

Abstract
Heat shock factor 1 (HSF1) is a stress-induced transcription factor that promotes expression of genes that protect mammalian cells from the lethal effects of severely elevated temperatures (>42°C). However, we recently showed that HSF1 is activated at a lower temperature (39.5°C) in T cells, suggesting that HSF1 may be important for preserving T cell function during pathogen-induced fever responses. To test this, we examined the role of HSF1 in clearance of Listeria monocytogenes, an intracellular bacterial pathogen that elicits a strong CD8+ T cell response in mice. Using temperature transponder microchips, we showed that the core body temperature increased approximately 2°C in L. monocytogenes-infected mice and that the fever response was maintained for at least 24 h. HSF1-deficient mice cleared a low-dose infection with slightly slower kinetics than did HSF1+/+ littermate controls but were significantly more susceptible to challenges with higher doses of bacteria. Surprisingly, HSF1-deficient mice did not show a defect in CD8+ T cell responses following sublethal infection. However, when HSF1-deficient mice were challenged with high doses of L. monocytogenes, increased levels of serum tumor necrosis factor alpha (TNF-α) and gamma interferon (IFN-γ) compared to those of littermate control mice were observed, and rapid death of the animals occurred within 48 to 60 h of infection. Neutralization of TNF-α enhanced the survival of HSF1-deficient mice. These results suggest that HSF1 is needed to prevent the overproduction of proinflammatory cytokines and subsequent death due to septic shock that can result following high-dose challenge with bacterial pathogens.
Heat shock factor 1 (HSF1) is a highly conserved transcriptional regulator that controls the expression of a variety of different gene products during heat shock and other environmental stresses (24, 33). Previous studies examining either the resistance of Caenorhabditis elegans to bacterial infection or the ability of mice to produce IgG have suggested that HSF1 may be important for generating protective immune responses (18, 38). In most cell types, the heat shock response that leads to trimerization and nuclear translocation of HSF1 occurs only during severe thermal stress, at temperatures at or above 42°C (32). However, in T cells, HSF1 can be activated at 39.5°C, a temperature that is within the range of a typical fever response (11).
Fever is induced during infection by Toll-like receptor (TLR) ligands that trigger the release of proinflammatory cytokines such as interleukin 1β (IL-1β), IL-6, and tumor necrosis factor alpha (TNF-α) and prostaglandin E2, which acts on the hypothalamus to temporarily increase the thermoregulatory set point of the body (7). In mice, fever has been characterized as an elevation of 1.3 to 2.1°C above the basal core body temperature (13). Although raising the body temperature to produce a fever is an energy-costly process, the evolutionary conservation of febrile responses suggests that fever is beneficial for the host and that the immune mechanisms designed to clear infections must be well adapted to functioning at elevated temperatures (15).
Since HSF1 is activated at much lower temperatures in T cells than in other cell types, we hypothesized that it might be needed to protect T cell function during infections that induce a fever response. In a previous in vitro study, we showed that T cells isolated from HSF1-deficient mice were unable to proliferate efficiently at physiologic fever temperature (39.5°C) (26). To further address this question in vivo, we chose to study the role of HSF1 during Listeria monocytogenes infection, since clearance of this intracellular bacterial pathogen is strictly dependent on protective T cell responses. Early resistance to listeriosis is mediated primarily by gamma interferon (IFN-γ) and TNF-α, and mice that lack either of these cytokines are highly susceptible to infection (3, 16, 28, 30). However, the activation and proliferation of CD8+ T cells that can lyse infected cells and release the bacteria from their protective intracellular niche are required for sterilizing immunity against L. monocytogenes.
Although fever is a commonly described symptom associated with human listeriosis (22, 40), the ability of L. monocytogenes to induce fever in small animal models has not been well characterized. One early study showed that the rectal temperature of rabbits increased by 2°C following injection with a partially purified toxin derived from L. monocytogenes and that this response peaked at 24 h postinjection (35). However, there are no reports in the literature describing the fever response in mice following L. monocytogenes infection.
In this paper, we monitored the core body temperature of mice infected with L. monocytogenes and found that both HSF1-deficient and wild-type mice maintain a fever response during the first 2 to 3 days postinfection. Surprisingly, we showed that CD8+ T cell responses were not impaired in HSF1-deficient mice during L. monocytogenes infection. However, HSF1−/− mice infected with at least 1 × 104 CFU of L. monocytogenes displayed increased lethality that appeared to be caused by misregulated production of proinflammatory cytokines such as TNF-α.
MATERIALS AND METHODS
Mice.
BALB/c/By/J and C57BL/6 mice were obtained from the Jackson Laboratory and housed under specific-pathogen-free conditions at the Division of Laboratory Animal Resources, University of Kentucky Medical Center. HSF1−/+ mice on a 129Sv × BALB/c background (42) were supplied to us as a breeding pair from Ivor Benjamin (University of Utah). HSF1 genotypes were determined by PCR analysis of tail DNA. HSF1−/− and HSF1+/+ littermates were used at 8 to 12 weeks of age, and the colony was maintained by breeding heterozygotes. Attempts to backcross the mice to either a BALB/c or C57BL/6 background resulted in a dramatic reduction in the number of viable HSF1−/− pups. This embryonic lethality, prevented by unknown modifier genes on the mixed background, is due to a defect in the production of the chorioallantoic placenta of the HSF1−/− embryos at about day 10 of gestation (42). All animal experiments were reviewed and approved by the University of Kentucky Institutional Animal Care and Use Committee.
Bacteria.
L. monocytogenes 10403s and the ovalbumin (OVA)-expressing derivative JJL-OVA (29) were grown in brain heart infusion (BHI) broth until early stationary phase, and then aliquots were prepared and frozen at −80°C and their titers were determined. The 50% lethal dose (LD50) for L. monocytogenes 10403s is ∼1 × 104 CFU in BALB/c mice and ∼2 × 105 CFU in C57BL/6 mice. Since the HSF1−/− and HSF1+/+ mice used in this study were on a mixed 129 × BALB/c background, an exact LD50 value could not readily be determined for these animals. However, a small-scale analysis of lethality in HSF1+/+ mice indicated that the lethal dose for L. monocytogenes 10403s in this mixed strain background was in the range of 8 × 103 to 2 × 104 CFU (data not shown).
Infection of mice.
An aliquot of L. monocytogenes was thawed on ice and then incubated in BHI broth with shaking at 37°C until early-exponential-phase growth was reached. Dilutions of bacteria prepared in phosphate-buffered saline (PBS) were used to infect mice by injecting a 200-μl volume intravenously (i.v.) into the lateral tail vein. The number of bacteria present in the inoculum was confirmed by plating serial dilutions on BHI agar (Difco) for each experiment. For challenge studies, mice were sacrificed at the indicated time points, and the spleens and livers were harvested aseptically and homogenized in 0.1% NP-40. Serial dilutions of the homogenates were plated on BHI agar, and the total number of CFU per organ was determined.
Core body temperature measurement.
Sterile microchip transponders (Bio Medic Data Systems, Inc.) were surgically implanted into the peritoneal cavities of mice at least 10 days before infection with L. monocytogenes. Temperature measurements were obtained by placing a radio frequency scanner (Bio Medic Data Systems model DAS 5007) near the abdomen of each mouse.
Western blotting.
Spleens were harvested, single-cell suspensions were prepared, and red blood cells (RBC) were lysed using ACK lysis buffer (0.15 M NH4Cl, 10 mM KHCO3, 0.1 mM EDTA). The cells were counted, and then the cell pellets were flash frozen in liquid nitrogen and stored overnight at −80°C. Whole-cell lysates containing both cytoplasmic and nuclear proteins were prepared using a buffer consisting of 25% glycerol, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 20 mM HEPES, 1 mM phenylmethylsulfonyl fluoride (PMSF), and 1 mM dithiothreitol (DTT). The lysates were centrifuged for 10 min at 15,000 × g, and the supernatants (5 × 106 cell equivalents) were separated on an 8% SDS-PAGE gel. Proteins were transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore) and then blocked with PBS-5% milk. Rat anti-mouse HSF1 monoclonal antibodies (clone 10H8; Enzo Life Sciences) were used at a 1:1,000 dilution followed by horseradish peroxidase (HRP)-conjugated rabbit anti-rat IgG polyclonal antibodies at a 1:5,000 dilution. For the actin control blot, the membrane was stripped and reprobed with HRP-conjugated anti-mouse actin monoclonal antibodies (clone C-2; Santa Cruz Biotech). HRP was detected using SuperSignal West Pico chemiluminescent substrate (Thermo Scientific).
ICS.
Single-cell suspensions of splenocytes harvested from infected mice were prepared, and the cells were treated with ammonium chloride to remove red blood cells (RBC). The cells were suspended in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), 1 mM glutamine, 1 mM sodium pyruvate, 1× minimal essential medium (MEM) nonessential amino acids, 50 μM 2-mercaptoethanol (2-ME), 100 U/ml penicillin, and 100 U/ml streptomycin (RP-10 medium) at a concentration of 1 × 107 cells/ml and plated in 24-well plates at 500 μl/well. Synthetic SIINFEKL peptide (Bio-Synthesis, Lewisville, TX) was added at a final concentration of 100 nM; phorbol myristate acetate (PMA) (10 ng/ml) plus ionomycin (1 μM) was used as a control. The cells were incubated for 4 h at 37°C in 5% CO2, and then IFN-γ intracellular cytokine staining (ICS) was performed using the Cytofix/Cytoperm kit with Golgi plug and antibodies (anti-T cell receptor β [anti-TCRβ], clone H57-597; anti-CD8α, clone 53-6.7; anti-IFN-γ, clone XMG1.2) from BD Biosciences. Fluorescence intensities were measured using a FACSCalibur flow cytometer, and analysis was performed using CellQuest software (BD Biosciences). Dead cells and monocytes were excluded using forward and side scatter gating, and at least 15,000 events in the lymphocyte gate were analyzed.
In vitro stimulation of BMDC.
Bone marrow-derived dendritic cells (BMDC) were generated by culturing bone marrow cells in the presence of granulocyte-macrophage colony-stimulating factor (GM-CSF) as described previously (23). Nonadherent cells harvested on day 8 of the culture were approximately 70 to 80% CD11c+ and had an immature phenotype (data not shown). The BMDC were resuspended in RP-10 at 2 × 106 cells/ml and stimulated with lipopolysaccharide (LPS) (Sigma) at a final concentration of 10 μg/ml. Culture supernatants were collected 6 h later, and the amounts of TNF-α and IL-6 present in each supernatant were determined by enzyme-linked immunosorbent assay (ELISA) (BD Biosciences).
Measurement of serum cytokine levels.
Tail vein venipuncture was used to collect peripheral blood directly into serum separator tubes (BD Biosciences). The blood was allowed to clot, and the serum was removed and frozen at −80°C. Serum cytokine levels were measured using a cytokine bead array (CBA) kit per the manufacturer's protocol (BD Biosciences).
In vivo neutralization of TNF-α.
Mice were injected (intravenously [i.v.]) with 1 μg of either anti-TNF-α antibody (clone 3-19.12; BD Biosciences) or an isotype control Armenian hamster IgG in 0.2 ml of PBS. Two hours later, mice were infected with 1 × 104 CFU of L. monocytogenes 10403s. Peripheral blood was collected 48 h postinfection (hpi) for serum cytokine analysis, and survival of the mice was monitored for 9 days.
Statistical analysis.
All statistical analyses were performed using Prism 4 for Macintosh software. Data were considered statistically significant if P values of <0.05 were obtained.
RESULTS
L. monocytogenes infection triggers an increase in core body temperature.
We previously showed that the transcription factor HSF1 was important for protecting T cell function when T cells were incubated at elevated temperatures within the range of a physiological fever response (26). To address the role of HSF1 during bacterial infection, we chose to study L. monocytogenes, since clearance of this intracellular bacterial pathogen is known to be strictly dependent on T cells (6, 19). Although the mouse model of systemic L. monocytogenes infection has been studied for decades, the time course of fever induction in Listeria-infected mice is not well established. To verify that L. monocytogenes did trigger a febrile response in mice, we used transponder implants to monitor the core body temperature of both BALB/c and C57BL/6 mice during systemic infection. Temperature measurements were recorded 1 day before, immediately prior to, and at various intervals for 3 days following intravenous infection with a sublethal dose of L. monocytogenes.
As shown in Fig. 1, core body temperatures increased following infection of either BALB/c or C57BL/6 mice. The peak of the fever response occurred approximately 46 h postinfection and resulted in a 2°C increase in body temperature. The baseline temperature of BALB/c mice was 36.5°C (±0.3°C) and increased to 38.4°C (±0.6°C), and the core body temperature of C57BL/6 mice increased from an average of 37.1°C (±0.5°C) to 39.2°C (±0.2°C). Within 3 days of bacterial challenge, the internal temperatures of both strains had returned to slightly above baseline values. Similar fever responses were observed in HSF1-deficient mice or HSF1+/+ littermate controls on a mixed BALB/c × 129Sv strain background (Fig. 1). The HSF1+/+ mice had a basal core temperature of 37.6°C (±0.8°C), and the HSF1-deficient animals started with an average temperature of 37.3°C (±0.5°C). In these animals, the peak of the fever response occurred approximately 40 h postinfection.
FIG. 1.

L. monocytogenes infection triggers a fever response in HSF1+/+ and HSF1−/− mice. (A) Temperature transponder microchips were implanted (intraperitoneally) in the indicated mouse strains, and baseline temperatures were recorded starting 10 days later. At t = 0, groups of BALB/c and C57BL/6 mice (n = 8) were infected (i.v.) with 3.9 × 103 CFU of L. monocytogenes strain 10403s. Groups of HSF1+/+ and HSF1−/− mice (n = 6) were infected (i.v.) with 6 × 103 CFU of L. monocytogenes strain JJL-OVA. The mean change in core body temperature ± standard deviation for each group of mice from one of two separate experiments is shown. (B) Splenocytes harvested 6 h after infection with 1 × 104 CFU of L. monocytogenes 10403s were used for Western blot analysis. Data from one of two mice used per group are shown.
Together, these results indicated that the core body temperature of mice infected with L. monocytogenes remained elevated for at least 24 h within the first few days of infection, a time period that could be critical for appropriate priming of Listeria-specific T cell responses. However, since previous in vitro studies used 39.5°C as the set point for fever temperature, and the highest core body temperatures that we recorded were 39°C for BALB/c mice and 39.4°C for C57BL/6 mice, we next performed Western blot analysis to verify that HSF1 expression levels were altered during infection of mice. We previously showed that HSF1 protein expression is upregulated in response to thermal stress (25), probably due to the increased stability of the activated trimer. As expected, no HSF1 expression was detected in the spleens of HSF1−/− mice. However, HSF1 expression increased greatly during L. monocytogenes infection of HSF1+/+ mice (Fig. 1B). These data indicated that the HSF1 pathway was activated in vivo during L. monocytogenes infection of mice.
CD8+ T cell activation is not impaired in HSF1-deficient mice.
We previously showed that HSF1-deficient T cells incubated at an elevated temperature (39.5°C) had an impaired ability to proliferate in response to anti-CD3/CD28 stimulation in vitro compared with T cells isolated from HSF1+/+ mice (26). Given these results, we predicted that L. monocytogenes-infected HSF1-deficient mice might have a decreased CD8+ T cell response because of the fever induced during the first few days of infection. To test this, we infected groups of Kb-expressing HSF1−/− and HSF1+/+ mice with L. monocytogenes JJL-OVA, a recombinant strain that expresses ovalbumin (29). Splenocytes were harvested 7 days later and stimulated in vitro with the ovalbumin-specific peptide SIINFEKL, and the number of antigen-reactive T cells present in the spleen was determined using intracellular cytokine staining for IFN-γ. Surprisingly, we found that the number of SIINFEKL-specific T cells had increased to 2 to 3% of all splenic CD8+ T cells in both HSF1+/+ and HSF1−/− mice (Fig. 2), a response that was comparable to results previously obtained 7 days after infection of C57BL/6 mice (20, 21). We also measured LLO91-99-specific T cell responses in the few animals that expressed Kd and saw no difference between HSF1-deficient and wild-type animals (data not shown). As a control, we also stimulated the cells with PMA plus ionomycin to find the maximal number of cells capable of secreting IFN-γ, but we did not find any significant difference between the two mouse strains after this treatment (Fig. 2). These results suggested that the thermoprotective functions of HSF1 were not required for CD8+ T cells to be activated and proliferate to normal levels during L. monocytogenes infection.
FIG. 2.
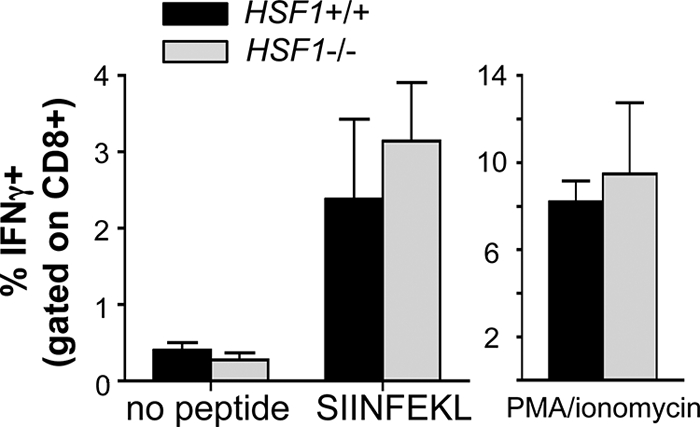
CD8+ T cell responses to L. monocytogenes are not impaired in HSF1-deficient mice. HSF1−/− and HSF1+/+ mice (n = 4) were infected with 6 × 103 CFU of L. monocytogenes strain JLL-OVA. Splenocytes were harvested 7 days postinfection and stimulated in vitro with either 100 nM SIINFEKL peptide, PMA plus ionomycin, or medium alone, and the percentage of IFN-γ+ cells (gated on CD8+ T cells) was determined by intracellular cytokine staining. Mean percentages of IFN-γ+ cells ± standard deviations for triplicate samples are shown. No significant differences (P < 0.5 with an unpaired t test) were observed when HSF1−/− and HSF1+/+ cells were compared. Data from one of two independent experiments are shown.
Clearance of primary sublethal L. monocytogenes infection is impaired in HSF1-deficient mice.
To investigate other possible effects of HSF1 on bacterial clearance, we infected HSF1+/+ and HSF1−/− mice with a sublethal dose of L. monocytogenes and determined the total number of bacteria present in the liver at 3 and 7 days postinfection. These time points were chosen based on the typical course of L. monocytogenes infection in C57BL/6 × DBA or C57BL/6 × BALB/c F1 mice, where the number of bacteria either reaches a peak or is beginning to decrease by 3 days postinfection, and the bacterial load is usually cleared completely by 7 days postinfection (6, 8). As shown in Fig. 3A, three out of five of the HSF1+/+ mice had reduced bacterial burdens in the liver within 3 days of infection, and we did not detect any L. monocytogenes in HSF1+/+ mouse livers at 7 days postinfection. In contrast, none of the HSF1-deficient mice had begun to clear the infection 3 days after challenge and there were still several hundred CFU present in the liver 7 days postinfection (Fig. 3A). These data suggested that HSF1−/− mice were capable of clearing a sublethal L. monocytogenes infection but that they did so with delayed kinetics.
FIG. 3.
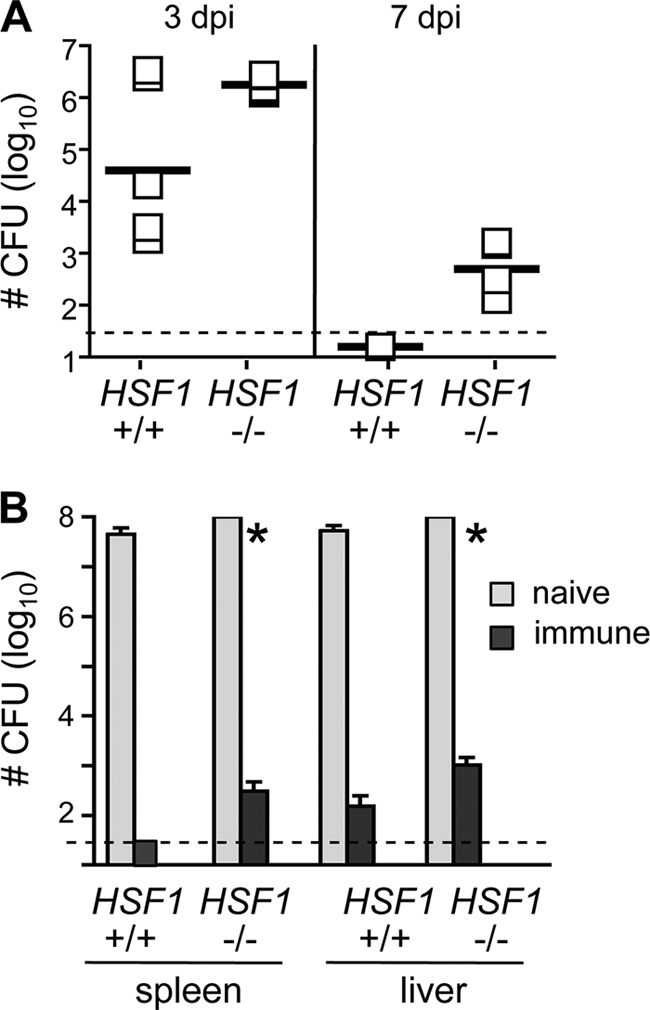
Clearance of primary L. monocytogenes infection is impaired in HSF1-deficient mice. (A) HSF1−/− and HSF1+/+ mice were infected (i.v.) with 4 × 103 CFU of L. monocytogenes 10403s. Groups of mice were sacrificed on days 3 and 7 postinfection, and the total number of CFU present in the liver was determined. Horizontal bars indicate the geometric means for each sample group. Although there was a trend toward decreased clearance in HSF1−/− mice compared with wild-type animals, none of the comparisons were statistically significant by a nonparametric test (Mann-Whitney). dpi, days postinfection. (B) Groups of mice (n = 4) were either infected (i.v.) with 4 × 103 CFU of L. monocytogenes 10403s (immune) or given PBS (naïve). Four weeks later, all of the mice were challenged with 7 × 104 CFU of L. monocytogenes 10403s. The total number of CFU per liver and spleen was determined 3 days postinfection. *, HSF1−/− mice were found dead at 3 days postinfection and CFU counts were obtained from tissues harvested from the dead animals. Mean values ± standard deviations are shown. Dashed lines indicate the limit of detection for each assay. For each plot, data from one of three separate experiments are shown.
To find out if immunization with a low dose of L. monocytogenes resulted in the generation of protective memory T cells that could clear a secondary lethal challenge, we infected groups of HSF1−/− and HSF1+/+ mice with 4 × 103 CFU of L. monocytogenes and then allowed the animals to recover from the infection. Control groups of mice received an injection of PBS. Four weeks later, all groups of mice were challenged with 7 × 104 CFU (a dose that represents ∼7 LD50 for BALB/c mice and ∼0.35 LD50 for C57BL/6 mice) and the total number of L. monocytogenes bacteria present in the liver and spleen was determined 3 days later. As shown in Fig. 3B, the L. monocytogenes-immune mice were readily able to clear the challenge dose, and we found only 102 CFU in the organs of either HSF1+/+ or HSF1−/− animals 3 days postinfection. Thus, HSF1-deficient mice generated protective immunity that was comparable to that of the wild-type littermate controls. The bacterial burden in the spleens and livers of the naïve animals was more than 4 logs greater than that observed in the immunized mice (Fig. 3B). Interestingly, all of the HSF1-deficient mice challenged with 7 × 104 CFU of L. monocytogenes died 3 days after infection, while the HSF1+/+ mice challenged with the same dose displayed signs of sickness but were still alive 3 days postinfection. These results suggested that HSF1-deficient mice were less resistant to high-dose L. monocytogenes infection than were HSF1+/+ littermate controls.
HSF1−/− mice succumb to lethal L. monocytogenes infection more rapidly than do HSF1+/+ mice.
The results presented above suggested that HSF1-deficient mice were capable of clearing a low-dose infection but were significantly more susceptible to challenge with a higher inoculum of L. monocytogenes. Although the mixed 129Sv × BALB/c background of the mice used in this study precluded the determination of a precise lethal dose, preliminary experiments indicated that the LD50 in HSF1+/+ mice was in the range of 8 × 103 to 2 × 104 CFU (data not shown), a dose that is similar to the LD50 of L. monocytogenes in BALB/c mice. To further characterize the response of HSF1-deficient mice to high-dose challenge with L. monocytogenes, we infected groups of mice preimplanted with temperature transponder microchips with 2 × 104 CFU and monitored survival over a 10-day period. As shown in Fig. 4A, half of the HSF1+/+ mice infected with this dose died 4 days postinfection, and the remaining HSF1+/+ animals recovered from the infection and survived till the end of the 10-day observation period. This suggests that 2 × 104 CFU was at or near the LD50 value for these animals. In contrast, HSF1-deficient mice infected with the same dose began dying 2 days postinfection, and by the third day, all of the mice in this group were dead (Fig. 4A). In both groups of animals, the core body temperature increased approximately 1°C during the first 24 h postinfection (Fig. 4B). However, while the fever continued to increase in HSF1+/+ mice, the core body temperature dropped significantly in HSF1-deficient mice during the second day of infection.
FIG. 4.
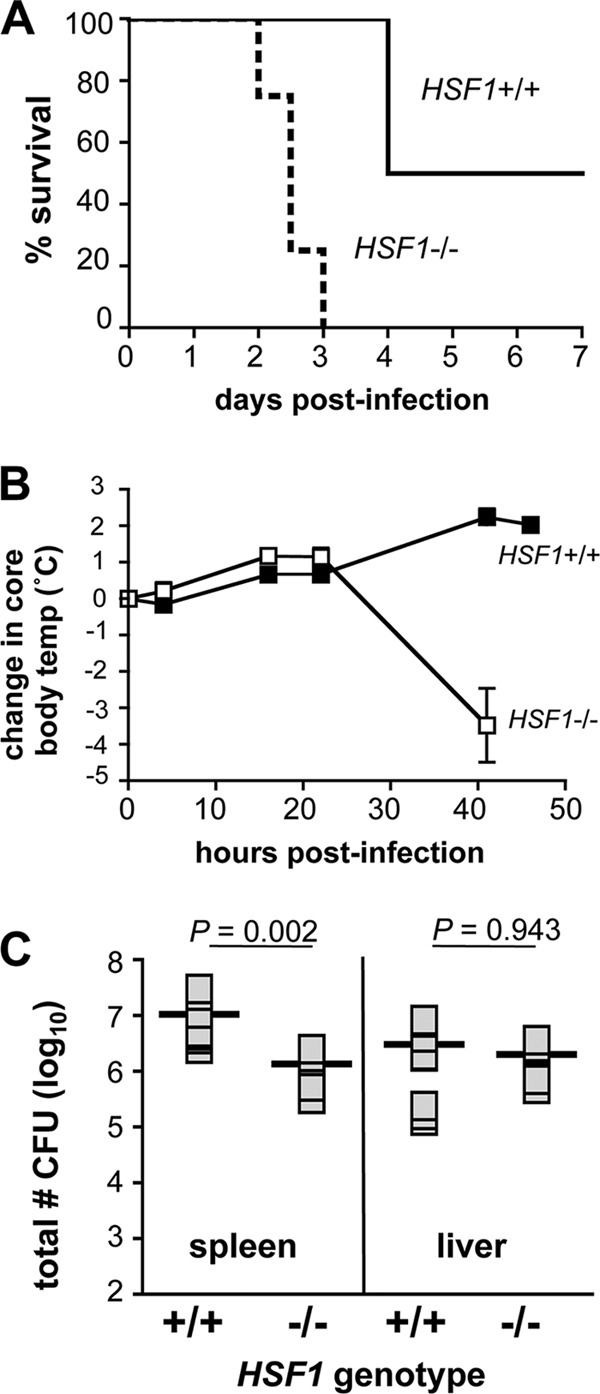
HSF1-deficient mice die more rapidly than do HSF1+/+ mice when given a dose of L. monocytogenes that is near the LD50. Temperature transponder microchips were implanted (intraperitoneally) in both HSF1−/− and HSF1+/+ mice (n = 6). Two weeks later, the mice were infected (i.v.) with 2 × 104 CFU of L. monocytogenes 10403s. (A) Survival of the infected animals was monitored for 10 days. (B) The core body temperature of the mice was determined at various intervals during the first 2 days of infection. Data shown represent the mean change in temperature from baseline values ± standard deviation for each group of mice. (C) In a separate experiment, groups of either HSF1+/+ (n = 8) or HSF1−/− (n = 6) mice were infected with 1 × 104 CFU of L. monocytogenes 10403s and the bacterial load in the spleen and liver was determined 48 hpi. Horizontal bars indicate the geometric mean for each sample group; means were compared by Mann-Whitney test, and P values for each comparison are indicated.
One explanation for the rapid death seen in HSF1-deficient mice could be that the mice were dying from overwhelming bacterial infection, due to an inability of the innate immune system to limit L. monocytogenes replication during the first 48 h following a high-dose challenge. To investigate this, we infected another group of animals with 2 × 104 CFU and sacrificed the mice at 48 h postinfection to determine the total bacterial burden in both the spleen and liver. As shown in Fig. 4C, the spleens and the livers of HSF1-deficient mice contained approximately 106 CFU 2 days postinfection. Although this is a large number of bacteria, previous studies have shown that either BALB/c or C57BL/6 mice can completely eliminate similar bacterial burdens that occur at the peak of infection (4, 17, 34). No significant difference in bacterial load was observed in the liver, and in the spleen, a greater number of L. monocytogenes bacteria was actually found in the wild-type animals (Fig. 4C). Thus, the increased lethality was not due to a greater bacterial load in HSF1-deficient mice.
HSF1-deficient mice have increased levels of TNF-α and IFN-γ in the serum 48 h after infection with a high dose of L. monocytogenes.
Previous studies have shown that during shock, the body temperature of mice often decreases by up to 5°C in the period immediately prior to death (31, 39). Thus, an alternate explanation for the rapid death observed in HSF1-deficient mice infected with at least 104 CFU was that the mice developed septic shock and died from multiple organ failure subsequent to the overproduction of proinflammatory cytokines such as TNF-α, IL-6, and IL-1β. Singh et al. previously showed that HSF1 could act as a repressor of TNF-α transcription in RAW 264.7 macrophages exposed to LPS and febrile temperatures (36, 37), a finding that supports the possibility that the TNF-α response may be misregulated during L. monocytogenes infection of HSF1-deficient mice. From other studies in our laboratory, we knew that bone marrow-derived dendritic cells treated with LPS secreted significantly more TNF-α into the culture medium than did HSF1+/+ cells (Fig. 5A). In contrast, no difference in LPS-induced IL-6 production was observed. These results suggested that HSF1-deficient cells would produce greater amounts of TNF-α after stimulation of TLR pathways by microbial ligands.
FIG. 5.
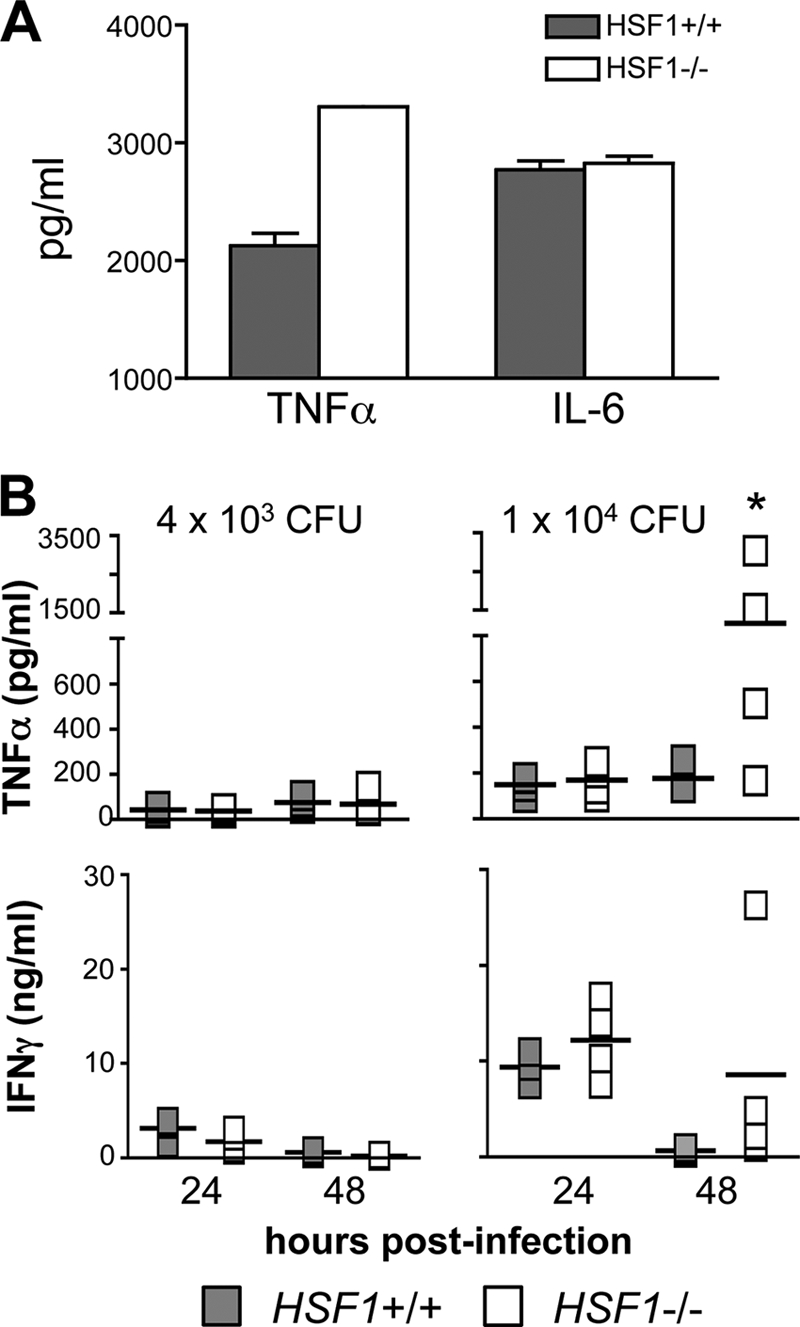
Serum TNF-α levels were increased in HSF1-deficient mice 48 h postinfection. (A) BMDC harvested from the indicated mouse strains were stimulated with LPS (10 μg/ml) in vitro, and the amounts of TNF-α and IL-6 present in the culture supernatant 6 h later were measured by ELISA. (B) HSF1−/− (white symbols) and HSF1+/+ (shaded symbols) mice were infected with either 4 × 103 or 1 × 104 CFU of L. monocytogenes 10403s. At 24 and 48 hpi blood was collected from the tail vein of each mouse and serum levels of TNF-α (top) and IFN-γ (bottom) were determined using a cytokine bead array (CBA) kit. Symbols indicate serum cytokine values for individual mice; mean values for each group are indicated by horizontal bars. *, mean level of serum TNF-α significantly greater than the average TNF-α level in HSF1+/+ mice (P = 0.0431) as determined by Mann-Whitney test. Representative data from one of three separate experiments are shown.
To find out if L. monocytogenes infection also caused a differential TNF-α response, we infected mice with either a sublethal dose (4 × 103 CFU) or a higher dose (1 × 104 CFU) of L. monocytogenes and determined the serum levels of TNF-α at both 24 and 48 h postinfection. We did not observe any difference in serum TNF-α concentrations at either time point in HSF1+/+ and HSF1−/− mice challenged with the lower dose of L. monocytogenes (Fig. 5B). Likewise, 24 h after infection with the higher challenge dose, both groups of mice showed a modest increase in TNF-α secretion. However, by 48 h postinfection, the serum TNF-α levels had continued to increase in HSF1-deficient animals but not in the HSF1+/+ littermate controls (Fig. 5B). We also examined serum IFN-γ concentrations in the same mice and found that IFN-γ levels were slightly elevated in HSF1-deficient mice compared to those in HSF1+/+ animals; however, this difference was not statistically significant. No differences were observed for serum levels of IL-2, IL-4, or IL-5 in these mice (data not shown). Together, these results suggested that HSF1 could be important for regulating TNF-α secretion at the peak of the fever response but only when the bacterial load had increased beyond a threshold level of ∼1 × 104 CFU.
Neutralization of TNF-α results in increased survival of HSF1−/− mice.
To investigate the possibility that overproduction of TNF-α resulted in the premature death of HSF1-deficient mice during high-dose L. monocytogenes infection, we used neutralizing antibodies against TNF-α and looked to see if we could prolong the survival of HSF1−/− mice. Since TNF-α is known to be required for clearance of L. monocytogenes (16, 30), it was important to identify an appropriate dose of neutralizing antibody that would reduce, but not eliminate, serum TNF-α. To do this, we injected either 1 μg or 20 μg of anti-TNF-α antibody into groups of HSF1+/+ and HSF1−/− mice 2 h before infecting the animals with L. monocytogenes. As shown in Fig. 6A, treatment with 1 μg of anti-TNF-α antibody reduced the serum TNF-α concentration of HSF1−/− mice to approximately the same level as that observed in wild-type animals 48 h postinfection. However, treatment with 20 μg of neutralizing antibody reduced serum TNF-α to levels that were 3-fold lower than those observed in the HSF1+/+ mice. Interestingly, treatment with 1 μg of TNF-α-neutralizing antibody also reduced serum IFN-γ levels in HSF1-deficient mice to values that were comparable to those for the wild-type animals (Fig. 6A).
FIG. 6.

Neutralization of TNF-α prolongs the survival of HSF1-deficient mice infected with a high dose of L. monocytogenes. Male HSF1+/+ (WT) and HSF1−/− (KO) mice were given (intraperitoneally) 1 μg of either anti-TNF-α or an isotype control antibody (Ab), and 2 h later all groups of mice were infected (i.v.) with 1 × 104 CFU of L. monocytogenes. (A) Blood was collected from the tail vein 48 hpi, and the amounts of TNF-α and IFN-γ present in the serum were determined. Representative data from one of four experiments are shown. (B) Pooled survival data from 4 separate experiments using male wild-type mice given isotype control Ab (n = 11), KO males given the isotype control (n = 13), or male KO mice given a TNF-neutralizing Ab (n = 14) are shown. Survival curves for the KO mouse sample groups were significantly different (P = 0.0082) as determined by log rank analysis of the Kaplan-Meier survival plots.
The pooled survival data from several small-scale TNF-α neutralization experiments are shown in Fig. 6B. In each experiment, one group of HSF1−/− mice received 1 μg of anti-TNF-α antibody prior to L. monocytogenes infection, and control groups of both wild-type and HSF1-deficient mice were given an isotype control antibody. As shown in previous experiments, it took 4 days until any deaths were observed in the HSF1+/+ group, and more than 60% of the mice survived infection with the challenge dose of ∼1 × 104 CFU. In contrast, the median survival time for HSF1-deficient mice given the isotype control antibody was 2.74 days. Pretreatment with the TNF-neutralizing antibody prolonged the median survival time for HSF1−/− mice to 5.05 days and resulted in an overall greater percentage of mice that were able to survive the infection (Fig. 6B). Thus, the single injection of anti-TNF-α antibody prolonged the survival of these animals by more than 2 days. These data indicated that neutralization of TNF-α could rescue HSF1−/− mice from rapid death following L. monocytogenes infection and suggested that HSF1 may be important for regulating TNF-α levels during bacterial infection.
DISCUSSION
TNF-α is a critical cytokine needed for clearance of L. monocytogenes infection. TNF-knockout mice demonstrate increased susceptibility to Listeria (16, 28, 30), and treatment with large quantities of neutralizing anti-TNF-α antibodies converted a sublethal L. monocytogenes infection into a lethal infection, characterized by undetectable levels of TNF-α in the serum and uncontrolled bacterial growth (27). However, systemic overproduction of TNF-α and other proinflammatory cytokines can rapidly lead to septic shock and death. Beutler et al. showed that partial neutralization of TNF could protect mice from the lethal effects of endotoxin exposure, a treatment that is known to induce a substantial TNF-α response (2). These studies indicate that the level of TNF-α produced during infection must be finely regulated to allow for bacterial clearance while avoiding lethality, and in fact, multiple mechanisms are known to be involved in the control of TNF-α activity (14, 41, 43). In this report, we show that the master stress regulator HSF1 also plays an important role in maintaining an appropriate balance of TNF-α production during infection with the Gram-positive intracellular bacterial pathogen L. monocytogenes.
TNF-α can be produced by a variety of cell types, including macrophages, dendritic cells, neutrophils, and both T and B lymphocytes. Grivennikov et al. showed that mice with a specific block in TNF-α expression in macrophages and neutrophils had drastically reduced serum levels of TNF-α during LPS-induced shock compared with wild-type mice (12). A lack of TNF-α-expressing lymphocytes did not affect the serum levels of TNF-α in those experiments. This suggests that macrophages and/or neutrophils are the critical cell type that must regulate TNF-α production during a fever response in order to prevent septic shock. In fact, the Hasday group showed that transcription of TNF-α was inhibited in a variety of different monocyte and macrophage cell types exposed to physiologic fever temperature (39.5°C) and that this transcriptional regulation was dependent on HSF1 (9, 10, 37).
Although HSF1 is thought of primarily as an activator of heat shock protein expression, previous studies have shown that HSF1 can also act as a transcriptional repressor when bound to heat shock elements upstream of genes encoding proinflammatory cytokines. For example, Cahill et al. showed that HSF1 repressed expression of IL-1β during heat shock of human monocyte-like cells (5). Full activation of HSF1 occurs in most cell types only during severe thermal stress (temperatures of >42°C); however, Singh et al. reported that HSF1 could be activated to an alternate DNA-binding form able to repress both TNF-α and IL-1β production in RAW 264.7 macrophages that were exposed to both fever-range temperature (39.5°C) and LPS (36). Since we observed increased serum TNF-α levels in HSF1-deficient mice infected with the Gram-positive pathogen L. monocytogenes during the time period that corresponded to peak fever temperatures, our results are consistent with their in vitro model and suggest that bacterial products other than LPS can also modulate HSF1 repressor activity.
The rapid death that we observed in HSF1-deficient mice infected with high doses of L. monocytogenes was similar to the mortality observed by Xiao et al. after injection with LPS (42), a treatment that is known to induce a large TNF-α response in mice. Although the TNF-α neutralization experiments that we performed strongly suggested that TNF-α was a key mediator in the rapid death of HSF1−/− mice, other cytokines and inflammatory mediators are likely to be involved as well. This is illustrated by the observation that the serum TNF-α levels of individual HSF1-deficient mice varied considerably following challenge with a high dose of L. monocytogenes, despite the fact that infection with that inoculum was uniformly fatal for HSF1-deficient mice. The animals used in these experiments were on a mixed 129Sv/BALB/c strain background, and our attempts to backcross the animals to either C57BL/6 or BALB/c mice failed. This suggests that there is at least one other gene present in the 129Sv strain that can partially compensate for a lack of HSF1. Presumably this gene product can reduce TNF-α production during the fever response to levels seen in wild-type animals. However, misregulation of other proinflammatory cytokines such as IL-1β may contribute more to the rapid death of those animals.
In this study, HSF1-deficient mice were fully capable of clearing a primary L. monocytogenes infection as long as the challenge dose was less than 1 × 104 CFU, and HSF1 regulatory function appeared to be required only when the mice were challenged with larger inocula. Since the TNF-α response during L. monocytogenes infection is typically proportionate to the challenge dose, we speculate that there is a threshold of Listeria-derived bacterial products that must be surpassed in order to trigger a serum TNF-α level high enough that would necessitate HSF1-mediated repression of TNF-α expression to prevent lethality during the peak of the fever response.
Although we previously showed that T cells harvested from HSF1-deficient mice were unable to proliferate efficiently in vitro when incubated at 39.5°C, in this study there was no significant difference in the antigen-specific CD8+ T cell response of HSF1−/− mice infected with L. monocytogenes compared with that of HSF1+/+ mice. We conclude that HSF1 is not required for normal activation of naïve T cells during primary infection or for the reactivation of memory T cells during secondary infection with L. monocytogenes. During primary L. monocytogenes infection, antigens are presented to T cells during the first 12 to 24 h postinfection, and after that time point, CD8+ T cells follow preprogrammed kinetics of activation, differentiation, and expansion, resulting in a peak number of antigen-specific T cells 7 days postinfection (1, 25). Since we show here that the fever response in Listeria-infected mice resulted in elevated temperatures that were maintained only between 24 and 72 h postinfection, the bulk of clonal expansion of the T cells probably occurred during a period in which there was no fever in vivo. It is not clear whether mice have a significant fever response during secondary L. monocytogenes infection; however, it seems likely that memory CD8+ T cells would be activated and begin proliferating before the core body temperature had risen significantly.
Acknowledgments
We thank Denise McElroy and Greg Baumann for technical assistance.
This work was supported by Public Health Service grants from the National Eye Institute (EY14060) to J.G.W. and from the National Center for Research Resources (P20 RR20171) to S.E.F.D.
Editor: J. N. Weiser
Footnotes
Published ahead of print on 18 October 2010.
REFERENCES
- 1.Badovinac, V. P., and J. T. Harty. 2002. CD8(+) T-cell homeostasis after infection: setting the ‘curve’. Microbes Infect. 4:441-447. [DOI] [PubMed] [Google Scholar]
- 2.Beutler, B., I. W. Milsark, and A. C. Cerami. 1985. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science 229:869-871. [DOI] [PubMed] [Google Scholar]
- 3.Buchmeier, N. A., and R. D. Schreiber. 1985. Requirement of endogenous interferon-gamma production for resolution of Listeria monocytogenes infection. Proc. Natl. Acad. Sci. U. S. A. 82:7404-7408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Busch, D. H., and E. G. Pamer. 1999. T lymphocyte dynamics during Listeria monocytogenes infection. Immunol. Lett. 65:93-98. [DOI] [PubMed] [Google Scholar]
- 5.Cahill, C. M., W. R. Waterman, Y. Xie, P. E. Auron, and S. K. Calderwood. 1996. Transcriptional repression of the prointerleukin 1beta gene by heat shock factor 1. J. Biol. Chem. 271:24874-24879. [PubMed] [Google Scholar]
- 6.Czuprynski, C. J., and J. F. Brown. 1990. Effects of purified anti-Lyt-2 mAb treatment on murine listeriosis: comparative roles of Lyt-2+ and L3T4+ cells in resistance to primary and secondary infection, delayed-type hypersensitivity, and adoptive transfer of resistance. Immunology 71:107-112. [PMC free article] [PubMed] [Google Scholar]
- 7.Dinarello, C. A. 2004. Infection, fever, and exogenous and endogenous pyrogens: some concepts have changed. J. Endotoxin Res. 10:201-222. [DOI] [PubMed] [Google Scholar]
- 8.Dunn, P. L., and R. J. North. 1991. Resolution of primary murine listeriosis and acquired resistance to lethal secondary infection can be mediated predominantly by Thy-1+ CD4- CD8- cells. J. Infect. Dis. 164:869-877. [DOI] [PubMed] [Google Scholar]
- 9.Ensor, J. E., E. K. Crawford, and J. D. Hasday. 1995. Warming macrophages to febrile range destabilizes tumor necrosis factor-alpha mRNA without inducing heat shock. Am. J. Physiol. 269:C1140-C1146. [DOI] [PubMed] [Google Scholar]
- 10.Fairchild, K. D., R. M. Viscardi, L. Hester, I. S. Singh, and J. D. Hasday. 2000. Effects of hypothermia and hyperthermia on cytokine production by cultured human mononuclear phagocytes from adults and newborns. J. Interferon Cytokine Res. 20:1049-1055. [DOI] [PubMed] [Google Scholar]
- 11.Gothard, L. Q., M. E. Ruffner, J. G. Woodward, O. K. Park-Sarge, and K. D. Sarge. 2003. Lowered temperature set point for activation of the cellular stress response in T-lymphocytes. J. Biol. Chem. 278:9322-9326. [DOI] [PubMed] [Google Scholar]
- 12.Grivennikov, S. I., A. V. Tumanov, D. J. Liepinsh, A. A. Kruglov, B. I. Marakusha, A. N. Shakhov, T. Murakami, L. N. Drutskaya, I. Forster, B. E. Clausen, L. Tessarollo, B. Ryffel, D. V. Kuprash, and S. A. Nedospasov. 2005. Distinct and nonredundant in vivo functions of TNF produced by T cells and macrophages/neutrophils: protective and deleterious effects. Immunity 22:93-104. [DOI] [PubMed] [Google Scholar]
- 13.Habicht, G. S. 1981. Body temperature in normal and endotoxin-treated mice of different ages. Mech. Ageing Dev. 16:97-104. [DOI] [PubMed] [Google Scholar]
- 14.Han, J., T. Brown, and B. Beutler. 1990. Endotoxin-responsive sequences control cachectin/tumor necrosis factor biosynthesis at the translational level. J. Exp. Med. 171:465-475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hasday, J. D., and I. S. Singh. 2000. Fever and the heat shock response: distinct, partially overlapping processes. Cell Stress Chaperones 5:471-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Havell, E. A. 1989. Evidence that tumor necrosis factor has an important role in antibacterial resistance. J. Immunol. 143:2894-2899. [PubMed] [Google Scholar]
- 17.Hiromatsu, K., Y. Yoshikai, G. Matsuzaki, S. Ohga, K. Muramori, K. Matsumoto, J. A. Bluestone, and K. Nomoto. 1992. A protective role of gamma/delta T cells in primary infection with Listeria monocytogenes in mice. J. Exp. Med. 175:49-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inouye, S., H. Izu, E. Takaki, H. Suzuki, M. Shirai, Y. Yokota, H. Ichikawa, M. Fujimoto, and A. Nakai. 2004. Impaired IgG production in mice deficient for heat shock transcription factor 1. J. Biol. Chem. 279:38701-38709. [DOI] [PubMed] [Google Scholar]
- 19.Kaufmann, S. H., and C. H. Ladel. 1994. Role of T cell subsets in immunity against intracellular bacteria: experimental infections of knock-out mice with Listeria monocytogenes and Mycobacterium bovis BCG. Immunobiology 191:509-519. [DOI] [PubMed] [Google Scholar]
- 20.Lefrancois, L., A. Marzo, and K. Williams. 2003. Sustained response initiation is required for T cell clonal expansion but not for effector or memory development in vivo. J. Immunol. 171:2832-2839. [DOI] [PubMed] [Google Scholar]
- 21.Loomis, W. P., and M. N. Starnbach. 2006. Chlamydia trachomatis infection alters the development of memory CD8+ T cells. J. Immunol. 177:4021-4027. [DOI] [PubMed] [Google Scholar]
- 22.Lorber, B. 1997. Listeriosis. Clin. Infect. Dis. 24:1-9. [DOI] [PubMed] [Google Scholar]
- 23.Lutz, M. B., N. Kukutsch, A. L. Ogilvie, S. Rossner, F. Koch, N. Romani, and G. Schuler. 1999. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods 223:77-92. [DOI] [PubMed] [Google Scholar]
- 24.McMillan, D. R., X. Xiao, L. Shao, K. Graves, and I. J. Benjamin. 1998. Targeted disruption of heat shock transcription factor 1 abolishes thermotolerance and protection against heat-inducible apoptosis. J. Biol. Chem. 273:7523-7528. [DOI] [PubMed] [Google Scholar]
- 25.Mercado, R., S. Vijh, S. E. Allen, K. M. Kerksiek, I. M. Pilip, and E. G. Pamer. 2000. Early programming of T cell populations responding to bacterial infection. J. Immunol. 165:6833-6839. [DOI] [PubMed] [Google Scholar]
- 26.Murapa, P., S. Gandhapudi, H. S. Skaggs, K. D. Sarge, and J. G. Woodward. 2007. Physiological fever temperature induces a protective stress response in T lymphocytes mediated by heat shock factor-1 (HSF1). J. Immunol. 179:8305-8312. [DOI] [PubMed] [Google Scholar]
- 27.Nakane, A., T. Minagawa, and K. Kato. 1988. Endogenous tumor necrosis factor (cachectin) is essential to host resistance against Listeria monocytogenes infection. Infect. Immun. 56:2563-2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pfeffer, K., T. Matsuyama, T. M. Kundig, A. Wakeham, K. Kishihara, A. Shahinian, K. Wiegmann, P. S. Ohashi, M. Kronke, and T. W. Mak. 1993. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 73:457-467. [DOI] [PubMed] [Google Scholar]
- 29.Pope, C., S.-K. Kim, A. Marzo, D. Masopust, K. Williams, J. Jiang, H. Shen, and L. LeFrancois. 2001. Regulation of the CD8 T cell response to Listeria monocytogenes infection. J. Immunol. 166:3402-3409. [DOI] [PubMed] [Google Scholar]
- 30.Rothe, J., W. Lesslauer, H. Lotscher, Y. Lang, P. Koebel, F. Kontgen, A. Althage, R. Zinkernagel, M. Steinmetz, and H. Bluethmann. 1993. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature 364:798-802. [DOI] [PubMed] [Google Scholar]
- 31.Saito, M., S. Nameda, N. N. Miura, Y. Adachi, and N. Ohno. 2008. SPG/IND-induced septic shock in a LPS-low responder strain, C3H/HeJ mice. Microb. Pathog. 44:402-409. [DOI] [PubMed] [Google Scholar]
- 32.Sarge, K. D., S. P. Murphy, and R. I. Morimoto. 1993. Activation of heat shock gene transcription by heat shock factor 1 involves oligomerization, acquisition of DNA-binding activity, and nuclear localization and can occur in the absence of stress. Mol. Cell. Biol. 13:1392-1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sarge, K. D., V. Zimarino, K. Holm, C. Wu, and R. I. Morimoto. 1991. Cloning and characterization of two mouse heat shock factors with distinct inducible and constitutive DNA-binding ability. Genes Dev. 5:1902-1911. [DOI] [PubMed] [Google Scholar]
- 34.Sasaki, T., M. Mieno, H. Udono, K. Yamaguchi, T. Usui, K. Hara, H. Shiku, and E. Nakayama. 1990. Roles of CD4+ and CD8+ cells, and the effect of administration of recombinant murine interferon gamma in listerial infection. J. Exp. Med. 171:1141-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Siddique, I. H., L. C. Ying, and B. B. Robinson. 1969. Hematological and febrile responses of rabbits to listerial hemolysins. Can. J. Comp. Med. 33:292-296. [PMC free article] [PubMed] [Google Scholar]
- 36.Singh, I. S., J. R. He, L. Hester, M. J. Fenton, and J. D. Hasday. 2004. Bacterial endotoxin modifies heat shock factor-1 activity in RAW 264.7 cells: implications for TNF-alpha regulation during exposure to febrile range temperatures. J. Endotoxin Res. 10:175-184. [DOI] [PubMed] [Google Scholar]
- 37.Singh, I. S., R. M. Viscardi, I. Kalvakolanu, S. Calderwood, and J. D. Hasday. 2000. Inhibition of tumor necrosis factor-alpha transcription in macrophages exposed to febrile range temperature. A possible role for heat shock factor-1 as a negative transcriptional regulator. J. Biol. Chem. 275:9841-9848. [DOI] [PubMed] [Google Scholar]
- 38.Singh, V., and A. Aballay. 2006. Heat-shock transcription factor (HSF)-1 pathway required for Caenorhabditis elegans immunity. Proc. Natl. Acad. Sci. U. S. A. 103:13092-13097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vlach, K. D., J. W. Boles, and B. G. Stiles. 2000. Telemetric evaluation of body temperature and physical activity as predictors of mortality in a murine model of staphylococcal enterotoxic shock. Comp. Med. 50:160-166. [PubMed] [Google Scholar]
- 40.Wing, E. J., and S. H. Gregory. 2002. Listeria monocytogenes: clinical and experimental update. J. Infect. Dis. 185:S18-S24. [DOI] [PubMed] [Google Scholar]
- 41.Xanthoulea, S., M. Pasparakis, S. Kousteni, C. Brakebusch, D. Wallach, J. Bauer, H. Lassmann, and G. Kollias. 2004. Tumor necrosis factor (TNF) receptor shedding controls thresholds of innate immune activation that balance opposing TNF functions in infectious and inflammatory diseases. J. Exp. Med. 200:367-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiao, X., X. Zuo, A. A. Davis, D. R. McMillan, B. B. Curry, J. A. Richardson, and I. J. Benjamin. 1999. HSF1 is required for extra-embryonic development, postnatal growth and protection during inflammatory responses in mice. EMBO J. 18:5943-5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu, W., J. S. Downey, J. Gu, F. Di Padova, H. Gram, and J. Han. 2000. Regulation of TNF expression by multiple mitogen-activated protein kinase pathways. J. Immunol. 164:6349-6358. [DOI] [PubMed] [Google Scholar]