorylation prevents cyclin B1 degradation by the proteasome.
Our results shown above (Fig. 6A and 11A) demonstrate that the lack of phosphorylation of p62 produces a reduction in cyclin B1 levels at the entry and exit from mitosis. Since cyclin B1 is ubiquitinated by the anaphase-promoting complex (APC) and subsequently degraded by the proteasome system, we hypothesized that the phosphorylation state of p62 could regulate cyclin B1 ubiquitylation, which would affect its stability. To test this hypothesis, HEK293 cells were cotransfected with MYC-tagged cyclin B1, HA-tagged ubiquitin, and FLAG-tagged p62WT or p62AA. Cell lysates were immunoprecipitated with anti-MYC antibody and immunoblotted with an anti-HA antibody to reveal the presence of ubiquitin-conjugated cyclin B1 species. Interestingly, our data showed that the expression of the nonphosphorylatable p62AA mutant led to enhanced ubiquitylation of cyclin B1 (Fig. 14A), suggesting that p62 phosphorylation protects cyclin B1 from ubiquitylation and subsequent degradation through the proteasome.
FIG. 14.

p62 phosphorylation prevents cyclin B1 degradation. (A) HEK293 cells were cotransfected with expression plasmids encoding FLAG-tagged p62WT or p62AA, HA-tagged ubiquitin, and MYC-tagged cyclin B1, and cell lysates were immunoprecipitated with MYC epitope-tagged antibody. The immunoprecipitates were analyzed by immunoblotting with an anti-HA antibody (left). Cell lysates were immunoblotted to detect HA, MYC, and FLAG expression (right). (B) HEK293 cells cotransfected with expression plasmids encoding FLAG-tagged p62WT or p62AA and MYC-tagged cyclin B1 were treated with 100 μg/ml of cycloheximide (CHX) for the indicated times. Ectopic cyclin B1 and ectopic p62 levels were detected by Western blot analysis using an anti-MYC antibody and an anti-FLAG antibody, respectively (left). Ectopic cyclin B1 levels were measured by using ImageJ software. The graph shows means ± standard deviations from three independent experiments (right).
To further test whether p62 phosphorylation is involved in the stability of cyclin B1, HEK293 cells were cotransfected with MYC-tagged cyclin B1, along with either FLAG-tagged p62WT or FLAG-tagged p62AA. The transfected cells were treated with cycloheximide (CHX), an inhibitor of protein synthesis, and cyclin B1 levels were analyzed by Western blotting. In cells expressing p62WT, cyclin B1 protein levels decreased by nearly 50% after 12 h of CHX treatment (Fig. 14B). In contrast, cyclin B1 protein levels were nearly completely degraded in the p62AA-expressing cells after 12 h of CHX treatment (Fig. 14B). These results demonstrate that p62 phosphorylation plays an important role in the stabilization of cyclin B1, consistent with its role in the control of cyclin B1 ubiquitylation.
DISCUSSION
The protein scaffold and signaling regulator p62 has been implicated in critical cellular functions such as bone homeostasis (2), obesity (20), and cancer (1) through the regulation of important signaling intermediaries like TRAF6/NF-κB (23), the atypical protein kinase C isoforms (23), caspase-8 (4), ERK1 (10), and the oxidative stress regulator Keap1 (7). p62 has also been shown to be targeted for degradation by autophagy, which presumably also degrades polyubiquitinated proteins that associate with p62 through its ubiquitin-associated (UBA) domain (8).
Here we show data on the regulation of p62 by, and its role in, the mitotic phase of the cell cycle. Our results demonstrate that p62 is phosphorylated at early mitosis by cdk1 and that this modification is critical for the optimal transition of cells through mitosis. We propose that, during mitosis, the phosphorylation of p62 by cdk1 at T269 and S272 is necessary for the maintenance of appropriate cyclin B1 levels, which leads to suitable cdk1 activity to allow cells to enter and exit that phase of the cell cycle in a timely manner. This is an important event because alterations in mitotic transit could influence cell proliferation and tumorigenesis. Interestingly, our data show that the levels of both cyclin B1 and cdk1 activities decay faster in p62 mutant cells than in p62WT cells upon removal of the nocodazole blockade. This is very important because it suggests that p62 phosphorylation serves to restrain the premature exit from mitosis by regulating cdk1/cyclin B1 activity, which is critical for appropriate cell proliferation (3, 9, 25). In support of this conclusion, the absence of p62 in cells delayed the exit from mitosis, which mimicked the phenotype of p62WT-expressing cells. It is very well documented that, at the end of mitosis, the machinery controlled by cdk1/cyclin B1 gets inactivated by degradation to allow for the mitotic exit. Interestingly, p62 is also inactivated by degradation, which is consistent with its role in that important cellular function. Future studies will determine the mechanisms controlling p62 degradation, including the potential involvement of the APC system. However, we present here compelling evidence that p62 phosphorylation is important for the control of cyclin B1 ubiquitylation, and therefore, it offers a mechanistic explanation for our observation that p62 phosphorylation regulates cyclin B1 stability. Our results suggest that the role of p62 is to promote cyclin B1 instability, which is inactivated by cdk1-mediated phosphorylation of p62. That would explain why overexpression of p62WT, which is phosphorylatable, promotes cyclin B1 stability (Fig. 14B), whereas that of p62AA decreases cyclin B1 levels (Fig. 14B), and why p62-deficient cells have higher levels of cyclin B1 (Fig. 13E), in that they would be deprived of the cyclin B1 destabilizing function of p62. This model would also explain the role of p62 at the exit of mitosis. At that stage of the cell cycle, p62WT is degraded prior to cyclin B1 degradation (Fig. 11A and B), but p62AA degradation is much slower than that of p62WT and cyclin B1 (Fig. 11A and B). We believe that this has important repercussions in the regulation of cyclin B1 levels at the exit of mitosis. If our model is correct and the role of unphosphorylated p62 is to promote cyclin B1 instability, which is blocked by cdk1-induced phosphorylation when cells abandon mitosis, as p62WT is degraded before cyclin B1, it should not really affect the kinetics of cyclin B1 degradation. But when it cannot be phosphorylated, like in the p62AA mutant, then cyclin B1 is more rapidly degraded (Fig. 11A and B). Therefore, p62AA has two important properties that influence cyclin B1 degradation during mitotic exit: (i) it induces cyclin B1 destabilization because it cannot be phosphorylated, and (ii) it is more stable, which further accelerates cyclin B1 degradation kinetics.
These observations unveil a novel role and mechanism of action for p62 that could have implications in tumorigenesis, since the finely tuned and adequately regulated control of mitosis is required to ensure proper control of cell division, an essential component of the tumorigenic process (18, 25). In keeping with this notion, recent results show that Ras tumorigenesis depends on dysfunctional mitotic machinery (12). Therefore, p62 phosphorylation by cdk1, and the proper control of mitosis transit exerted by phosphorylated p62, should be relevant for the proliferation of Ras-transformed cells and for Ras-induced tumorigenicity. In fact, our data shown here demonstrate that Ras-transformed cells expressing the unphosphorylatable form of p62 proliferate faster and produce more tumors in a Ras-induced tumorigenesis assay than the Ras transformants expressing p62WT. These findings have to be put in the context of our previous observations, demonstrating that p62 KO EFs showed dramatically reduced survival when transformed by the Ras oncogene (1). Under those conditions, the prevailing mechanism is the inability of Ras to control NF-κB in the absence of p62, which leads to increased ROS production and cell death (1). However, under the conditions shown here, whereby cells express p62 that cannot be phosphorylated by cdk1/cyclin B1, the major role of p62 is to restrain the proliferation of Ras transformants by being phosphorylated by the mitotic complex of cdk1/cyclin B1. Interestingly, p62 is overexpressed in a myriad of tumor types, and due to its complex structure and ability to interact with different signaling regulators, it is likely to use a variety of mechanisms to promote cell transformation, including the proper control of mitosis by cdk1-mediated phosphorylation. Therefore, p62 emerges as a critical node for the control of cell survival and transformation through its ability to impinge not only on cell survival but also on mitotic pathways.
Acknowledgments
This work is funded by National Institutes of Health grants R01AI072581 (to J.M.), R01CA132847 (to J.M.), R01DK088107 (to J.M.), and R01CA134530 (to M.T.D.-M.), by a Greater Cincinnati Foundation award (to J.M.) and by Department of Defense grant DoD-PC080441 (to M.T.D.-M.).
We are grateful for skilled research assistance by Lyndsey Bolanos. We thank Maryellen Daston for editing the manuscript and Glenn Doerman for the artwork.
We dedicate this paper in memoriam to our beloved colleague Marie W. Woston.
Footnotes
Published ahead of print on 25 October 2010.
REFERENCES
- 1.Duran, A., J. F. Linares, A. S. Galvez, K. Wikenheiser, J. M. Flores, M. T. Diaz-Meco, and J. Moscat. 2008. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell 13:343-354. [DOI] [PubMed] [Google Scholar]
- 2.Duran, A., M. Serrano, M. Leitges, J. M. Flores, S. Picard, J. P. Brown, J. Moscat, and M. T. Diaz-Meco. 2004. The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis. Dev. Cell 6:303-309. [DOI] [PubMed] [Google Scholar]
- 3.Holland, A. J., and D. W. Cleveland. 2009. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat. Rev. Mol. Cell Biol. 10:478-487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jin, Z., Y. Li, R. Pitti, D. Lawrence, V. C. Pham, J. R. Lill, and A. Ashkenazi. 2009. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell 137:721-735. [DOI] [PubMed] [Google Scholar]
- 5.Joshi, J., P. J. Fernandez-Marcos, A. Galvez, R. Amanchy, J. F. Linares, A. Duran, P. Pathrose, M. Leitges, M. Canamero, M. Collado, C. Salas, M. Serrano, J. Moscat, and M. T. Diaz-Meco. 2008. Par-4 inhibits Akt and suppresses Ras-induced lung tumorigenesis. EMBO J. 27:2181-2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kastan, M. B., and J. Bartek. 2004. Cell cycle checkpoints and cancer. Nature 432:316-323. [DOI] [PubMed] [Google Scholar]
- 7.Komatsu, M., H. Kurokawa, S. Waguri, K. Taguchi, A. Kobayashi, Y. Ichimura, Y. S. Sou, I. Ueno, A. Sakamoto, K. I. Tong, M. Kim, Y. Nishito, S. Iemura, T. Natsume, T. Ueno, E. Kominami, H. Motohashi, K. Tanaka, and M. Yamamoto. 2010. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 12:213-223. [DOI] [PubMed] [Google Scholar]
- 8.Komatsu, M., S. Waguri, M. Koike, Y. S. Sou, T. Ueno, T. Hara, N. Mizushima, J. Iwata, J. Ezaki, S. Murata, J. Hamazaki, Y. Nishito, S. Iemura, T. Natsume, T. Yanagawa, J. Uwayama, E. Warabi, H. Yoshida, T. Ishii, A. Kobayashi, M. Yamamoto, Z. Yue, Y. Uchiyama, E. Kominami, and K. Tanaka. 2007. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131:1149-1163. [DOI] [PubMed] [Google Scholar]
- 9.Kops, G. J., B. A. Weaver, and D. W. Cleveland. 2005. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer 5:773-785. [DOI] [PubMed] [Google Scholar]
- 10.Lee, S. J., P. T. Pfluger, J. Y. Kim, R. Nogueiras, A. Duran, G. Pages, J. Pouyssegur, M. H. Tschoep, M. T. Diaz-Meco, and J. Moscat. 2010. A functional role for the p62-ERK1 axis in the control of energy homeostasis and adipogenesis. EMBO Rep. 11:226-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindqvist, A., V. Rodriguez-Bravo, and R. H. Medema. 2009. The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J. Cell Biol. 185:193-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luo, J., M. J. Emanuele, D. Li, C. J. Creighton, M. R. Schlabach, T. F. Westbrook, K. K. Wong, and S. J. Elledge. 2009. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 137:835-848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malumbres, M., and M. Barbacid. 2009. Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 9:153-166. [DOI] [PubMed] [Google Scholar]
- 14.Malumbres, M., and M. Barbacid. 2005. Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 30:630-641. [DOI] [PubMed] [Google Scholar]
- 15.Moscat, J., and M. T. Diaz-Meco. 2009. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 137:1001-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moscat, J., M. T. Diaz-Meco, A. Albert, and S. Campuzano. 2006. Cell signaling and function organized by PB1 domain interactions. Mol. Cell 23:631-640. [DOI] [PubMed] [Google Scholar]
- 17.Musacchio, A., and E. D. Salmon. 2007. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 8:379-393. [DOI] [PubMed] [Google Scholar]
- 18.Nigg, E. A. 2001. Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell Biol. 2:21-32. [DOI] [PubMed] [Google Scholar]
- 19.Peters, J. M. 2006. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat. Rev. Mol. Cell Biol. 7:644-656. [DOI] [PubMed] [Google Scholar]
- 20.Rodriguez, A., A. Duran, M. Selloum, M. F. Champy, F. J. Diez-Guerra, J. M. Flores, M. Serrano, J. Auwerx, M. T. Diaz-Meco, and J. Moscat. 2006. Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab. 3:211-222. [DOI] [PubMed] [Google Scholar]
- 21.Sanchez, P., G. De Carcer, I. V. Sandoval, J. Moscat, and M. T. Diaz-Meco. 1998. Localization of atypical protein kinase C isoforms into lysosome-targeted endosomes through interaction with p62. Mol. Cell. Biol. 18:3069-3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santamaria, D., C. Barriere, A. Cerqueira, S. Hunt, C. Tardy, K. Newton, J. F. Caceres, P. Dubus, M. Malumbres, and M. Barbacid. 2007. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 448:811-815. [DOI] [PubMed] [Google Scholar]
- 23.Sanz, L., M. T. Diaz-Meco, H. Nakano, and J. Moscat. 2000. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J. 19:1576-1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vecchione, A., G. Baldassarre, H. Ishii, M. S. Nicoloso, B. Belletti, F. Petrocca, N. Zanesi, L. Y. Fong, S. Battista, D. Guarnieri, R. Baffa, H. Alder, J. L. Farber, P. J. Donovan, and C. M. Croce. 2007. Fez1/Lzts1 absence impairs Cdk1/Cdc25C interaction during mitosis and predisposes mice to cancer development. Cancer Cell 11:275-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weaver, B. A., and D. W. Cleveland. 2005. Decoding the links between mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell death. Cancer Cell 8:7-12. [DOI] [PubMed] [Google Scholar]