

Abstract
In this review, we provide an overview of the methods employed in four recent studies that described novel methods for computational prediction of secreted effectors from type III and IV secretion systems in Gram-negative bacteria. We present the results of these studies in terms of performance at accurately predicting secreted effectors and similarities found between secretion signals that may reflect biologically relevant features for recognition. We discuss the Web-based tools for secreted effector prediction described in these studies and announce the availability of our tool, the SIEVE server (http://www.sysbep.org/sieve). Finally, we assess the accuracies of the three type III effector prediction methods on a small set of proteins not known prior to the development of these tools that we recently discovered and validated using both experimental and computational approaches. Our comparison shows that all methods use similar approaches and, in general, arrive at similar conclusions. We discuss the possibility of an order-dependent motif in the secretion signal, which was a point of disagreement in the studies. Our results show that there may be classes of effectors in which the signal has a loosely defined motif and others in which secretion is dependent only on compositional biases. Computational prediction of secreted effectors from protein sequences represents an important step toward better understanding the interaction between pathogens and hosts.
Bacterial pathogens secrete numerous proteins, called effectors, which promote virulence by interacting with the host network and environment. In many Gram-negative bacteria, specialized type III and type IV secretion systems that allow injection of these effector proteins directly into the host cell cytoplasm have evolved (11, 20). Effectors are generally known to be targeted for secretion by a sequence in their N termini, for type III, or C termini, for type IV, that has no easily identifiable pattern. Recently, we and others have described machine learning data integration methods to accurately identify proteins secreted by both secretion systems (4, 9, 41, 65).
Type III secretion systems are believed to have originated from flagella (49) and are essential for virulence in most studied pathogens. This machinery is a needlelike structure that spans the inner and outer membranes (20, 34, 85) and allows injection of protein effectors directly into the cytoplasm of the eukaryotic host cell (19). Since the pore is quite narrow (∼25 to 30 Å), it is believed that secreted proteins must pass through in a largely unfolded state, in some cases mediated by chaperone proteins (1). Each system has a repertoire of effector proteins that enact the virulence program of the bacteria by directly interacting with a number of host cell signaling pathways (19).
Type III secreted effectors are thought to have two possibly overlapping N-terminal domains which mediate secretion. Residues 1 to 25 form an N-terminal secretion signal that is highly variable in sequence (44) and, in some cases, is highly tolerant of mutations (63). In Yersinia pestis, the observations that effectors with frameshift mutations were still secreted and that synonymous mutations could abolish secretion led to the conclusion that the 5′ mRNA sequences of some effector genes are responsible for targeting (3, 61, 70). However, it has been demonstrated that the amino acid sequence is responsible for targeting in other cases (23, 30, 63). In many effectors, a chaperone binding domain spans from residues 15 to 30 to around residue 100 and allows a cognate chaperone protein to bind the effector (38). Removal of either domain prevents the effector from being targeted to the secretion system and subsequent secretion or translocation through the type III secretion system (40, 44, 71).
Most of the core components of the type III secretion system are conserved between species (55), and several lines of evidence indicate that the targeting mechanisms employed by the system may also be conserved. The first is that, in some cases, type III secretion systems can export proteins bearing secretion signals from other bacteria (18, 24, 62). The second is that a recently discovered class of type III secretion inhibitor can block secretion in multiple species—Y. pestis, Chlamydia trachomatis, and Salmonella enterica serovar Typhimurium (5, 29, 45, 48, 53)—though the mechanism of inhibition is unclear (48). It has been shown from available structures of effectors bound to their cognate chaperones that the structure of this interaction is conserved across species (39). The computational approaches reviewed here also strongly suggest that features of the secretion signal are conserved within and between organisms.
In all but one of the type III effector structures determined by X-ray diffraction (XRD)-based methods, the N-terminal 15 to 35 residues were reported to be unstructured (17, 37, 39) or were not included in the protein used for crystallization (21). Unstructured N-terminal regions in crystal structures are the result of diffuse electron density that cannot be associated with the sequence. Studies showing that some N-terminal secretion signals are highly tolerant of mutations (63) but are not mRNA encoded indicate that some signals may be dependent upon a truly unstructured state for recognition (16). Our recent structural characterization of N-terminal type III secretion signal peptides found that these were unstructured in solution, supporting this idea (8).
Though there are related families of type III effectors (43, 74), more than 75% of effectors have no detectable sequence similarity to other known effectors. Approaches based on sequence similarity and on genomic location in pathogenicity islands have been used to identify many currently known effectors (15, 43). Most recently, homology with known effectors has been used to greatly expand the estimated number of secreted effectors in pathogenic Escherichia coli O157:H7 (76). This finding indicates that there may be a large number of unknown effectors in pathogenic bacteria with type III or IV secretion systems, even in well-studied organisms like S. Typhimurium, Legionella pneumophila, and Y. pestis. General features of the protein sequence have also been used to the same end, focused on the N-terminal secretion signal. In the plant pathogen Pseudomonas syringae, amino acid biases and patterns, including amphipathicity, exposed polar residues, and a net negative charge in the N-terminal secretion signal, were used to identify novel effectors (25, 26, 58), though these same criteria do not seem to work as well in other species (58). Detection of common promoter elements has also been used to identify novel effectors in P. syringae (77), but this approach is limited to known and detectable motifs. Finally, detecting chaperone genes using a number of genomic criteria and then identifying effectors based on their genomic proximity to the chaperone has been used successfully to identify novel effectors (56). However, we and others have found these approaches to be ineffective in identifying secreted effectors in general (4, 65).
Experimental data associated with the recognition of secretion signals on effector proteins transported by the type IV secretion system remain difficult to elucidate. Unlike the conserved type III secretion platform, type IV secretion has been separated into two distinct classes, T4a and T4b (13). The T4a secretion pathway is exemplified by the VirB translocation system in the plant pathogen Agrobacterium tumefaciens. This translocation pathway is related to conjugal transfer machines that support delivery of nucleic acid-protein hybrids from donor to recipient cell (14). In A. tumefaciens, a mobilizing relaxase, VirD2, functions to nick DNA at an origin of transfer and covalently bind to a single strand (57). The hybrid is recognized by a membrane-bound coupling receptor that supports translocation (12). Secretion information in the VirD2-DNA hybrid was determined to be confined to the C-terminal end of the VirD2 protein component (79). Importantly, studies revealed that additional proteins were recognized and transported into host cells via the virB type IV secretion system, independent of DNA association (69, 78). The effector protein VirF was subjected to positional mutagenesis and revealed a consensus motif in the C terminus implicating positively charged amino acid residues important for substrate recognition (79). Indeed, the majority of bacterial pathogens requiring type IV secretion systems for virulence harbor genes that are conserved with those of the virB system in A. tumefaciens. Mutagenesis of conserved positively charged residues in the C terminus of the effector protein CagA in the human pathogen Helicobacter pylori did not impair T4 secretion, suggesting that additional recognition elements are present (28).
The T4b secretion pathway represents a more sophisticated secretion platform, based on the requirement for at least double the protein components required for effector protein translocation into host cells (13, 66). Currently, genomic information suggests that two intracellular pathogens, Legionella pneumophila and Coxiella burnetii, use T4b secretion for transport of effectors into host cells. L. pneumophila uses a type IV secretion system termed “Dot/Icm” to support pathogenesis in human phagocytes. To date, over 140 effector proteins have been demonstrated to be transported into host cells by this transporter (9). Analysis of confirmed effectors revealed little conservation in primary amino acid sequence. However, it is now clear that the majority of Dot/Icm substrates harbor secretion information in their C-terminal ends (42, 46). Additionally, chaperone complexes recognize patterns on effectors that are distinct from the C-terminal translocation signal, adding a level of complexity to Dot/Icm substrate recognition (6, 10, 51).
The first Dot/Icm-dependent effector protein described was RalF (47). Analysis of the C terminus revealed a 20-amino acid (aa) segment sufficient for translocation of a Cya reporter into host cells (46). Nagai et al. (46) hypothesized that Dot/Icm effector proteins harbored a conserved hydrophobic residue at the −3 position relative to the carboxyl end. In RalF, substitution of either serine or threonine for valine abrogated T4 secretion (46). Additionally, a positive charge in the RalF C terminus was dispensable for substrate recognition, unlike with T4a translocation. Substitution for this hydrophobic residue in other known effectors has not translated to abrogation of secretion similar to that of RalF (E. D. Cambronne laboratory, unpublished observations). The crystal structure of the RalF protein revealed that the C-terminal 20 amino acids were disordered, suggesting that a lack of secondary structure might contribute to recognition of substrates by the Dot/Icm transporter (2).
Kubori et al. developed a search algorithm based on positional features found in known effector proteins that allowed them to successfully identify 19 novel effector proteins in L. pneumophila (33). This feature parameter was applied to the study performed by Burstein et al. (9). It is likely that a combination of physical features of positional amino acids and the lack of secondary structure in the C-terminal ends of Dot/Icm effector proteins provide the necessary secretion signatures.
Recently, several groups, including ours, have independently described computational methods to identify type III and type IV secreted effectors on the basis of protein sequence information (4, 9, 41, 65). These approaches all involve data integration methods that use machine learning techniques to train on sets of known secreted effectors. The methods all accurately identify secreted substrates of type III (4, 41, 65) or type IV (9) systems in a range of bacteria. Machine learning describes a set of computational approaches in which a method is “trained” on a set of known examples to learn the relationships between the input features from each example that allow the best discrimination between the positive and negative example sets (Fig. 1). Data of different kinds, for example, sequence information and structural properties of a protein, can easily be integrated using such methods to provide better and more explanatory models. Three main elements go into the construction of a machine learning approach to classification (52, 75, 84). The first is the algorithm itself; for example, support vector machines (SVMs), artificial neural networks (ANNs), and Bayesian classifiers provide different ways to learn classifiers from input data. The second element is the features used as input to the algorithm. The features are numeric representations of various attributes of the proteins or genes used as examples. These include amino acid composition, G+C content, taxonomic distribution, and others that were used in the studies reviewed here. The last element is the training data, made up of positive and negative examples for the desired class of proteins, in this case secreted effectors. Important to the training data is the method used to examine the performance of the approach with examples that were not used in the training set, for example, cross-validation techniques or validation on other well-characterized data sets. Machine learning classification approaches have been used in a large number of biological applications, including detection of remote homology between proteins (67, 68, 82), detection of binding sites and posttranslational modification sites (7, 59, 60) from genomic features, and prediction of secondary structure and disordered regions in proteins (27, 80, 81).
FIG. 1.
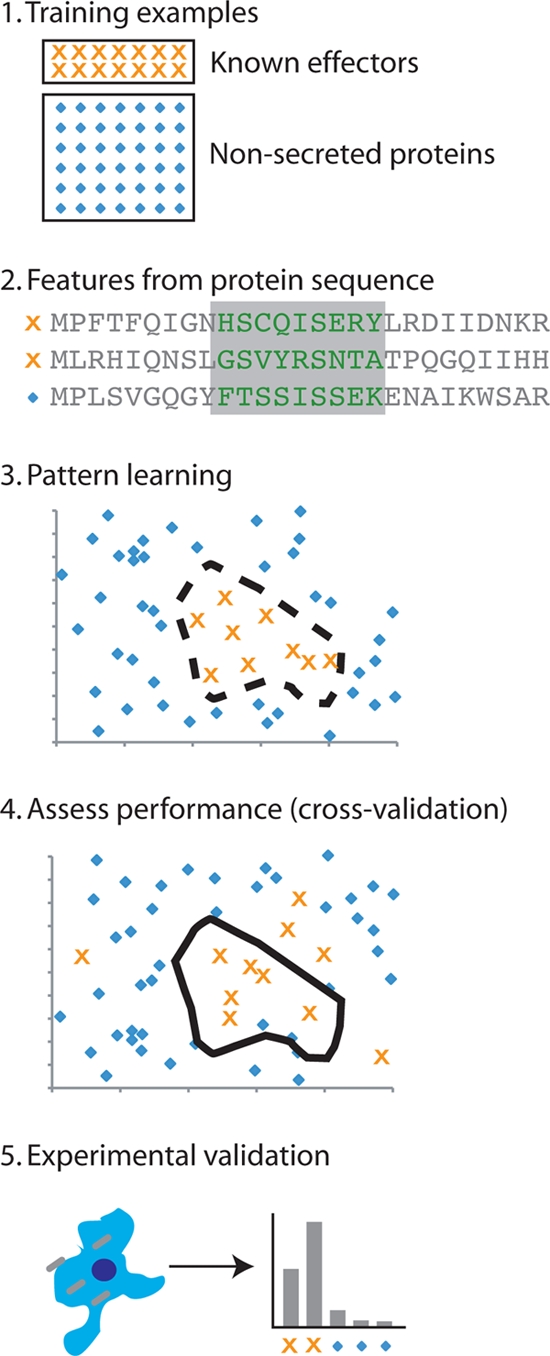
Machine learning approaches to secreted effector identification. Each of the methods described in this review follows a similar process. Step 1, sets of known secreted effectors (positive examples) and proteins that are not secreted or assumed to be not secreted (negative examples) are chosen. Step 2, features of the protein sequence (e.g., amino acid conservation, sequence, phylogenetic distribution, etc.) are derived from all proteins and transformed into a numeric representation. Step 3, a machine learning algorithm (e.g., a support vector machine) learns to discriminate the positive examples from negative examples in a high-dimensional space formed by the chosen protein features. Step 4, the performance of the approach is assessed by applying the model learned in step 3 to independent examples that were not included in the training. Step 5, experimental validation must then be applied to finally determine whether or not a protein is secreted.
DESCRIPTION OF APPROACHES TO IDENTIFICATION OF SECRETED EFFECTORS FROM TYPE III AND TYPE IV SYSTEMS
Three papers were published recently by Arnold et al. (4), Löwer and Schneider (41), and Samudrala et al. (65) that describe the application of machine learning approaches to the identification of type III secreted substrates; in addition, a fourth study by Burstein et al. (9) reported the use of such approaches to the prediction of type IV secreted effectors. We first briefly describe the similarities and differences between approaches. We compare the methods and protein features used, the training and testing data sets used, and finally, the prediction accuracy reported and important conclusions drawn by each study.
COMPARISON OF METHODS AND PROTEIN FEATURES USED TO CLASSIFY SECRETED EFFECTORS
The selection of features from effector proteins for use as input in the classification method is very important to the outcomes of the study, and the choice of classification algorithm can have a significant impact as well. In our study (65), we used an SVM to classify proteins on the basis of five protein features: amino acid composition in the N-terminal 30 residues, two measures of protein evolutionary conservation, the G+C content of the cognate gene, the phylogenetic distribution of similar proteins in more than 50 other organisms, and the amino acid sequence for the N-terminal 30 residues. Burstein et al. (9) used a similarly broad range of input features to classify type IV effectors: similarity to known effectors and proteins in eukaryotes, taxonomic distribution, identities of neighboring genes, G+C content, and presence of two regulatory elements. Also, a search for a set of previously described C-terminal secretion signal patterns was included in a second-round filter to improve results (33). In that study (33), Kubori et al. used these as input to SVM, an artificial neural network (ANN), two Bayesian methods, and a voting method that classified proteins on the basis of the results from all four methods. In contrast, the other two type III classification studies used a set focused on the protein sequence: Arnold et al. (4) used amino acid composition and di- and tripeptide pattern frequency in a number of different machine learning algorithms, and Löwer and Schneider (41) used a sliding window of primary protein sequence as input to both SVM and ANN algorithms.
COMPARISON OF EXAMPLES USED FOR TRAINING AND VALIDATION OF SECRETED EFFECTOR CLASSIFICATION
The process of training a classification algorithm involves learning patterns of positive examples (e.g., known secreted effectors) that distinguish them from the negative examples (e.g., nonsecreted proteins). The nature of this approach is to define the best classifier based on the examples used for training. Unfortunately, this can often lead to a situation in which the classifier works very well on the training data but is unable to generalize, that is, accurately classify other examples that were not used in the training set. In machine learning, this problem is typically addressed using cross-validation approaches in which a set of the training data is left out of the process and used later to assess the “true” performance of the method. To get an accurate estimate of performance, this process is repeated a number of times using different sets of training examples in each iteration. This process can still provide a biased estimate of performance of a method since the examples are still drawn from the same set, but it is often the best approach to address a difficult issue when there are few positive examples known.
Our study used two relatively well-characterized bacteria for training and validation: the animal pathogen S. Typhimurium and the plant pathogen Pseudomonas syringae (65). Both organisms have experimentally characterized sets of type III secreted effectors that are approximately the same size, about 35 to 40 effectors each. For negative training examples, we used all proteins in each organism that are not positive examples. This establishes a conservative training set, since some of the examples in our negative set may be undiscovered secreted effectors (at least five were; see below), but it is a “real-world” set that should provide a good, though conservative, estimate of performance. Additionally, any method of filtering negative examples to provide a more confidently nonsecreted set might introduce significant biases in the data set that could render classification trivial. Imagine that we filtered the negative examples to include only metabolic enzymes. These proteins are very different from secreted effectors, in general, and many trivial classification methods would perform very well on this set but not provide accurate identification of new effectors. We used two validation approaches. For the results presented in the paper, we simply trained on the examples in one organism and assessed performance on the set of examples from the other organism, after first eliminating significantly similar sequences from the set. As an alternate approach, we used a leave-one-out cross-validation method in which each positive example was excluded from training in turn and performance was evaluated on it and a representative set of negative examples.
In the two remaining type III secreted effector classification studies, a large set of experimentally characterized secreted effectors was assembled from published studies (76) and databases. Because the organisms we chose for our study (65) are two of the best characterized in terms of type III secretion, both of these example sets had significant overlap with our positive example sets. In studies by both Löwer and Schneider (41) and Arnold et al. (4), negative example sets were constructed and used in approximately equal numbers to positive examples: 1:1, positive to negative, by Löwer and Schneider and 1:2, positive to negative, by Arnold et al. By comparison, the ratio in our study (65) was approximately 1:120, positive to negative, which is close to the naturally occurring ratio in the two organisms examined. One potential problem with using a training set with equal numbers of positive and negative examples in cross-validation is that it can artificially inflate performance estimates because the number of false-positive classifications is proportional to the number of examples classified. So applying these methods to all proteins in an organism may result in a large number of false-positive identifications. Interestingly, Löwer and Schneider (41) included proteins known to be secreted by other secretion systems as negative examples for training in order to provide a method capable of discriminating type III secreted proteins from other secreted proteins. Both studies used cross-validation to obtain performance estimates. Also in both studies, the C-terminal portions of positive examples were used as negative examples in independent control experiments, which provides a good control for potential biases, for example, in amino acid composition, that are not specific to secretion.
Finally, Burstein et al. (9) focused on just L. pneumophila and, thus, used the known effectors for positive examples and L. pneumophila proteins that are not shared with Escherichia coli as a set of negative examples in a 1:5 positive-to-negative ratio. A potential issue with this choice of negative examples is that the features used included information based on evolutionary relationships, which could cause the classification algorithm to choose a trivial, but effective, classification. However, this does not appear to have affected their final results, based on their experimental validation results. Because their positive examples (known secreted effectors) are not likely to be shared with E. coli, this approach does not introduce significant artificial bias between their positive- and negative-example proteins that would be exploited by the classification algorithms. Their approach was iterative, and so they included effectors identified in the previous round for each new training iteration, which seems to provide better results, although they did not analyze this in their paper.
COMPARISON OF RESULTS AND CONCLUSIONS FROM SECRETED EFFECTOR CLASSIFICATION STUDIES
Given the different approaches and methods of validation, it is difficult to compare the performances of the studies directly. Here we compare the results reported in each of the studies, and below we examine the performances of the three type III secreted effector prediction methods on an independent data set. Three of the studies used a measure of classification performance called the receiver operator characteristic (ROC) area under the curve (AUC). The ROC plots sensitivity against specificity, with an AUC of 1.0 representing perfect classification (all the positive and negative examples classified correctly) and an AUC of 0.5 being equivalent to random chance (64). Our study (65) reported an ROC AUC of 0.95 when the SVM was trained on S. Typhimurium and tested on P. syringae or 0.96 when trained on P. syringae and tested on S. Typhimurium. These are very good results but are limited in breadth because we chose to focus on only two bacteria and did not examine the method's performance with other bacteria. Burstein et al. reported an ROC AUC of 0.98 for classification of type IV effectors, which is extremely good (9). Additionally, this is the only study of the four that performed experimental validation. In total, 40 of the predictions made were validated, significantly expanding the known secreted effector repertoire in L. pneumophila. The experimental results reported indicate that the estimate of performance made using a cross-validation approach is close to the true performance of the method. One caveat is that only 50 proteins with the highest scores were selected for validation, and it remains unknown how many effectors might be missed by this approach (i.e., the false-negative percentage). This could be addressed by validating a sample of predictions with a range of scores. Arnold et al. reported ROC AUCs in a range of 0.85 to 0.87 for their entire test set (4). These results are significant, and though they are not perfect, they represent a test of a much larger set of proteins from many different organisms. The study by Löwer and Schneider was also the only one to use a measure of classification other than the ROC (41). This study used the Matthew's correlation coefficient (MCC) and reported a maximum correlation of 0.63. Since MCC values range from perfect correlation at 1.0 to random classification equivalence at 0, the results can be very roughly compared to the ROC results by scaling the MCC to 0.82, which agrees well with the results reported by Arnold et al (4). As mentioned above, one potential caveat with the results from both Arnold et al. (4) and Löwer and Schneider (41) is that they may produce more false positives in a real-world application than indicated by the performance estimates, due to the limited number of negative examples used for training and cross-validation. However, our independent test of these methods in this review (see below) suggests that this difference in training approaches does not have a significant impact on false-positive prediction rates.
Each of these studies also performed additional analysis using the developed models to investigate the nature of the secretion signal (Table 1). Our study (65) reported that the information contained in the signal extends only to around residue 30 and that the first 15 residues are most important for classification in both S. Typhimurium and P. syringae effectors. Furthermore, we identified a significant sequence pattern in this region that was similar between effectors in the two bacteria. The sequence pattern was enriched in serine and isoleucine residues and depleted in leucine residues at several positions. Additionally, the pattern agrees with some other patterns and biases previously described for secreted effectors from various organisms. In contrast, Arnold et al. concluded that there is no position-specific sequence motif that identifies secreted effector signal sequences (4). They postulate that the primary determinant of secretion is based only on nonpositional amino acid composition in this region. This conclusion was partially based on the failure of traditional sequence alignment methods to detect a motif in this region, something we observed as well. In addition, they analyzed point mutations and frame shifts in effector and noneffector sequences and determined that most secretion signal sequences are robustly tolerant of point mutations but are sensitive to frameshift mutations. We discuss this difference in “Conclusions and Future Prospects” below. Arnold et al. (4) performed several other analyses of the secretion signal and found that these regions are significantly enriched in serine, threonine, and proline residues and that leucine is depleted in this region, again agreeing well with our findings. Finally, they predicted secondary structure for these regions and found that they were generally enriched in random coil, indicating a possible lack of structure. Arnold et al. (4) determined that the secretion signal information was contained in the first 30 residues in animal pathogens, though extended to 50 residues in plant pathogens. They also concluded that the first 15 residues provided the maximum discriminatory power. Löwer and Schneider also investigated the length of the secretion signal by its information content in classification and found that the optimal length for classification was 30 residues (41). Both of these findings closely agree with ours and indicate that the signal for type III secretion is located in the first 30 or so residues, with a particularly important region in the first 15 residues.
TABLE 1.
Important features of type III and type IV secretion signalsa
| Important feature | Identification by: |
||||
|---|---|---|---|---|---|
| Arnold et al. (4) | Löwer and Schneider (41) | Samudrala et al. (65) | Burstein et al. (9) | Previous observations | |
| Positional sequence pattern | No | Yesa | Yes | Yes | Yes |
| Disordered structural motif | Yes | Yes | Yes | ||
| Charged residue biases | Yes | Yesa | Yes | Yes | |
| Serine/threonine bias | Yes | Yesa | Yes | Yes | |
| Proline bias | Yes | Yesa | No | No | |
| Hydrophobic bias | Yes | Yesa | Yes | Yes | |
| Terminal 20 to 30 residues important | Yes | Yes | Yes | Yes | Yes |
| G+C gene content bias | Yes | Yes | Yes | ||
| Taxonomic distribution | Yes | Yes | Yes | ||
The study results implied the indicated importance but did not specifically call it out.
Though the C-terminal type IV secretion signal detected by Burstein et al. is evolutionarily distinct from the N-terminal type III secretion signal, similar approaches were employed to characterize its composition (9). Those authors found that the C-terminal 20 residues of type IV effectors had amino acid compositional biases that were positional, i.e., the preference for particular types of amino acids varies over the length of the secretion signal. Specifically, they found that negatively charged residues (aspartic acid and glutamic acid) were depleted at C-terminal positions 4 to 6 but favored at positions 8 to 18, hydrophobic residues were depleted at positions 8 to 12, and aliphatic hydroxyl group-bearing residues (serine and threonine) were favored at positions 3 to 11. These biases mirror the biases noted in the type III effector prediction studies to a certain extent: an enrichment of serine and threonine residues in the N termini of type III secretion signals and depletion of hydrophobic residues, particularly leucines (4, 65). Negatively charged residues were found to be depleted in type III secretion signals (4), which is seen in the very C-terminal region of type IV secretion signals. These similarities (Table 1), and the fact that both type III and type IV secretion signals seem to be disordered, suggest that recognition of secretion signals from both systems may have very similar requirements and may involve similar mechanisms.
IS THE TYPE III SECRETION SIGNAL DEPENDENT ON ORDER OF AMINO ACID RESIDUES?
As discussed above, a major point of discrepancy between the findings reported by our study (65) and those of Arnold et al. (4) is that of whether the secretion signal is primarily based on general amino acid composition or if there is a positional importance, similar to that found in more traditional sequence motifs. For the purposes of this discussion, we refer to the former possibility as sequence order independence (SOI) and the latter as sequence order dependence (SOD). We were interested in examining this difference using our model and reasoned that SOI would result in randomly scrambled secretion signals having scores equal to or better than the wild-type signals. Accordingly, we generated 500 random sequence permutations of each signal in our set of secreted effectors from S. Typhimurium and then analyzed these sequences by our method, SIEVE (SVM-based identification and evaluation of virulence effectors), trained on P. syringae effectors (65), by the method described by Arnold et al., EffectiveT3, trained on plant pathogens (4), and by the method of Löwer and Schneider (using a reduced number of sequences to accommodate their server) (41). We then considered effectors in which 30% or less of the scores from the scrambled sequences exceeded the score for wild-type sequences to be SOD. Surprisingly, we found that SIEVE and EffectiveT3 predicted identical ratios of SOI to SOD effectors, 35%, though the two methods did not classify all effectors identically. The method of Löwer and Schneider (41) produced a ratio of 62% SOI to SOD effectors, which was much higher. All three methods agreed in categorizing 10 of the effectors, including InvJ and SipB, as sequence order dependent. These effectors are both SOD members and have small, well-defined sequences that are necessary for secretion (31, 63), indicating that they are indeed sequence order dependent. Additionally, we have generated two randomly scrambled variants of the SseJ secretion signal that were predicted with high (SseJ-H) and low (SseJ-L) SIEVE scores (8). We examined these for secretion experimentally and found that both were secreted but that the SseJ-L was secreted at very low levels, consistent with our predictions. The difference in ability to direct secretion of three sequences (SseJ, SseJ-H, and SseJ-L) demonstrates that the secretion signal for SseJ is SOD.
These observations suggest that there may be at least two different types of type III secretion signals in S. Typhimurium, one that is dependent on a degenerate SOD motif, as suggested in our study (65), and another that is more dependent on the general amino acid composition in the N-terminal 30 residues. It is certainly possible that these differences are due to details of the computational models rather than the underlying biology, and further computational and experimental investigation is under way to help clarify this difference.
APPLICATIONS AND EXPERIMENTAL VALIDATION OF SECRETED EFFECTOR PREDICTIONS
Pathogens have evolved complicated and multifaceted means to manipulate host networks in order to evade host defenses and establish productive infections. In pathogens with type III or IV secretion systems, this is largely accomplished through the actions of secreted effectors. Thus, knowledge about the repertoire of secreted effectors for a pathogen can help define the interaction between the pathogen and host. A detailed understanding of these interactions is necessary to begin to develop systems biology models of pathogen-host interactions. Additionally, these interactions may represent novel targets for development of therapeutic intervention. Unlike traditional antibiotics that aim to eliminate the bacteria and thus exert a strong selective pressure on the development of resistance, therapies based on targeting the pathogen-host interaction could potentially avoid this problem.
Two primary applications of methods to identify secreted effectors from protein sequence are represented in the papers reviewed here: identification of novel effectors in well-studied pathogens and identification of secreted effectors in relatively uncharacterized pathogens. Both our study (65) and that of Burstein et al. (9) used these methods to identify novel effectors in existing, well-studied pathogens. Surprisingly, both studies identified a number of high-confidence predictions for novel secreted effectors. Burstein et al. (9) reported experimental validation of 40 novel effectors in L. pneumophila, increasing the number of known effectors by nearly 50%. Their analysis also validated their performance estimates, showing a positive predictive value [(true-positive predictions)/(true-positive predictions + false-positive predictions)] of 80%. We have recently completed a proteomics-based experimental discovery with validation in macrophage cells of several novel type III secreted effectors from S. Typhimurium (see the report by Niemann et al. [50]). Below, we show that SIEVE predicts four out of five of these novel effectors with an overall accuracy of 88%. Arnold et al. (4) and Löwer and Schneider (41) both applied their methods to predict secreted effectors across a large number of genomes, many of them uncharacterized. Both studies reported that significant portions of the encoded proteomes investigated were predicted to be secreted. Arnold et al. (4) examined 739 bacterial and archeal encoded proteomes and found that between 2% and 7% of all proteins were predicted to be secreted in organisms with identified type III secretion systems. Löwer and Schneider (41) examined 705 proteobacterial encoded proteomes and found that 11.5% of these were predicted to be secreted, though this percentage was not dependent on the presence of a type III secretion system. In addition, our study (65) predicted secreted effectors in the genetically intractable C. trachomatis. Though we presented evidence that these were reasonable predictions, it remains unclear if our predictions will be useful. We have since predicted secreted effectors in the animal pathogen Mannheimia hemolytica (35) and show evidence for the first time that this pathogen may have a type III secretion system (36).
Finally, these methods can be used to further define the nature of the secretion signal from type III and type IV effectors and to characterize the evolutionary relationships between these systems. For example, Arnold et al. (4) present an interesting analysis of the evolutionary history of the secretion signal that suggests that that evolution may be an example of convergent sequence adaptation. This model fits well with the known properties of the secretion signal, which do not share strong sequence similarity within or across organisms.
AVAILABILITY OF METHODS FOR SECRETED EFFECTOR PREDICTION
As discussed above, an important application of these methods is the prediction of secreted effector repertoires in uncharacterized bacterial genomes. The availability and ease of use are both important considerations for biologists interested in using these predictive methods (Table 2). In this section, we briefly discuss the use of the tool described by Arnold et al., EffectiveT3 (4), and the method described by Löwer and Schneider (41) and for the first time describe a Web-based tool to make predictions based on the algorithm we described previously (65), SIEVE (http://www.sysbep.org/sieve/). We then present a side-by-side comparison of predictions from each of the three tools on a set of experimental validations we have recently completed for S. Typhimurium.
TABLE 2.
Availability and features of secreted effector prediction methods
| Method | URL | Maximum no. of sequences | Training data set | Reported AUC | Validation (%) |
|---|---|---|---|---|---|
| EffectiveT3 | http://www.effectors.org | 10,000 | Pan-genome | 0.87 | 88 |
| Löwer and Schneider (41) | http://gecco.org.chemie.uni-frankfurt.de | 50 | Pan-genome | 0.82 | 77 |
| SIEVE | http://www.sysbep.org/sieve | None | S. Typhimurium/P. syringae | 0.95 | 88 |
| Burstein et al. (9) | None | NA | L. pneumophila | 0.98 | NAa |
NA, not available.
TYPE III SECRETED EFFECTOR PREDICTION USING EffectiveT3
The EffectiveT3 tool is available as a Web tool or a Java-based stand-alone program at http://www.chlamydiaedb.org, though the Web server can be reached directly at http://www.effectors.org (4). The Web tool requires the user to upload a FASTA-format protein sequence file with the query sequences. Several classification models are available for selection, and the user can set a threshold for the confidence of the predictions to be returned. Results are returned as a table of sequence identifiers, a probability based on the algorithm, and a classification label (positive, negative, or short or invalid sequence). The maximum number of sequences that can be uploaded and processed at one time by the Web tool is 10,000, which is sufficient for prediction of entire bacterial genomes.
TYPE III SECRETED EFFECTOR PREDICTION USING THE METHOD DESCRIBED BY LÖWER AND SCHNEIDER
The paper by Löwer and Schneider (41) also describes a Web interface to their prediction algorithm at http://www.modlab.de, though the type III secreted effector prediction submission form can be reached directly at http://gecco.org.chemie.uni-frankfurt.de/T3SS_prediction/T3SS_prediction.html. As with EffectiveT3, the Web server requires the input of a FASTA-format protein sequence file and allows the selection of their SVM or ANN model and modification of several parameters (length of sequence window considered and a threshold for results from the ANN). The maximum number of sequences that can be processed in one submission is 50, which significantly limits the utility of the Web tool for predicting secreted effectors from complete proteomes.
TYPE III SECRETED EFFECTOR PREDICTION USING SIEVE
Since the publication of our paper (65), we have released a public Web tool for prediction of type III secreted effectors, which we describe here. The SIEVE Web server is freely available with a brief user registration at http://www.sysbep.org/sieve. Our algorithm is slower than that used by EffectiveT3 since we use features that require a large sequence similarity search using BLAST. To accommodate the efficient processing of large sets of protein sequences, for example, whole genome sequences, we use ScalaBLAST, a parallel implementation of BLAST (54), running on a 68-processor cluster. ScalaBLAST is used to generate the phylogenetic profile necessary for input into the SVM model. Using this framework for a submitted FASTA file with 7,230 protein sequences (from the genome of Burkholderia pseudomallei), SIEVE finished in 28 min, versus 6 h 51 min running on a single processor.
The SIEVE algorithm requires only the input of a protein sequence or sequences (in FASTA format). It generates several sets of features based on the input sequence, as described previously (65), to classify the protein(s) using the available SVM-based model. The amino acid composition in the N-terminal 30 residues and the N-terminal sequence itself are converted to vectors. A parallel BLAST search is run using the previously described ScalaBLAST (54), and a simple phylogenetic profile is constructed and converted to a vector. Two features described previously, G+C content and evolutionary conservation, are not included in these models because their removal has little impact on the overall performance of the algorithm. Removing these two features entirely did not change the performance of the approach from our previously reported results (65).
The feature set is used for classification using an SVM model described previously (65). This model is trained on type III secreted substrates from S. Typhimurium and P. syringae (STMpPSY model). The user is then alerted by e-mail that the job has finished and is presented with a list of input sequence identifiers with SVM discriminant scores and associated probabilities for each protein sequence. Based on our previous analysis (65), as well as more recent validation of predictions, the probabilities returned are conservative but provide a way to prioritize candidate effectors.
We have used the SIEVE Web server to provide predictions of type III secreted effectors for several Gram-negative enteropathogens being studied by the Center for Systems Biology of EnteroPathogens (http://www.sysbep.org). We provide the results of this analysis on the center's website at http://www.sysbep.org/data/SIEVE. These predictions provide a valuable resource for the community and insight into the biology of these important enteropathogens.
COMPARISON OF PREDICTIONS BETWEEN ALGORITHMS
We were interested in seeing how each of these three algorithms would perform on the same set of proteins, so we tested a set of proteins from S. Typhimurium that we recently discovered and experimentally validated (50). This set has a range of SIEVE confidence values and includes 5 secreted proteins and 12 nonsecreted proteins, as validated by a CyaA fusion assay (22). The sequences corresponding to these examples were submitted to each of the three servers discussed, and the results were ranked according to the scores returned by the server. We then examined the accuracy (percentage of true-positive predictions plus true-negative predictions out of all predictions made) of each method by considering the top 4 predictions positive for each method. The results are quite consistent: SIEVE (65) made 15/17 correct predictions (accuracy, 88%), and EffectiveT3 (4) and the method described by Löwer and Schneider (41) both made 11/17 correct predictions (accuracy, 65%). Given the small size of the test set, these results can be considered equivalent, and further testing on larger, independent data sets will provide better estimates of performance. Though their performances were similar, the methods differed in the specific proteins that were correctly or incorrectly classified (Table 3). SIEVE predicted 4/5 secreted proteins correctly, whereas EffectiveT3 and the Löwer and Schneider (41) method both predicted 2/5 secreted proteins correctly. Interestingly, all methods predict that SrfN should not be secreted by the type III secretion system, but our CyaA fusion assay results show that it is secreted. In this case, we have evidence that SrfN is secreted via an alternative mechanism (Yoon et al., submitted for publication). Also, all three methods predict that PipA is secreted, but our results, using cya′ fusions to test secretion to animal cells, show that it is not secreted. This raises the possibility that PipA may be alternatively regulated and that it may be secreted under the right conditions or to different cell types than we tested. These results show that all methods are comparable in terms of accuracy and further suggest that combining the three approaches, possibly in a voting scheme (like the one used by Burstein et al. [9]) could provide a better predictive method.
TABLE 3.
Validation resultsa
| Open reading frame | Gene | Validation result (rank of prediction) by: |
|||
|---|---|---|---|---|---|
| SIEVE | EffectiveT3 | Löwer and Schneider 41 | CyaA fusion assay | ||
| STM2585A | 2 | 11 | 13 | 1 | |
| STM2585 | 3 | 11 | 14 | 2 | |
| STM2139 | 5 | 1 | 2 | 3 | |
| PSLT037 | spvD | 1 | 1 | 1 | 4 |
| STM0082 | srfN | 14 | 11 | 12 | 5 |
| STM3762 | cigR | 7 | 6 | 11 | 6 |
| STM1087 | pipA | 4 | 3 | 3 | 7 |
| STM1599 | pdgL | 6 | 4 | 10 | 8 |
| STM1809 | 8 | 11 | 17 | 9 | |
| STM1513 | 9 | 11 | 7 | 10 | |
| STM1633 | 10 | 9 | 9 | 11 | |
| STM0211 | yaeH | 11 | 7 | 5 | 12 |
| STM1121 | ymdF | 12 | 8 | 4 | 13 |
| STM3392 | yhdV | 13 | 11 | 16 | 14 |
| STM4082 | yiiQ | 15 | 10 | 6 | 15 |
| STM3595 | 16 | 11 | 15 | 16 | |
| STM1548 | 17 | 5 | 8 | 17 | |
Bold values indicate the top five predictions.
CONCLUSIONS AND FUTURE PROSPECTS
Correct identification of novel secreted effectors using protein sequence is merely a first step toward more complete characterization of the complex pathogen-host interaction. The studies reviewed here (4, 41, 65) suggest that type III secretion signals are similar across many different bacteria. These similarities include amino acid composition biases that are located in the N-terminal 30 residues of the type III secreted effectors. Our comparison of the three methods reviewed indicate that they can predict secreted effectors with comparable accuracies. The results provide only a little insight into what the potential mechanisms of secretion signal recognition might be. An existing hypothesis based on structural studies is that this region may be disordered, and this idea is supported by secondary-structure predictions made by Arnold et al. (4), our structural characterization of several secretion signal peptides (8), and our computational analysis discussed here. A better understanding of the secretion signal could lead to novel therapeutic agents that specifically target virulence effectors and to methods of delivering proteins with therapeutic (32) or other (83) uses.
Another aspect of effectors important to the understanding of pathogen-host interactions is their function both in terms of their host binding partners/targets and their mode of action. The studies reviewed do not provide insight into these aspects, but similar approaches based on machine learning may be a way to begin to make predictions. A considerable problem in this area is that bacterial pathogens have evolved many molecular mimics to interact with host pathways. These proteins share little sequence similarity with their host counterpart (e.g., GTPase interacting proteins) but are structurally and functionally similar (72, 73) and are therefore difficult to characterize by examining primary protein sequence. Because of this, they are good candidates for machine learning classification methods that can consider more general features of protein sequences that might correlate with their function and can learn similarities from known examples. However, there are relatively few effectors in any particular class that have been characterized, making this a difficult prospect at present.
Acknowledgments
This work was supported by the National Institute of Allergy and Infectious Diseases, NIH/DHHS, through interagency agreement Y1-AI-8401-01, by the Biomolecular Systems Initiative under the Laboratory Directed Research and Development Program at the Pacific Northwest National Laboratory (PNNL), a multiprogram national laboratory operated by Battelle for the U.S. Department of Energy under contract DE-AC06-76RL01830, by the Department of Energy Science Undergraduate Laboratory Internship to D.S., by NIH grant RO1 AI022933 to F.H., by Medical Research Foundation of Oregon grant MRF810 to E.D.C., by National Science Foundation grant DBI 0217241 to R.S., and by a Searle Scholars Program grant to R.S.
Editor: H. L. Andrews-Polymenis
Footnotes
Published ahead of print on 25 October 2010.
REFERENCES
- 1.Akeda, Y., and J. E. Galan. 2005. Chaperone release and unfolding of substrates in type III secretion. Nature 437:911-915. [DOI] [PubMed] [Google Scholar]
- 2.Amor, J. C., J. Swails, X. Zhu, C. R. Roy, H. Nagai, A. Ingmundson, X. Cheng, and R. A. Kahn. 2005. The structure of RalF, an ADP-ribosylation factor guanine nucleotide exchange factor from Legionella pneumophila, reveals the presence of a cap over the active site. J. Biol. Chem. 280:1392-1400. [DOI] [PubMed] [Google Scholar]
- 3.Anderson, D. M., and O. Schneewind. 1997. A mRNA signal for the type III secretion of Yop proteins by Yersinia enterocolitica. Science 278:1140-1143. [DOI] [PubMed] [Google Scholar]
- 4.Arnold, R., S. Brandmaier, F. Kleine, P. Tischler, E. Heinz, S. Behrens, A. Niinikoski, H. W. Mewes, M. Horn, and T. Rattei. 2009. Sequence-based prediction of type III secreted proteins. PLoS Pathog. 5:e1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bailey, L., A. Gylfe, C. Sundin, S. Muschiol, M. Elofsson, P. Nordstrom, B. Henriques-Normark, R. Lugert, A. Waldenstrom, H. Wolf-Watz, and S. Bergstrom. 2007. Small molecule inhibitors of type III secretion in Yersinia block the Chlamydia pneumoniae infection cycle. FEBS Lett. 581:587-595. [DOI] [PubMed] [Google Scholar]
- 6.Bardill, J. P., J. L. Miller, and J. P. Vogel. 2005. IcmS-dependent translocation of SdeA into macrophages by the Legionella pneumophila type IV secretion system. Mol. Microbiol. 56:90-103. [DOI] [PubMed] [Google Scholar]
- 7.Bhardwaj, N., and H. Lu. 2007. Residue-level prediction of DNA-binding sites and its application on DNA-binding protein predictions. FEBS Lett. 581:1058-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buchko, G. W., G. Niemann, E. S. Baker, M. E. Belov, R. D. Smith, F. Heffron, J. N. Adkins, and J. E. McDermott. 2010. A multi-pronged search for a common structural motif in the secretion signal of Salmonella enterica serovar Typhimurium type III effector proteins. Mol. Biosyst. 6:2448-2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burstein, D., T. Zusman, E. Degtyar, R. Viner, G. Segal, and T. Pupko. 2009. Genome-scale identification of Legionella pneumophila effectors using a machine learning approach. PLoS Pathog. 5:e1000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cambronne, E. D., and C. R. Roy. 2007. The Legionella pneumophila IcmSW complex interacts with multiple Dot/Icm effectors to facilitate type IV translocation. PLoS Pathog. 3:e188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cambronne, E. D., and C. R. Roy. 2006. Recognition and delivery of effector proteins into eukaryotic cells by bacterial secretion systems. Traffic 7:929-939. [DOI] [PubMed] [Google Scholar]
- 12.Cascales, E., and P. J. Christie. 2004. Definition of a bacterial type IV secretion pathway for a DNA substrate. Science 304:1170-1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cascales, E., and P. J. Christie. 2003. The versatile bacterial type IV secretion systems. Nat. Rev. Microbiol. 1:137-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christie, P. J., K. Atmakuri, V. Krishnamoorthy, S. Jakubowski, and E. Cascales. 2005. Biogenesis, architecture, and function of bacterial type IV secretion systems. Annu. Rev. Microbiol. 59:451-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deng, W., J. L. Puente, S. Gruenheid, Y. Li, B. A. Vallance, A. Vazquez, J. Barba, J. A. Ibarra, P. O'Donnell, P. Metalnikov, K. Ashman, S. Lee, D. Goode, T. Pawson, and B. B. Finlay. 2004. Dissecting virulence: systematic and functional analyses of a pathogenicity island. Proc. Natl. Acad. Sci. U. S. A. 101:3597-3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dunker, A. K., C. J. Brown, J. D. Lawson, L. M. Iakoucheva, and Z. Obradovic. 2002. Intrinsic disorder and protein function. Biochemistry 41:6573-6582. [DOI] [PubMed] [Google Scholar]
- 17.Evdokimov, A. G., D. E. Anderson, K. M. Routzahn, and D. S. Waugh. 2001. Unusual molecular architecture of the Yersinia pestis cytotoxin YopM: a leucine-rich repeat protein with the shortest repeating unit. J. Mol. Biol. 312:807-821. [DOI] [PubMed] [Google Scholar]
- 18.Frithz-Lindsten, E., A. Holmstrom, L. Jacobsson, M. Soltani, J. Olsson, R. Rosqvist, and A. Forsberg. 1998. Functional conservation of the effector protein translocators PopB/YopB and PopD/YopD of Pseudomonas aeruginosa and Yersinia pseudotuberculosis. Mol. Microbiol. 29:1155-1165. [DOI] [PubMed] [Google Scholar]
- 19.Galan, J. E., and A. Collmer. 1999. Type III secretion machines: bacterial devices for protein delivery into host cells. Science 284:1322-1328. [DOI] [PubMed] [Google Scholar]
- 20.Galan, J. E., and H. Wolf-Watz. 2006. Protein delivery into eukaryotic cells by type III secretion machines. Nature 444:567-573. [DOI] [PubMed] [Google Scholar]
- 21.Gazi, A. D., S. N. Charova, N. J. Panopoulos, and M. Kokkinidis. 2009. Coiled-coils in type III secretion systems: structural flexibility, disorder and biological implications. Cell Microbiol. 11(5):719-729. [DOI] [PubMed] [Google Scholar]
- 22.Geddes, K., M. Worley, G. Niemann, and F. Heffron. 2005. Identification of new secreted effectors in Salmonella enterica serovar Typhimurium. Infect. Immun. 73:6260-6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh, P. 2004. Process of protein transport by the type III secretion system. Microbiol. Mol. Biol. Rev. 68:771-795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ginocchio, C. C., and J. E. Galan. 1995. Functional conservation among members of the Salmonella typhimurium InvA family of proteins. Infect. Immun. 63:729-732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greenberg, J. T., and B. A. Vinatzer. 2003. Identifying type III effectors of plant pathogens and analyzing their interaction with plant cells. Curr. Opin. Microbiol. 6:20-28. [DOI] [PubMed] [Google Scholar]
- 26.Guttman, D. S., B. A. Vinatzer, S. F. Sarkar, M. V. Ranall, G. Kettler, and J. T. Greenberg. 2002. A functional screen for the type III (Hrp) secretome of the plant pathogen Pseudomonas syringae. Science 295:1722-1726. [DOI] [PubMed] [Google Scholar]
- 27.Hirose, S., K. Shimizu, S. Kanai, Y. Kuroda, and T. Noguchi. 2007. POODLE-L: a two-level SVM prediction system for reliably predicting long disordered regions. Bioinformatics 23:2046-2053. [DOI] [PubMed] [Google Scholar]
- 28.Hohlfeld, S., I. Pattis, J. Puls, G. V. Plano, R. Haas, and W. Fischer. 2006. A C-terminal translocation signal is necessary, but not sufficient for type IV secretion of the Helicobacter pylori CagA protein. Mol. Microbiol. 59:1624-1637. [DOI] [PubMed] [Google Scholar]
- 29.Hudson, D. L., A. N. Layton, T. R. Field, A. J. Bowen, H. Wolf-Watz, M. Elofsson, M. P. Stevens, and E. E. Galyov. 2007. Inhibition of type III secretion in Salmonella enterica serovar Typhimurium by small-molecule inhibitors. Antimicrob. Agents Chemother. 51:2631-2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karavolos, M. H., M. Wilson, J. Henderson, J. J. Lee, and C. M. A. Khan. 2005. Type III secretion of the Salmonella effector protein SopE is mediated via an N-terminal amino acid signal and not an mRNA sequence. J. Bacteriol. 187:1559-1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim, B. H., H. G. Kim, J. S. Kim, J. I. Jang, and Y. K. Park. 2007. Analysis of functional domains present in the N-terminus of the SipB protein. Microbiology 153:2998-3008. [DOI] [PubMed] [Google Scholar]
- 32.Konjufca, V., S. Y. Wanda, M. C. Jenkins, and R. Curtiss III. 2006. A recombinant attenuated Salmonella enterica serovar Typhimurium vaccine encoding Eimeria acervulina antigen offers protection against E. acervulina challenge. Infect. Immun. 74:6785-6796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kubori, T., A. Hyakutake, and H. Nagai. 2008. Legionella translocates an E3 ubiquitin ligase that has multiple U-boxes with distinct functions. Mol. Microbiol. 67:1307-1319. [DOI] [PubMed] [Google Scholar]
- 34.Kubori, T., A. Sukhan, S. I. Aizawa, and J. E. Galan. 2000. Molecular characterization and assembly of the needle complex of the Salmonella typhimurium type III protein secretion system. Proc. Natl. Acad. Sci. U. S. A. 97:10225-10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lawrence, P. K., W. Kittichotirat, R. E. Bumgarner, J. E. McDermott, D. R. Herndon, D. P. Knowles, and S. Srikumaran. 2010. Genome sequences of Mannheimia haemolytica serotype A2: ovine and bovine isolates. J. Bacteriol. 192:1167-1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lawrence, P. K., W. Kittichotirat, J. E. McDermott, and R. E. Bumgarner. 2010. A three-way comparative genomic analysis of Mannheimia haemolytica isolates. BMC Genomics 11:535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee, C. C., M. D. Wood, K. Ng, C. B. Andersen, Y. Liu, P. Luginbuhl, G. Spraggon, and F. Katagiri. 2004. Crystal structure of the type III effector AvrB from Pseudomonas syringae. Structure 12:487-494. [DOI] [PubMed] [Google Scholar]
- 38.Lee, S. H., and J. E. Galan. 2004. Salmonella type III secretion-associated chaperones confer secretion-pathway specificity. Mol. Microbiol. 51:483-495. [DOI] [PubMed] [Google Scholar]
- 39.Lilic, M., M. Vujanac, and C. E. Stebbins. 2006. A common structural motif in the binding of virulence factors to bacterial secretion chaperones. Mol. Cell 21:653-664. [DOI] [PubMed] [Google Scholar]
- 40.Lloyd, S. A., A. Forsberg, H. Wolf-Watz, and M. S. Francis. 2001. Targeting exported substrates to the Yersinia TTSS: different functions for different signals? Trends Microbiol. 9:367-371. [DOI] [PubMed] [Google Scholar]
- 41.Löwer, M., and G. Schneider. 2009. Prediction of type III secretion signals in genomes of gram-negative bacteria. PLoS One 4:e5917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luo, Z. Q., and R. R. Isberg. 2004. Multiple substrates of the Legionella pneumophila Dot/Icm system identified by interbacterial protein transfer. Proc. Natl. Acad. Sci. U. S. A. 101:841-846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miao, E. A., and S. I. Miller. 2000. A conserved amino acid sequence directing intracellular type III secretion by Salmonella typhimurium. Proc. Natl. Acad. Sci. U. S. A. 97:7539-7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Michiels, T., and G. R. Cornelis. 1991. Secretion of hybrid proteins by the Yersinia Yop export system. J. Bacteriol. 173:1677-1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muschiol, S., L. Bailey, A. Gylfe, C. Sundin, K. Hultenby, S. Bergstrom, M. Elofsson, H. Wolf-Watz, S. Normark, and B. Henriques-Normark. 2006. A small-molecule inhibitor of type III secretion inhibits different stages of the infectious cycle of Chlamydia trachomatis. Proc. Natl. Acad. Sci. U. S. A. 103:14566-14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagai, H., E. D. Cambronne, J. C. Kagan, J. C. Amor, R. A. Kahn, and C. R. Roy. 2005. A C-terminal translocation signal required for Dot/Icm-dependent delivery of the Legionella RalF protein to host cells. Proc. Natl. Acad. Sci. U. S. A. 102:826-831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nagai, H., J. C. Kagan, X. Zhu, R. A. Kahn, and C. R. Roy. 2002. A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science 295:679-682. [DOI] [PubMed] [Google Scholar]
- 48.Negrea, A., E. Bjur, S. E. Ygberg, M. Elofsson, H. Wolf-Watz, and M. Rhen. 2007. Salicylidene acylhydrazides that affect type III protein secretion in Salmonella enterica serovar typhimurium. Antimicrob. Agents Chemother. 51:2867-2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nguyen, L., I. T. Paulsen, J. Tchieu, C. J. Hueck, and M. H. Saier, Jr. 2000. Phylogenetic analyses of the constituents of type III protein secretion systems. J. Mol. Microbiol. Biotechnol. 2:125-144. [PubMed] [Google Scholar]
- 50.Niemann, G. S., R. N. Brown, J. K. Gustin, A. Stufkens, A. S. Shaikh-Kidwai, J. Li, J. E. McDermott, H. M. Brewer, A. Schepmoes, R. D. Smith, J. N. Adkins, and F. Heffron. 2011. Discovery of novel secreted virulence factors from Salmonella enterica serovar Typhimurium by proteomic analysis of culture supernatants. Infect. Immun. 79:33-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ninio, S., D. M. Zuckman-Cholon, E. D. Cambronne, and C. R. Roy. 2005. The Legionella IcmS-IcmW protein complex is important for Dot/Icm-mediated protein translocation. Mol. Microbiol. 55:912-926. [DOI] [PubMed] [Google Scholar]
- 52.Noble, W. S. 2006. What is a support vector machine? Nat. Biotechnol. 24:1565-1567. [DOI] [PubMed] [Google Scholar]
- 53.Nordfelth, R., A. M. Kauppi, H. A. Norberg, H. Wolf-Watz, and M. Elofsson. 2005. Small-molecule inhibitors specifically targeting type III secretion. Infect. Immun. 73:3104-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oehmen, C., and J. Nieplocha. 2006. ScalaBLAST: a scalable implementation of BLAST for high performance data-intensive bioinformatics analysis. IEEE Trans. Parallel Distrib. Syst. 17:740-749. [Google Scholar]
- 55.Pallen, M. J., S. A. Beatson, and C. M. Bailey. 2005. Bioinformatics, genomics and evolution of non-flagellar type-III secretion systems: a Darwinian perspective. FEMS Microbiol. Rev. 29:201-229. [DOI] [PubMed] [Google Scholar]
- 56.Panina, E. M., S. Mattoo, N. Griffith, N. A. Kozak, M. H. Yuk, and J. F. Miller. 2005. A genome-wide screen identifies a Bordetella type III secretion effector and candidate effectors in other species. Mol. Microbiol. 58:267-279. [DOI] [PubMed] [Google Scholar]
- 57.Pansegrau, W., F. Schoumacher, B. Hohn, and E. Lanka. 1993. Site-specific cleavage and joining of single-stranded DNA by VirD2 protein of Agrobacterium tumefaciens Ti plasmids: analogy to bacterial conjugation. Proc. Natl. Acad. Sci. U. S. A. 90:11538-11542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Petnicki-Ocwieja, T., D. J. Schneider, V. C. Tam, S. T. Chancey, L. Shan, Y. Jamir, L. M. Schechter, M. D. Janes, C. R. Buell, X. Tang, A. Collmer, and J. R. Alfano. 2002. Genomewide identification of proteins secreted by the Hrp type III protein secretion system of Pseudomonas syringae pv. tomato DC3000. Proc. Natl. Acad. Sci. U. S. A. 99:7652-7657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Petrova, N. V., and C. H. Wu. 2006. Prediction of catalytic residues using Support Vector Machine with selected protein sequence and structural properties. BMC Bioinform. 7:312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Plewczynski, D., A. Tkacz, A. Godzik, and L. Rychlewski. 2005. A support vector machine approach to the identification of phosphorylation sites. Cell. Mol. Biol. Lett. 10:73-89. [PubMed] [Google Scholar]
- 61.Ramamurthi, K. S., and O. Schneewind. 2005. A synonymous mutation in Yersinia enterocolitica yopE affects the function of the YopE type III secretion signal. J. Bacteriol. 187:707-715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rosqvist, R., S. Hakansson, A. Forsberg, and H. Wolf-Watz. 1995. Functional conservation of the secretion and translocation machinery for virulence proteins of yersiniae, salmonellae and shigellae. EMBO J. 14:4187-4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Russmann, H., T. Kubori, J. Sauer, and J. E. Galan. 2002. Molecular and functional analysis of the type III secretion signal of the Salmonella enterica InvJ protein. Mol. Microbiol. 46:769-779. [DOI] [PubMed] [Google Scholar]
- 64.Salzberg, S. L. 1997. On comparing classifiers: pitfalls to avoid and recommended approach. Data Min. Knowl. Discov. 1:317-328. [Google Scholar]
- 65.Samudrala, R., F. Heffron, and J. E. McDermott. 2009. Accurate prediction of secreted substrates and identification of a conserved putative secretion signal for type III secretion systems. PLoS Pathog. 5:e1000375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Segal, G., M. Feldman, and T. Zusman. 2005. The Icm/Dot type-IV secretion systems of Legionella pneumophila and Coxiella burnetii. FEMS Microbiol. Rev. 29:65-81. [DOI] [PubMed] [Google Scholar]
- 67.Shah, A. R., C. S. Oehmen, J. Harper, and B. J. Webb-Robertson. 2007. Integrating subcellular location for improving machine learning models of remote homology detection in eukaryotic organisms. Comput. Biol. Chem. 31:138-142. [DOI] [PubMed] [Google Scholar]
- 68.Shah, A. R., C. S. Oehmen, and B. J. Webb-Robertson. 2008. SVM-HUSTLE—an iterative semi-supervised machine learning approach for pairwise protein remote homology detection. Bioinformatics 24:783-790. [DOI] [PubMed] [Google Scholar]
- 69.Simone, M., C. A. McCullen, L. E. Stahl, and A. N. Binns. 2001. The carboxy-terminus of VirE2 from Agrobacterium tumefaciens is required for its transport to host cells by the virB-encoded type IV transport system. Mol. Microbiol. 41:1283-1293. [DOI] [PubMed] [Google Scholar]
- 70.Sorg, J. A., N. C. Miller, and O. Schneewind. 2005. Substrate recognition of type III secretion machines—testing the RNA signal hypothesis. Cell Microbiol. 7:1217-1225. [DOI] [PubMed] [Google Scholar]
- 71.Sory, M. P., A. Boland, I. Lambermont, and G. R. Cornelis. 1995. Identification of the YopE and YopH domains required for secretion and internalization into the cytosol of macrophages, using the cyaA gene fusion approach. Proc. Natl. Acad. Sci. U. S. A. 92:11998-12002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stebbins, C. E., and J. E. Galan. 2000. Modulation of host signaling by a bacterial mimic: structure of the Salmonella effector SptP bound to Rac1. Mol. Cell 6:1449-1460. [DOI] [PubMed] [Google Scholar]
- 73.Stebbins, C. E., and J. E. Galan. 2001. Structural mimicry in bacterial virulence. Nature 412:701-705. [DOI] [PubMed] [Google Scholar]
- 74.Stevens, M. P., A. Friebel, L. A. Taylor, M. W. Wood, P. J. Brown, W.-D. Hardt, and E. E. Galyov. 2003. A Burkholderia pseudomallei type III secreted protein, BopE, facilitates bacterial invasion of epithelial cells and exhibits guanine nucleotide exchange factor activity. J. Bacteriol. 185:4992-4996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tarca, A. L., V. J. Carey, X. W. Chen, R. Romero, and S. Draghici. 2007. Machine learning and its applications to biology. PLoS Comput. Biol. 3:e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tobe, T., S. A. Beatson, H. Taniguchi, H. Abe, C. M. Bailey, A. Fivian, R. Younis, S. Matthews, O. Marches, G. Frankel, T. Hayashi, and M. J. Pallen. 2006. An extensive repertoire of type III secretion effectors in Escherichia coli O157 and the role of lambdoid phages in their dissemination. Proc. Natl. Acad. Sci. U. S. A. 103:14941-14946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vencato, M., F. Tian, J. R. Alfano, C. R. Buell, S. Cartinhour, G. A. DeClerck, D. S. Guttman, J. Stavrinides, V. Joardar, M. Lindeberg, P. A. Bronstein, J. W. Mansfield, C. R. Myers, A. Collmer, and D. J. Schneider. 2006. Bioinformatics-enabled identification of the HrpL regulon and type III secretion system effector proteins of Pseudomonas syringae pv. phaseolicola 1448A. Mol. Plant Microbe Interact. 19:1193-1206. [DOI] [PubMed] [Google Scholar]
- 78.Vergunst, A. C., B. Schrammeijer, A. den Dulk-Ras, C. M. de Vlaam, T. J. Regensburg-Tuink, and P. J. Hooykaas. 2000. VirB/D4-dependent protein translocation from Agrobacterium into plant cells. Science 290:979-982. [DOI] [PubMed] [Google Scholar]
- 79.Vergunst, A. C., M. C. van Lier, A. den Dulk-Ras, T. A. Stuve, A. Ouwehand, and P. J. Hooykaas. 2005. Positive charge is an important feature of the C-terminal transport signal of the VirB/D4-translocated proteins of Agrobacterium. Proc. Natl. Acad. Sci. U. S. A. 102:832-837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang, Y., Z. Xue, and J. Xu. 2006. Better prediction of the location of alpha-turns in proteins with support vector machine. Proteins 65:49-54. [DOI] [PubMed] [Google Scholar]
- 81.Weathers, E. A., M. E. Paulaitis, T. B. Woolf, and J. H. Hoh. 2004. Reduced amino acid alphabet is sufficient to accurately recognize intrinsically disordered protein. FEBS Lett. 576:348-352. [DOI] [PubMed] [Google Scholar]
- 82.Webb-Robertson, B. J., C. Oehmen, and M. Matzke. 2005. SVM-BALSA: remote homology detection based on Bayesian sequence alignment. Comput. Biol. Chem. 29:440-443. [DOI] [PubMed] [Google Scholar]
- 83.Widmaier, D. M., D. Tullman-Ercek, E. A. Mirsky, R. Hill, S. Govindarajan, J. Minshull, and C. A. Voigt. 2009. Engineering the Salmonella type III secretion system to export spider silk monomers. Mol. Syst. Biol. 5:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wilkinson, D. J. 2007. Bayesian methods in bioinformatics and computational systems biology. Brief. Bioinform. 8:109-116. [DOI] [PubMed] [Google Scholar]
- 85.Yip, C. K., T. G. Kimbrough, H. B. Felise, M. Vuckovic, N. A. Thomas, R. A. Pfuetzner, E. A. Frey, B. B. Finlay, S. I. Miller, and N. C. Strynadka. 2005. Structural characterization of the molecular platform for type III secretion system assembly. Nature 435:702-707. [DOI] [PubMed] [Google Scholar]