

Abstract
MyD88-deficient mice were previously shown to have increased susceptibility to Bacillus anthracis infection relative to wild-type animals. To determine the mechanism by which MyD88 protects against B. anthracis infection, knockout mice were challenged with nonencapsulated, toxigenic B. anthracis or with anthrax toxins. MyD88-deficient mice had increased susceptibility to B. anthracis and anthrax lethal toxin but not to edema toxin. Lethal toxin alone induced marked multifocal intestinal ulcers in the knockout animals, compromising the intestinal epithelial barrier. The resulting enteric bacterial leakage in the knockout animals led to peritonitis and septicemia. Focal ulcers and erosion were also found in MyD88-heterozygous control mice but with far lower incidence and severity. B. anthracis infection also induced a similar enteric bacterial septicemia in MyD88-deficient mice but not in heterozygous controls. We show that lethal toxin and B. anthracis challenge induce bacteremia as a result of intestinal damage in MyD88-deficient mice. These results suggest that loss of the intestinal epithelial barrier and enteric bacterial septicemia may contribute to sensitizing MyD88-deficient mice to B. anthracis and that MyD88 plays a protective role against lethal toxin-induced impairment of intestinal barrier.
Bacillus anthracis is the causative agent of anthrax and kills the host animal through the actions of two toxins. Edema toxin (ET) and lethal toxin (LT) are crucial virulence factors composed of protective antigen (PA) and either edema factor (EF) or lethal factor (LF). PA binds to anthrax toxin receptors and translocates LF and EF into cells (42). The two anthrax toxin receptors, tumor endothelium marker 8 (TEM8) and capillary morphogenesis protein 2 (CMG2) (6, 23, 34), are widely expressed in tissues (5, 14), making many organs potential targets for the anthrax toxins. LF is a metalloprotease that cleaves mitogen-activated protein kinase (MAPK) kinases (10), thereby inactivating MAPK signaling pathways and altering the immune function of many immune cells, including macrophages, dendritic cells, neutrophils, and epithelial cells (2, 11, 21, 29, 40). EF is a potent calmodulin-dependent adenylate cyclase which increases cyclic AMP (cAMP) levels in cells (22). The comparison of B. anthracis mutants lacking individual toxin components suggests that LF is essential for inducing host death (31).
MyD88 is the crucial adaptor protein for interleukin-1 (IL-1) receptor and all Toll-like receptors (TLRs) except TLR3 (17, 18). TLRs are crucial to innate immunity and pathogen-associated molecular pattern recognition. These receptors act as sensors for a wide range of stimuli including lipopolysaccharides, lipoproteins, and unmethylated cytidine-guanosine dinucleotide (CpG) motifs in DNA (3). To date, 11 human TLRs and 13 mouse TLRs have been identified. Because TLR and IL-1 receptor functions are impaired in MyD88-deficient mice, these mice have a higher susceptibility to many pathogens (examples include Staphylococcus aureus and Klebsiella pneumonia [9, 36]). In an inhalational anthrax mouse model, MyD88-deficient mice were found to be more susceptible than wild-type animals (16). IL-1 receptor-deficient mice were also shown to have increased susceptibility to B. anthracis (41). Thus, MyD88 signaling pathways play a protective role against B. anthracis infection although the exact mechanism for the protective function is not known.
MyD88 has also been shown to play a role in intestinal homeostasis (33). Enteric bacteria continuously activate intestinal epithelial cells through MyD88-dependent signaling pathways, resulting in the production of protective factors such as hsp70, IL-6, KC-1, and tumor necrosis factor alpha (TNF-α) (33). These MyD88-dependent protective responses are crucial for intestinal epithelial cell repair and homeostasis. Previous reports demonstrated that a high proportion of sublethally irradiated mice infected with B. anthracis develop polymicrobial septicemia with enteric bacteria (7, 8). This suggests that B. anthracis may disrupt intestinal epithelial barrier function, allowing bacterial translocation from the intestinal tract. In this study we show that MyD88 plays a protective role against intestinal barrier disruption induced by both B. anthracis infection and LT. We find that LT injection causes severe intestinal damage in MyD88-deficient mice, which results in enteric bacterial septicemia. B. anthracis infection also induces a polymicrobial bacteremia with enteric bacteria, likely as a result of the actions of LT. Our data show that MyD88-dependent signaling pathways are required to protect the intestinal epithelial barrier from anthrax lethal toxin during B. anthracis infection.
MATERIALS AND METHODS
Spores and toxins.
Spores were prepared from the nonencapsulated toxigenic Ames 35 (A35) strain which is missing the pXO2 plasmid, and the Ames 33 (A33) strain which is missing both the pXO1 and pXO2 virulence plasmids (32), as previously described (15). Briefly, bacteria were grown at 37°C for 3 days on Schaeffer's sporulation agar, and spores were purified from plates with multiple washes using ice-cold distilled water. Residual vegetative cells were killed by heat (65°C for 30 min). Spore purity was confirmed by phase-contrast microscopy, and no vegetative bacteria were found in any field. Spore quantification was performed using a Petroff-Hausser counting chamber. LF and PA were purified from B. anthracis, and EF was purified from Escherichia coli as previously described (35, 39). The LF used in these studies was a recombinant version having a nonnative HMAGG N-terminal sequence (13).
Animal experiments.
MyD88-deficient mice backcrossed to C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME) for over eight generations were kindly provided by Tod J. Merkel (Food and Drug Administration, Bethesda, MD) (1). Male and female mice were used at 7 to 16 weeks of age; sex ratios and age distributions did not differ significantly between experimental groups. Heterozygous littermates were used as controls in all experiments except infections with A35 B. anthracis spores because wild-type littermates and heterozygous mice were equally sensitive to spore infection. In toxin challenge studies, mice were injected with LT intraperitoneally (i.p.). Toxin dosage refers to the amount of LF or EF given with an equal amount of PA (i.e., 100 μg of LT is 100 μg of LF plus 100 μg of PA). In spore challenge experiments, mice were injected subcutaneously (s.c.) in the scruff of the neck. In some studies, spleens were removed aseptically and homogenized in phosphate-buffered saline (PBS), and blood was collected by cardiac puncture. To distinguish B. anthracis from other bacteria, spleen or blood specimens were serially diluted in PBS and plated separately on both brain heart infusion (BHI) plates and modified MacConkey (without crystal violet) agar plates (BD, Franklin Lakes, NJ). MacConkey agar medium without crystal violet allows growth of enteric bacteria but inhibits B. anthracis. B. anthracis was distinguished from other bacteria by colony morphology on BHI plates. For histological analyses, organs (stomach, duodenum, small intestine, cecum, and large intestine) were harvested at 24, 48, and 72 h after toxin injection. Tissues were fixed in 10% neutral buffered formalin (Sigma, St. Louis, MO), embedded in paraffin, and stained with hematoxylin and eosin (H&E) by Histoserv Inc. (Gaithersburg, MD). A board-certified veterinary pathologist analyzed all samples. All animal studies were performed in accordance with protocols approved by the National Institute of Allergy and Infectious Diseases Animal Care and Use Committee.
Cytotoxicity assay.
Cytotoxicity assays were performed as previously reported (28). Briefly, bone marrow-derived macrophages (BMDMs) from mice were cultured in differentiation medium consisting of 30% L929 cell-conditioned medium in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum, 10 mM HEPES, and 50 μg/ml gentamicin (all obtained from Invitrogen, Carlsbad, CA). Cells were seeded in 96-well plates 7 days after differentiation and were treated with LT for 8 or 24 h. Cell viability was assessed by MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl tetrazolium bromide] (USB Corporation, Cleveland, OH) assay. Percent viabilities were calculated relative to untreated controls.
Statistical analyses.
Comparisons of bacteria counts were carried out using a Mann-Whitney test. A log rank test was used for comparison of survival times. All analyses were performed using GraphPad Prism, version 5 (GraphPad System Inc., San Diego, CA).
RESULTS
MyD88-deficient mice have increased susceptibility to subcutaneous anthrax infection.
Previous studies have shown that MyD88-deficient mice are more susceptible to anthrax infection through the inhalational route (16). To determine whether this increased susceptibility extends to s.c. infections, we infected MyD88-deficient mice and their heterozygous or wild-type littermates with toxigenic A35 spores (Fig. 1, left panel). The MyD88-deficient mice were significantly more susceptible than their heterozygous littermates. Heterozygous mice were as susceptible as wild-type littermates. Mice infected with the nontoxigenic Ames 33 strain were completely resistant to infection (Fig. 1, right panel), verifying that factors encoded by the pXO1 virulence plasmid (which also contains the anthrax toxin genes) are required for lethality in spore-infected MyD88-deficient mice.
FIG. 1.

MyD88-deficient mice are more susceptible to subcutaneous infection. (Left) Wild-type mice (n = 11), MyD88-heterozygous mice (n = 26), and MyD88-deficient mice (n = 27) were infected with A35 spores (1 × 107, s.c.). The P value for comparison of MyD88-deficient mice and -heterozygous mice is <0.0001 by a log rank test. (Right) MyD88-heterozygous mice (n = 4) and MyD88-deficient mice (n = 4) were infected with A33 toxin-deficient spores (1 × 108, s.c.). Mice were monitored for 7 days after infection.
MyD88 deficiency sensitizes mice to LT.
To test whether MyD88-deficient mice are altered in sensitivity to anthrax toxins, we challenged the knockout mice and heterozygous controls with ET and LT. MyD88-deficient mice succumbed to LT significantly earlier than MyD88-heterozygous mice (Fig. 2A). In contrast, ET challenge showed no significant difference in time-to-death (TTD) values between knockout and control animals (Fig. 2B).
FIG. 2.

MyD88-deficient mice have increased susceptibility to LT but not ET. (A) MyD88-heterozygous and MyD88-deficient mice were injected intraperitoneally with 100 μg of LT (n = 34/group). (B) MyD88-heterozygous (n = 20) mice and MyD88-deficient mice (n = 19) were injected intraperitoneally with 75 μg of ET. Mice were monitored for 7 days. The P value for the comparison of curves in panel A is <0.0001. Curves in panels B are not significantly different (P > 0.05).
MyD88 deficiency does not affect macrophage sensitivity to LT.
Our previous studies indicated that sensitivity of mouse macrophages to LT, despite having no effect on overall animal susceptibility to toxin, can result in a more rapid time to death (27). To determine whether MyD88-deficient mice, which have the C57BL/6J background, have altered macrophage sensitivity, we tested BMDMs from these mice for LT sensitivity. MyD88-deficient macrophages showed similar sensitivity to LT as control cells from heterozygous littermates (Fig. 3), and both were resistant over 8 h of toxin treatment, succumbing by 24 h. This result suggested that the increased susceptibility of the knockout strain was unlikely to be attributable to macrophage sensitivity differences.
FIG. 3.
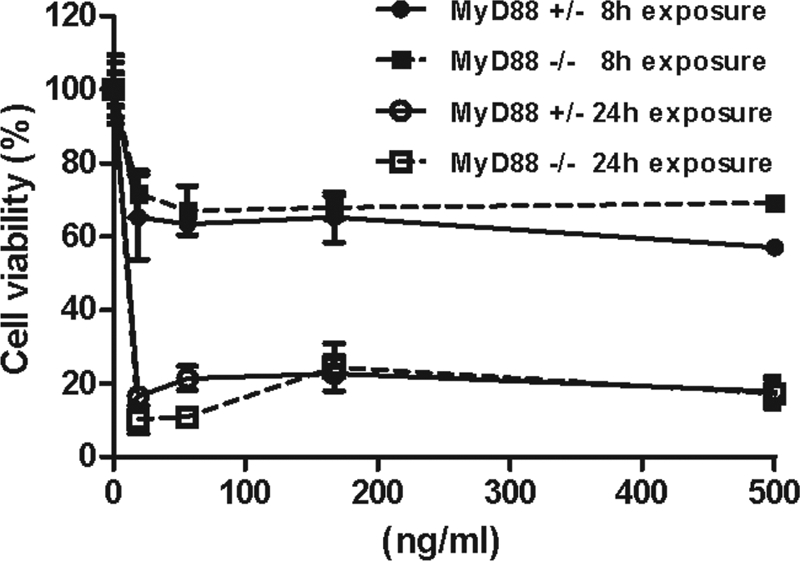
MyD88 deficiency does not sensitize macrophages to LT. BMDMs were treated with various doses of LT for 8 or 24 h, and viability was assessed by MTT staining. Percent viability is expressed relative to untreated controls. Data represent the mean ± the standard error of the mean of three independent experiments. There is no significant difference between MyD88-deficient and heterozygous macrophages.
LT induces bacteremia in MyD88-deficient mice.
Previous studies have shown that intestinal homeostasis is disrupted in MyD88-deficient mice (33). Similarly, sublethally irradiated mice infected with B. anthracis die earlier than sham-irradiated mice, due to a polymicrobial bacteremia caused by enteric bacteria translocation to the blood (7, 8). Our earlier studies of LT-treated mice also showed a low incidence of bloody intestinal content in a limited number of mice (25). We hypothesized that bacteremia caused by enteric bacterial leakage from the gastrointestinal (GI) tract following toxin challenge could occur in MyD88-deficient mice and result in increased susceptibility to LT. Spleens and blood from mice injected with LT were collected and plated to enumerate bacteria. We found that MyD88-deficient mice had significantly higher numbers of bacteria than control mice in both the spleen (Fig. 4A) and blood (Fig. 4B) following LT treatment. MyD88-deficient control mice that were not treated with LT did not have any bacteria in the blood or spleen (data not shown). To exclude the possibility that the localized high concentration of LT in the abdominal cavity during i.p. injection caused bacterial translocation, we also injected mice intravenously with LT and found the same high numbers of bacteria in the spleen and blood from all MyD88-deficient mice, but no bacteria was found in organs from any heterozygous controls (data not shown). Therefore, we conclude that LT-induced bacterial translocation occurs independent of the toxin administration route. Isolated bacteria were identified as K. pneumonia and various Enterococcus species. These species are common enteric flora, confirming that they originated from the GI tract. Thus, LT causes significant enteric bacterial translocation in MyD88-deficient mice.
FIG. 4.

LT treatment of MyD88-deficient mice induces bacteremia. Bacterial counts from homogenized spleen (A) and blood (B) samples collected 48 h after LT treatment (n = 10/group) are shown. Data represent the mean ± the standard error of the mean. Significant differences (P < 0.05) between MyD88-heterozygous mice and MyD88-deficient mice are indicated with an asterisk (*).
Histopathological analyses of intestines of LT-treated mice.
Bacterial translocation from the intestinal lumen usually occurs due to immune deficiency, intestine barrier disruption, or overgrowth of bacteria in the intestine (4). MyD88-deficient mice do not have a higher commensal bacterial load than wild-type mice (38), and despite being immunodeficient, these mice did not evidence spontaneous bacteremia due to intestinal leakage (data not shown). Therefore, the disruption of the intestinal barrier observed in LT-treated knockout mice is a result of LT-mediated events. To investigate the effect of LT on intestines, organ samples of LT-treated mice were collected at 24, 48, or 72 h after toxin administration. Untreated MyD88-deficient and -heterozygous controls did not display any intestinal pathology. MyD88-deficient mice showed marked damage to the intestinal epithelium by 48 h after LT treatment, and the severity of pathology increased at 72 h. Extensive multifocal ulcers and submucosal edema were noted in both the small and large intestines (Fig. 5A to C). Bacteria traversed the epithelium in some locations and could be identified within intestinal epithelial cells (Fig. 5D). Some ulcers extended through the muscularis to the serosa and the peritoneal surface (Fig. 5E). Acute inflammatory cell infiltrates comprised primarily of neutrophils were present in the ulcerated intestinal segments and within the serosa extending to the peritoneal surface (Fig. 5B, C, and E). Epithelial necrosis and atrophy of small intestinal villi were prominent in the affected segments (Fig. 5E). Vascular bacterial thrombi were present within lymphatics and venules in association with focally extensive ulcers (Fig. 5F). Only one of five MyD88-deficient mice did not have multifocal ulcers. In contrast, heterozygous controls had no intestinal pathology at 24 and 48 h. By 72 h, two of five MyD88-heterozygous mice had one or two focal ulcers in colon or cecum, and one animal had epithelial erosion in the small intestine (Fig. 5G and H). Compared to the multifocal ulceration in the MyD88-deficient mice, findings in heterozygous animals indicated that LT challenge induced a less extensive ulceration of intestines and colon and that MyD88 was required for protection against extensive intestinal epithelial damage induced by the toxin. The ulceration in the MyD88-deficient mice, accompanied by enteric bacterial peritonitis and resultant cytokine-mediated shock, likely contributed to increased animal sensitivity and a more rapid death.
FIG. 5.

Histopathological changes in intestines of LT-treated mice. MyD88-deficient mice (A to F) and MyD88-heterozygous mice (G and H) were treated intraperitoneally with 100 μg of LT. (A) Multifocal ulcers in MyD88-deficient mice shown in cross-section of colon (arrowheads) and prominent submucosal edema (arrow) (magnification, ×25). (B) Magnified view of one of five ulcers shown in panel A. Bacteria (*) are visible within the lamina propria of the ulcerated segment of colon (magnification, ×200). (C) Two foci in small intestine of MyD88-deficient mouse with bacteria (*) evident within lamina propria (magnification, ×100). (D) Bacteria present within cytoplasm of small intestinal epithelial cells from the eroded region in panel C (magnification, ×630). (E) Ulcer in ileum of MyD88-deficient mouse extended through the serosa to the peritoneal surface, with bacteria evident in the peritoneal fat (magnification, ×100). (F) Bacteria (*) in a lymphatic vessel adjacent to a colonic ulcer in an MyD88-deficient mouse (magnification, ×630). (G) Focal erosion and ulceration in small intestine of a heterozygous control mouse (magnification, ×200). (H) Cecal ulcer with bacteria (*) and associated inflammatory infiltrate within the lamina propria of a heterozygous mouse (magnification, ×100).
MyD88-deficient mice infected with A35 spores show polymicrobial bacteremia.
To determine whether B. anthracis spore infection also caused enteric bacteremia, we infected mice with 1 × 107 spores/mouse (s.c.) and collected spleen and blood samples 48 h after infection. To enumerate enteric bacteria, MacConkey medium without crystal violet was used. MacConkey agar components inhibit B. anthracis growth while the omission of crystal violet allows growth of enterococci, staphylococci, and most other enteric species. B. anthracis bacteria were enumerated on BHI plates using their unique colony morphology. As shown in Fig. 6, heterozygous controls had no bacterial growth in spleen or blood. However, both B. anthracis (detected on BHI agar) (Fig. 6A and C) and enteric bacteria (detected on MacConkey agar) (Fig. 6B and D) were found in MyD88-deficient mice. MyD88-deficient mice had significantly higher CFU counts of B. anthracis in spleens than heterozygous mice (P < 0.05). Although the differences in enteric bacteria numbers between heterozygous mice and MyD88-deficient mice were not significant in either the blood (P = 0.14) or the spleen (P = 0.07), 50% of MyD88-deficient mice had enteric bacteria, whereas enteric bacteria were never found in infected heterozygous mice. Thus, B. anthracis infection, similar to LT treatment, also induced bacterial translocation from the GI tract in MyD88-deficient mice.
FIG. 6.

B. anthracis infection induces enteric bacteremia. Mice (n = 6/group) were infected with 1 × 107 spores/mouse. Homogenized spleen and blood samples collected at 48 h after infection were plated on BHI plates or MacConkey agar without crystal violet. Data represent the mean ± the standard error of the mean. Significant differences (P < 0.05) between heterozygous mice and MyD88-deficient mice are indicated by an asterisk.
DISCUSSION
Mixed bacterial infection has been described in monkeys challenged with B. anthracis as well as in irradiated mice (7, 8, 37). In the monkey model, unidentified cocci were observed in heart tissue (37). In irradiated mice, enteric bacteria were isolated from spleens following infection with B. anthracis. Toxin-deficient strains did not result in this enteric bacterial leakage (7). These reports suggested that B. anthracis infection could cause bacterial translocation from the intestinal tract due to actions of the anthrax toxins.
In this study, we showed that bacterial translocation of enteric bacteria occurs extensively in MyD88-deficient mice injected with LT or infected with B. anthracis. LT also induced intestinal damage in some heterozygous control animals but with far less severity than in the knockout mice, indicating that MyD88 plays a protective role against LT effects in the GI tract. The striking severity of multifocal ulcerations induced by LT in small and large intestines in MyD88-deficient mice indicated that signaling pathways involving this adaptor play a protective role against the LT-induced damage to the intestinal epithelium. In MyD88-deficient mice, ulceration and disruption of the intestinal barrier allowed access of bacteria from the GI tract lumen to the blood and the peritoneum, leading to septicemia and bacterial peritonitis. Ulcer incidence was minimal in heterozygous control animals, was not multifocal, and did not cause peritonitis. These findings show that the intestine can be targeted by anthrax LT. Our results may explain the red intestinal contents that were seen in our previous studies of LT-treated BALB/cJ and C57BL/6J mice (25) although no evidence of intestinal hemorrhage or necrosis was detected by light microscopy at 48 h in those studies. In this study, the single focal ulcers in heterozygous animals were seen only after 72 h of toxin treatment.
The higher burden of B. anthracis in MyD88-deficient mice found in our studies also indicates a role for receptors which signal through this adaptor protein in the immune response against B. anthracis. Although TLR2 recognizes heat-killed B. anthracis, TLR2-deficient mice do not exhibit increased susceptibility to B. anthracis (16). MyD88 is also an adaptor protein for signaling through the IL-1 receptor. IL-1 receptor knockout mice have been shown to be more susceptible to B. anthracis (41), and thus the reduced signaling through this receptor may account for the reduced resistance to infection seen in MyD88-deficient mice.
Immune suppression can lead to loss of intestinal barrier function (19, 20). Spontaneous bacterial translocation does not occur in MyD88-deficient mice, and thus the action of LT is required for the bacterial leakage reported here. LT is known to inhibit the function of T cells, B cells, macrophages, and neutrophils (26). The effects of LT on these cells could be sufficient to result in loss of the intestinal barrier function (12, 30). Furthermore, the MAPK pathways targeted by LT in intestinal epithelial cells are also known to play a role in repair of damaged epithelium (24). This loss of protective pathways combined with MyD88 deficiency, which is known to reduce the expression of factors which are protective for the intestinal epithelium, such as heat shock proteins, TNF-α, IL-6, and KC-1 (33), may be sufficient to explain the loss of barrier function observed in our studies. Further study is needed to determine the molecular mechanisms by which MyD88 protects the intestinal barrier from LT.
In conclusion, we show that the intestine can be targeted by LT and that MyD88 plays a role in protecting the intestinal barrier against the actions of this toxin in mice. This finding represents a new mechanism by which the MyD88 signaling pathways play a protective role against B. anthracis infection.
Acknowledgments
We thank Rasem Fattah for protein purification. We also thank Zachary Newman for help with manuscript and figure preparation.
This research was supported by the Intramural Research Program of the NIH, National Institute of Allergy and Infectious Diseases.
Editor: A. J. Bäumler
Footnotes
Published ahead of print on 25 October 2010.
REFERENCES
- 1.Adachi, O., T. Kawai, K. Takeda, M. Matsumoto, H. Tsutsui, M. Sakagami, K. Nakanishi, and S. Akira. 1998. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 9:143-150. [DOI] [PubMed] [Google Scholar]
- 2.Agrawal, A., J. Lingappa, S. H. Leppla, S. Agrawal, A. Jabbar, C. Quinn, and B. Pulendran. 2003. Impairment of dendritic cells and adaptive immunity by anthrax lethal toxin. Nature 424:329-334. [DOI] [PubMed] [Google Scholar]
- 3.Akira, S., S. Uematsu, and O. Takeuchi. 2006. Pathogen recognition and innate immunity. Cell 124:783-801. [DOI] [PubMed] [Google Scholar]
- 4.Berg, R. D. 1999. Bacterial translocation from the gastrointestinal tract. Adv. Exp. Med. Biol. 473:11-30. [DOI] [PubMed] [Google Scholar]
- 5.Bonuccelli, G., F. Sotgia, P. G. Frank, T. M. Williams, C. J. de Almeida, H. B. Tanowitz, P. E. Scherer, K. A. Hotchkiss, B. I. Terman, B. Rollman, A. Alileche, J. Brojatsch, and M. P. Lisanti. 2005. Anthrax toxin receptor (ATR/TEM8) is highly expressed in epithelial cells lining the toxin's three sites of entry (lung, skin, and intestine). Am. J. Physiol. Cell Physiol. 288:C1402-C1410. [DOI] [PubMed] [Google Scholar]
- 6.Bradley, K. A., J. Mogridge, M. Mourez, R. J. Collier, and J. A. Young. 2001. Identification of the cellular receptor for anthrax toxin. Nature 414:225-229. [DOI] [PubMed] [Google Scholar]
- 7.Brook, I., T. B. Elliott, R. A. Harding, S. S. Bouhaouala, S. J. Peacock, G. D. Ledney, and G. B. Knudson. 2001. Susceptibility of irradiated mice to Bacillus anthracis Sterne by the intratracheal route of infection. J. Med. Microbiol. 50:702-711. [DOI] [PubMed] [Google Scholar]
- 8.Brook, I., A. Germana, D. E. Giraldo, T. D. Camp-Hyde, D. L. Bolduc, M. A. Foriska, T. B. Elliott, J. H. Thakar, M. O. Shoemaker, W. E. Jackson, and G. D. Ledney. 2005. Clindamycin and quinolone therapy for Bacillus anthracis Sterne infection in 60Co-gamma-photon-irradiated and sham-irradiated mice. J. Antimicrob. Chemother. 56:1074-1080. [DOI] [PubMed] [Google Scholar]
- 9.Cai, S., S. Batra, L. Shen, N. Wakamatsu, and S. Jeyaseelan. 2009. Both TRIF- and MyD88-dependent signaling contribute to host defense against pulmonary Klebsiella infection. J. Immunol. 183:6629-6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duesbery, N. S., C. P. Webb, S. H. Leppla, V. M. Gordon, K. R. Klimpel, T. D. Copeland, N. G. Ahn, M. K. Oskarsson, K. Fukasawa, K. D. Paull, and G. F. Vande Woude. 1998. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 280:734-737. [DOI] [PubMed] [Google Scholar]
- 11.Friedlander, A. M., R. Bhatnagar, S. H. Leppla, L. Johnson, and Y. Singh. 1993. Characterization of macrophage sensitivity and resistance to anthrax lethal toxin. Infect. Immun. 61:245-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gautreaux, M. D., E. A. Deitch, and R. D. Berg. 1994. T lymphocytes in host defense against bacterial translocation from the gastrointestinal tract. Infect. Immun. 62:2874-2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gupta, P. K., M. Moayeri, D. Crown, R. J. Fattah, and S. H. Leppla. 2008. Role of N-terminal amino acids in the potency of anthrax lethal factor. PLoS One 3:e3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanks, S., S. Adams, J. Douglas, L. Arbour, D. J. Atherton, S. Balci, H. Bode, M. E. Campbell, M. Feingold, G. Keser, W. Kleijer, G. Mancini, J. A. McGrath, F. Muntoni, A. Nanda, M. D. Teare, M. Warman, F. M. Pope, A. Superti-Furga, P. A. Futreal, and N. Rahman. 2003. Mutations in the gene encoding capillary morphogenesis protein 2 cause juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am. J. Hum. Genet. 73:791-800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu, H., Q. Sa, T. M. Koehler, A. I. Aronson, and D. Zhou. 2006. Inactivation of Bacillus anthracis spores in murine primary macrophages. Cell. Microbiol. 8:1634-1642. [DOI] [PubMed] [Google Scholar]
- 16.Hughes, M. A., C. S. Green, L. Lowchyj, G. M. Lee, V. K. Grippe, M. F. Smith, Jr., L. Y. Huang, E. T. Harvill, and T. J. Merkel. 2005. MyD88-dependent signaling contributes to protection following Bacillus anthracis spore challenge of mice: implications for Toll-Like receptor signaling. Infect. Immun. 73:7535-7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawai, T., O. Adachi, T. Ogawa, K. Takeda, and S. Akira. 1999. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11:115-122. [DOI] [PubMed] [Google Scholar]
- 18.Kawai, T., and S. Akira. 2007. TLR signaling. Semin. Immunol. 19:24-32. [DOI] [PubMed] [Google Scholar]
- 19.Koh, A. Y., J. R. Kohler, K. T. Coggshall, R. N. Van, and G. B. Pier. 2008. Mucosal damage and neutropenia are required for Candida albicans dissemination. PLoS Pathog. 4:e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koh, A. Y., G. P. Priebe, and G. B. Pier. 2005. Virulence of Pseudomonas aeruginosa in a murine model of gastrointestinal colonization and dissemination in neutropenia. Infect. Immun. 73:2262-2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lehmann, M., D. Noack, M. Wood, M. Perego, and U. G. Knaus. 2009. Lung epithelial injury by B. anthracis lethal toxin is caused by MKK-dependent loss of cytoskeletal integrity. PLoS One 4:e4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leppla, S. H. 1982. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc. Natl. Acad. Sci. U. S. A. 79:3162-3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu, S., D. Crown, S. Miller-Randolph, M. Moayeri, H. Wang, H. Hu, T. Morley, and S. H. Leppla. 2009. Capillary morphogenesis protein-2 is the major receptor mediating lethality of anthrax toxin in vivo. Proc. Natl. Acad. Sci. U. S. A. 106:12424-12429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malago, J. J., J. F. Koninkx, and J. E. van Dijk. 2002. The heat shock response and cytoprotection of the intestinal epithelium. Cell Stress Chaperones 7:191-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moayeri, M., D. Haines, H. A. Young, and S. H. Leppla. 2003. Bacillus anthracis lethal toxin induces TNF-α-independent hypoxia-mediated toxicity in mice. J. Clin. Invest. 112:670-682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moayeri, M., and S. H. Leppla. 2009. Cellular and systemic effects of anthrax lethal toxin and edema toxin. Mol. Aspects Med. 30:439-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moayeri, M., N. W. Martinez, J. Wiggins, H. A. Young, and S. H. Leppla. 2004. Mouse susceptibility to anthrax lethal toxin is influenced by genetic factors in addition to those controlling macrophage sensitivity. Infect. Immun. 72:4439-4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Newman, Z. L., S. H. Leppla, and M. Moayeri. 2009. CA-074Me protection against anthrax lethal toxin. Infect. Immun. 77:4327-4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O'Brien, J., A. Friedlander, T. Dreier, J. Ezzell, and S. H. Leppla. 1985. Effects of anthrax toxin components on human neutrophils. Infect. Immun. 47:306-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Owens, W. E., and R. D. Berg. 1980. Bacterial translocation from the gastrointestinal tract of athymic (nu/nu) mice. Infect. Immun. 27:461-467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pezard, C., P. Berche, and M. Mock. 1991. Contribution of individual toxin components to virulence of Bacillus anthracis. Infect. Immun. 59:3472-3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pomerantsev, A. P., R. Sitaraman, C. R. Galloway, V. Kivovich, and S. H. Leppla. 2006. Genome engineering in Bacillus anthracis using Cre recombinase. Infect. Immun. 74:682-693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rakoff-Nahoum, S., J. Paglino, F. Eslami-Varzaneh, S. Edberg, and R. Medzhitov. 2004. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118:229-241. [DOI] [PubMed] [Google Scholar]
- 34.Scobie, H. M., G. J. Rainey, K. A. Bradley, and J. A. Young. 2003. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. U. S. A. 100:5170-5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soelaiman, S., B. Q. Wei, P. Bergson, Y. S. Lee, Y. Shen, M. Mrksich, B. K. Shoichet, and W. J. Tang. 2003. Structure-based inhibitor discovery against adenylyl cyclase toxins from pathogenic bacteria that cause anthrax and whooping cough. J. Biol. Chem. 278:25990-25997. [DOI] [PubMed] [Google Scholar]
- 36.Takeuchi, O., K. Hoshino, and S. Akira. 2000. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J. Immunol. 165:5392-5396. [DOI] [PubMed] [Google Scholar]
- 37.Twenhafel, N. A., E. Leffel, and M. L. Pitt. 2007. Pathology of inhalational anthrax infection in the African green monkey. Vet. Pathol. 44:716-721. [DOI] [PubMed] [Google Scholar]
- 38.Vaishnava, S., C. L. Behrendt, A. S. Ismail, L. Eckmann, and L. V. Hooper. 2008. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc. Natl. Acad. Sci. U. S. A. 105:20858-20863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Varughese, M., A. Chi, A. V. Teixeira, P. J. Nicholls, J. M. Keith, and S. H. Leppla. 1998. Internalization of a Bacillus anthracis protective antigen-c-Myc fusion protein mediated by cell surface anti-c-Myc antibodies. Mol. Med. 4:87-95. [PMC free article] [PubMed] [Google Scholar]
- 40.Warfel, J. M., A. D. Steele, and F. D'Agnillo. 2005. Anthrax lethal toxin induces endothelial barrier dysfunction. Am. J. Pathol. 166:1871-1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu, C. C., M. Sabet, T. Hayashi, R. Tawatao, J. Fierer, D. A. Carson, D. G. Guiney, and M. Corr. 2008. In vivo efficacy of a phosphodiester TLR-9 aptamer and its beneficial effect in a pulmonary anthrax infection model. Cell. Immunol. 251:78-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Young, J. A., and R. J. Collier. 2007. Anthrax toxin: receptor-binding, internalization, pore formation, and translocation. Annu. Rev. Biochem. 76:243-265. [DOI] [PubMed] [Google Scholar]