

Abstract
Binding of the complement inhibitor factor H (fH) to the surface of Neisseria meningitidis is critical for evasion of innate host defenses. The meningococcal vaccine candidate factor H-binding protein (fHbp) serves as an fH ligand. We prepared 16 recombinant fHbp natural sequence variants. By enzyme-linked immunosorbent assay (ELISA), the variants from a New Zealand epidemic strain (fHbp ID 14) and from an endemic United Kingdom strain (ID 15) showed 10-fold lower fH binding than a reference fHbp from an epidemic Norwegian strain (ID 1). By surface plasmon resonance, association rate constants (ka) for fHbp ID 14 and 15 were similar to those for ID 1, but dissociation rate constants (kd) were 4- to 10-fold higher than those for ID 1. To determine the effect of fH affinity on fHbp fitness, we prepared isogenic mutants of strain H44/76 that expressed fHbp ID 1, 14, or 15. By flow cytometry, mutants expressing fHbp ID 14 or 15 had lower fH binding than ID 1. When incubated in plasma or blood of nonimmune donors, all three mutants showed similar increases in CFU/ml. In contrast, an isogenic fHbp knockout mutant, which grew well in broth, was rapidly killed in plasma or blood. Thus, although fHbp expression was required for survival of strain H44/76 in blood or plasma, expression of two natural fHbp sequence variants with lower fH affinity had minimal or no effect on nonimmune clearance. One reason may be the high fH concentrations in normal serum, which favor saturation of fH binding to fHbp, even when dissociation rates varied over 10-fold.
Neisseria meningitidis is a Gram-negative bacterium that commonly inhabits the human nasopharynx as a harmless commensal organism. Certain strains possess virulence factors that contribute to penetration of host cellular barriers (12) and evasion of immunity. These can lead to invasion and replication of the organism in the bloodstream (15) and sepsis and meningitis (24). Factor H-binding protein (fHbp), formerly known as GNA1870 (17, 27) or LP2086 (7, 28), is a sparse lipoprotein that binds the complement-inhibitor protein, factor H (fH), to the bacterial cell surface (16). Binding of fH leads to inhibition of the alternative complement pathway and enhances the ability of the organism to survive in nonimmune human blood (16, 20, 22, 26).
Amino acid sequence variants of fHbp have been classified by phylogenetic analyses into two subfamilies, designated A and B, by Fletcher et al. (7) or three variant groups described by Masignani et al. (17). Subfamily A contains variant groups 2 and 3 (v.2 and v.3), and subfamily B contains variant group 1 (v.1). More recently, Beernink and Granoff determined that fHbp had a modular architecture consisting of five variable segments, each derived from one of two genetic lineages (3). Nearly all prevalent disease-causing meningococcal isolates had fHbp sequence variants that could be classified into one of six modular groups, designated I to VI, based on different combinations of the variable modular segments (19). Four of the six modular groups (approximately 40% of all sequence variants) were natural chimeric proteins with segments derived from different lineages.
Little information is available on the effect of fHbp sequence variation on binding to fH. In one study, a representative fHbp from each of the three variant groups described by Masignani et al. (17) (i.e., modular groups I, II, and VI) was reported to bind fH (16), but binding of fH to fHbp variants from other modular groups has not been reported. In the present study, we prepared recombinant fHbps representative of sequence variants from different modular groups or expressed by prevalent disease-causing strains. Given the importance of fH binding on evasion of innate immune defenses, we were surprised by large differences in fH binding by different sequence variants. To investigate the effect of fH binding on fHbp fitness, we prepared isogenic mutants expressing different amounts of fHbp, or expressing fHbp variants with lower or higher fH binding, and compared their relative abilities to survive in nonimmune human blood or plasma.
MATERIALS AND METHODS
Cloning, expression, and purification of fHbp proteins.
Genes encoding 16 fHbp amino acid sequence variants (Table 1) were cloned into Escherichia coli expression plasmid pET21b as described previously (17). The fHbp genes in modular group I were amplified by PCR using forward and reverse primers v.1 NdeI and v.1 XhoI, respectively; those encoding modular group II proteins were amplified using primers v.2 NdeI and v.3 HindIII; and those from modular group IV were amplified using primers v.2 NdeI and v.1 XhoI. The corresponding genes encoding modular groups III and V through IX proteins were amplified using primers v.2 NdeI and v.2 XhoI. The plasmids were transformed into E. coli strain BL21(DE3), and 1-liter cultures were grown at 37°C to an optical density at 600 nm (OD600) of 0.5 and induced for 3 h. The proteins were purified by affinity chromatography using Ni-nitrilotriacetic acid (NTA) agarose (Qiagen, Valencia, CA) under native conditions using the manufacturer's protocols. Fractions containing fHbp were dialyzed against phosphate-buffered saline (PBS) (Roche, Indianapolis, IN) and stored in aliquots at −30°C prior to use.
TABLE 1.
Characteristics of factor H-binding protein variants
| fHbp IDa | Subfamilyb | Variant groupc | Modular groupd | Percentage among group B disease isolatese |
fH-binding ratiof | ||
|---|---|---|---|---|---|---|---|
| United States | United Kingdom | France | |||||
| 1 | B | 1 | I | 29 | 4 | 20 | 1.0 |
| 4 | B | 1 | I | 7 | 24 | 7 | 0.3 |
| 6 | B | 1 | I | 0 | 0 | 0 | 0.7 |
| 13 | B | 1 | I | 5 | 10 | 2 | 0.4 |
| 14 | B | 1 | I | 5 | 7 | 23 | 0.1 |
| 15 | B | 1 | IV | 0.3 | 22 | 6 | 0.1 |
| 55 | B | 1 | IV | 0 | 0 | 0 | 1.6 |
| 22 | A | 2 | III | 0.3 | 0.2 | 0.4 | 2.9 |
| 19 | A | 2 | VI | 13 | 8 | 19 | 1.9 |
| 77 | A | 2 | VI | 0 | 0 | 0 | 1.6 |
| 28 | A | 3 | II | 0 | 0 | 0 | 1.0 |
| 175 | A | 3 | IX | 0 | 0 | 0 | 0.8 |
| 45 | A | 3 | V | 0.3 | 4 | 1 | 5.2 |
| 79 | A | 3 | V | 0 | 0 | 0 | 3.0 |
| 67 | A | 3 | VIII | 0 | 0 | 0 | 1.8 |
| 207 | A/B | 1/2 | VII | 0 | 0 | 0 | 1.0 |
fHbp ID from the fHbp database at http://pubmlst.org/neisseria/fHbp/.
Variant group as defined by Masignani et al. (17).
From population-based surveillance of group B disease in the United States (excluding an epidemic in Oregon), the United Kingdom, and France (18). For fHbp variants not observed in any of the three countries listed, the source isolates came from other countries (18).
Ratio of the respective concentrations of purified fH to obtain an OD405 of 1.0 with fHbp ID 1 compared to that of the test fHbp as measured by ELISA. Ratios above 1 indicate higher binding; ratios below 1 indicate lower binding.
Construction of mutant meningococcal strains.
Capsular group B strain NZ98/254 was used to create fHbp knockout (KO) (13, 26) or overexpressed mutants as previously described (26). For the inactivated mutant, the fHbp gene was replaced by an erythromycin resistance cassette carried on pBSΔgna1870erm (17). For overexpression, the NZ98/254 fHbp KO strain was transformed with plasmid pComP1523-fHbp ID 14 that integrated into the chromosome and expressed fHbp under the control of the strong promoter from gene nmb1523 (13, 26).
Capsular group B strain H44/76 naturally expresses fHbp in the variant 1 (v.1) group (ID 1, as classified in the fHbp database at http://pubmlst.org/neisseria/fHbp/). For preparation of H44/76 mutants that expressed fHbp v.1 variants with low or high fH binding, we first transformed the organism with a PCR-amplified gene from group B strain M1239 encoding fHbp ID 28 in the v.3 group. Transformants with recombinant v.3 fHbp were selected by adding 50% human complement and 100 μg/ml of anti-fHbp monoclonal antibodies (MAbs) JAR 4 and 5, which together were bactericidal against strains with fHbp v.1 but not v.3 (4). Transformants were screened for the loss of the JAR 5 epitope by a modified whole-cell colony blot procedure using anti-fHbp MAb JAR 5. Binding of JAR 5 was detected by anti-IgG2b IRDye secondary antibody (Rockland Immunochemicals, Gilbertsville, PA). JAR 5-negative clones were isolated and sequenced to confirm in-frame integration of the recombinant fHbp v.3 gene. The H44/76 fHbp v.3 mutant strain was subsequently transformed to express the original fHbp amino acid sequence variant ID 1 or one of two low fH-binding sequence variants, ID 14 and 15 (both in the v.1 group). In brief, forward and reverse primers were designed to amplify by PCR the fHbp open reading frames from wild-type strains MC58 (ID 1), NZ98/254 (ID 14), and NM452 (ID 15). Transformants with recombinant v.1 fHbp were selected as described above except that anti-fHbp MAbs JAR 13 and 31 (4), which together were bactericidal against the mutant strain with fHbp v.3, were used instead of JAR 4 and JAR 5. Transformants were screened for expression of fHbp v.1 by colony blotting with the anti-fHbp v.1 MAb JAR 5, and the sequences of the respective v.1 genes were verified as described above.
Binding of fH to recombinant proteins.
Microtiter plates were coated with purified His6-tagged recombinant fHbp antigens (2 μg/ml) as previously described (4). Serial dilutions were made of purified, full-length human fH (Complement Technology, Inc., Tyler, TX) or, as a control for binding of the protein to the well, mouse anti-fHbp MAbs specific for fHbp in the variant 1 (JAR 5) or variant 2 or 3 (JAR 31) group (2, 4, 27). After being washed, bound fH was detected with sheep anti-fH polyclonal antiserum (1:2,000 dilution; LifeSpan Biosciences, Seattle, WA) followed by anti-sheep IgG conjugated with alkaline phosphatase (1:5,000 dilution; Sigma-Aldrich, St. Louis, MO). Bound anti-fHbp MAbs were detected by anti-mouse IgG conjugated with alkaline phosphatase.
Factor H binding kinetics.
The kinetics of fH binding to fHbp were measured by surface plasmon resonance using a Biacore X/100 instrument (GE Healthcare, Piscataway, NJ). Purified human fH (Calbiochem, San Diego, CA) was coupled to a CM-5 biosensor chip (GE Healthcare) to a density of about 2,000 response units. Recombinant fHbp variants, which were purified as described previously (2, 17), were flowed over the fH-bound chip at six concentrations ranging from 0.016 to 0.5 μM, each performed at least three times. After the off-rates were measured for 10 min for each analyte injection, complete regeneration of the surface was achieved with one 60-s injection of 100 mM glycine and 3 M NaCl, pH 2.0. The interaction affinity, as described by the equilibrium dissociation constant (KD), was determined locally by fitting to the kinetic simultaneous kd/ka model (kd, dissociation rate; ka, association rate) using a model of equimolar stoichiometry.
Flow cytometry.
Binding of human fH or mouse anti-fHbp antibodies to the surface of live meningococci was measured by indirect fluorescence flow cytometry, which was performed as described previously (26, 27). Controls in the assay included a mouse MAb, SEAM 12, which is specific for the group B capsular polysaccharide (8), or, as a negative control, sera from mice immunized with Freund's adjuvant without a vaccine antigen.
Survival of N. meningitidis in human blood and plasma.
The ability of N. meningitidis mutants to survive and grow in human blood or plasma was tested in a 96-well microtiter plate format as described elsewhere (19a, 25). The study was approved by the Institutional Review Board of Children's Hospital & Research Center at Oakland. In brief, after obtaining written informed consent, blood from a healthy adult was obtained using a syringe containing recombinant hirudin (lepirudin, 28 μg/ml final concentration; Baxter Healthcare, Deerfield, IL) as the anticoagulant to avoid the effects of heparin on activation of complement proteins (23). To each well of the microtiter plate were added 90 μl of plasma or blood and 10 μl of PBS buffer containing approximately 800 CFU of bacteria. All assays were performed in triplicate. The microtiter plates were incubated for 4 h at 37°C in 5% CO2 on a MS 3 digital minishaker (IKA, Wilmington, NC) with agitation at 500 oscillations per min. Samples were removed from the wells, and serial dilutions were spread on chocolate agar plates, which were incubated at 37°C in 5% CO2.
RESULTS
Differences in fH binding among fHbp sequence variants.
We prepared recombinant proteins from fHbp genes encoding 16 amino acid sequence variants representative of proteins from different modular groups and/or prevalent group B strains (Table 1). The relatedness of the different proteins with respect to percent differences in amino acid identity is illustrated in the phylogram in Fig. 1. Seven of the fHbp sequence variants were assigned to the variant group 1 as described by Masignani et al. (17), three to the variant group 2, and five to the variant group 3. One sequence variant, ID 207, was located between variant groups 1 and 2.
FIG. 1.

Phylogram of 16 fHbp natural amino acid sequence variants investigated for fH binding (Table 1). Each sequence variant is identified by a number assigned in the fHbp peptide database at http://pubmlst.org/neisseria/fHbp/. The ratio of fH binding of each variant relative to ID 1 is shown in parentheses (Table 1). Ratios above 1 indicate higher binding; ratios below 1 indicate lower binding. The phylogram was constructed using the server at http://www.phylogeny.fr (6) as described previously (3). The scale bar represents 5 changes per 100 amino acid residues.
Each of the recombinant proteins was adsorbed to wells of a microtiter plate, and we measured concentration-dependent binding of soluble human fH by ELISA. The respective results were compared to that for fHbp ID 1, which served as a positive control. Two of the proteins, fHbp ID 14 and 15, showed 10-fold-lower concentration-dependent binding than fHbp ID 1 (Fig. 2 A). In contrast, binding of an anti-fHbp MAb to each of these proteins was indistinguishable from one another, which showed that comparable amounts of the respective recombinant proteins were adsorbed to the wells (Fig. 2B). Among the different sequence variants, there also were examples of proteins with higher fH binding than fHbp ID 1, for example, fHbp ID 79 and 45 (Fig. 2C and D). The respective ratios of fH binding for each of the variants compared with fHbp ID 1 are summarized in Table 1 and Fig. 1.
FIG. 2.

(A and C) Binding of human fH to recombinant fHbp as measured by ELISA. (A) ID 1 protein was encoded by an fHbp v.1 gene from strain H44/76 and served as a positive control. The ID 14 and 15 v.1 proteins showed ∼10-fold lower binding than ID 1. (C) ID 28 was encoded by an fHbp v.3 gene from strain M1239 and served as a positive control. ID 79 and 45 v.3 proteins showed ∼4- and 10-fold higher fH binding, respectively, than ID 28. (B and D) Binding of control anti-fHbp MAbs to the respective fHbp variants. (B) JAR 5 (specific for fHbp v.1 proteins) binding to the respective fHbp variants in panel A. (D) JAR 31 (specific for fHbp v.2 and v.3) binding to the respective fHbp variants in panel C.
In surface plasmon resonance experiments, there were no significant differences between the respective fH association rates (ka) of fHbp ID 14 and 15, which showed lower fH binding by ELISA, and that of fHbp ID 1 (Table 2). The dissociation rates (kd), however, of fHbp ID 14 and 15 were 10- and 4-fold higher, respectively, than that of fHbp ID 1 (P ≤ 0.01, Table 2). The resulting KD of ID 14 was ∼8-fold higher than that of ID 1 (P = 0.058), and that of ID 15 was ∼4-fold higher than that of ID 1 (P = 0.036).
TABLE 2.
Binding constants for binding of fH to different fHbp sequence variants as determined by surface plasmon resonancea
| fHbp IDb | Mean ± SE |
||
|---|---|---|---|
| kac | kdd | KDe | |
| 1 | (1.43 ± 0.21) × 105 | (0.65 ± 0.13) × 10−2 A | (0.45 ± 0.09) × 10−7 D |
| 14 | (2.23 ± 0.28) × 105 | (7.16 ± 1.39) × 10−2 B | (3.50 ± 1.15) × 10−7 E |
| 15 | (1.63 ± 0.18) × 105 | (2.64 ± 0.33) × 10−2 C | (1.71 ± 0.39) × 10−7 F |
ka, association rate constant (1/Ms); kd, dissociation rate constant (1/s); KD, equilibrium dissociation constant (kd/ka); where M = molar and s = time in seconds. Data shown are from three independent measurements for each fHbp sequence variant.
fHbp identification number from fHbp database at http://pubmlst.org/neisseria/fHbp/ (see also Table 1).
Respective ka values are not different from each other (P > 0.05 by t test).
Respective kd values by t test: A versus B, P = 0.01; A versus C, P = 0.002; and B versus C, P = 0.03.
Respective KD values by t test: D versus E, P = 0.058; D versus F, P = 0.036; E versus F, P = 0.22.
The 21 fH contact residues inferred from the crystal structure of fHbp ID 1 complexed with a fragment of fH (21) are shown in Table S1 in the supplemental material. All of the contact residues are in either segment A, C, or E, as defined by the modular architecture described previously (3). None of these individual differences is specific for low fH binding because they also occurred in variants with high fH binding.
Effect of overexpression of fHbp ID 14 on survival of N. meningitidis strain NZ98/254 in human blood.
fHbp ID 14, which had relatively low fH affinity, was naturally expressed by an invasive group B meningococcal strain (NZ98/254) isolated from a patient during an epidemic in New Zealand (1). In a previous study, an fHbp knockout mutant of NZ98/254 could survive in human blood (26) whereas fHbp knockout mutants prepared from other isolates such as a group B epidemic strain (H44/76) from Norway were rapidly killed by human blood (26). To determine the effect of increased expression of fHbp ID 14 on the growth of strain NZ98/254 in human plasma or blood, we used an NZ98/254 mutant engineered to have increased expression of its endogenous fHbp ID 14 (26). By flow cytometry, the mutant bacteria showed increased binding by an anti-fHbp antiserum prepared against recombinant fHbp ID 14, compared with binding to the parental wild-type strain (Fig. 3 B). The NZ98/254 mutant with increased fHbp ID 14 expression also showed increased fH binding to the bacterial surface (Fig. 3C). When incubated in nonimmune human blood or plasma, the mutant with increased bound fH showed increases in CFU/ml during the 4 h of incubation that were similar to that of the wild-type strain (Fig, 3E and F, respectively). As previously observed, the corresponding fHbp KO mutant also showed good survival, which was not significantly different from that of the wild-type strain or mutant with increased fHbp expression. Thus, for a strain that did not have an absolute requirement for fHbp expression for survival in human plasma or blood, overexpression of fHbp ID 14 in strain NZ98/254 did not confer a survival advantage.
FIG. 3.

(A to C) Binding of human fH on the surface of mutants of live encapsulated N. meningitidis strain NZ98/254 as determined by indirect fluorescence flow cytometry. A wild-type (WT) strain, an fHbp knockout mutant (fHbp KO), and a mutant with overexpressed fHbp (OE fHbp) were incubated with mouse anti-capsular polysaccharide MAb (5 μg/ml) (A), polyclonal anti-fHbp ID 14 antiserum (1:1,000) (B), or human fH (10 μg/ml) (C). (D to F) Survival of strain NZ98/254 in broth (D), nonimmune human blood (donor 2) (E), or plasma (F). Symbols represent medians of three replicate values. Error bars representing ranges are too small to be visible on the log10 y axis. Similar respective results were observed when the respective wild-type and mutant strains were incubated in plasma from a second donor (donor 1; data not shown).
Effect of expression of fHbp variants with lower fH binding on survival of strain H44/76 mutants in human blood.
In a previous study, strain H44/76 (fHbp ID 1) required fHbp expression for survival in human blood or plasma (26). To determine the effect of fH affinity on survival of H44/76, we prepared three fHbp mutants, ID 14 and 15 with low fH binding (Table 1) and a mutant complemented with ID 1 with high fH binding. The three H44/76 mutants were constructed by transformation of the wild-type strain with PCR fragments and selection by survival in human complement in the presence of variant-specific MAbs. Because the incubation in human complement may have selected for other changes in these strains, we compared fHbp protein expression and fH binding by the wild-type and ID 1-complemented mutant and survival of the two strains in human plasma.
By flow cytometry, the respective binding of anti-capsular polysaccharide MAbs and binding of mouse polyclonal anti-fHbp ID 1 antiserum to bacterial cells from the two strains were indistinguishable (see Fig. S1 in the supplemental material, panels A and B, respectively). The mutant and complemented strains also showed no significant differences in binding of fH (Fig. S1, panel C), or growth in plasma samples from two donors (Fig. S1, panels E and F). Thus, by these criteria, the wild-type strain and the fHbp ID 1-complemented mutant were indistinguishable.
The three mutants with ID 14 or 15 or complemented ID 1 had similar amounts of capsule (Fig. 4 A) and fHbp (Fig. 4B) detected on their surfaces by flow cytometry. In contrast, binding of fH (10 μg/ml) to the mutants with fHbp ID 14 or 15 was on average 2.6-fold (range of 2.2 to 3.0) or 5.6-fold (range of 3.7 to 7.5) lower, respectively, than that of the H44/76-complemented mutant expressing fHbp ID 1 (Fig. 4C).
FIG. 4.
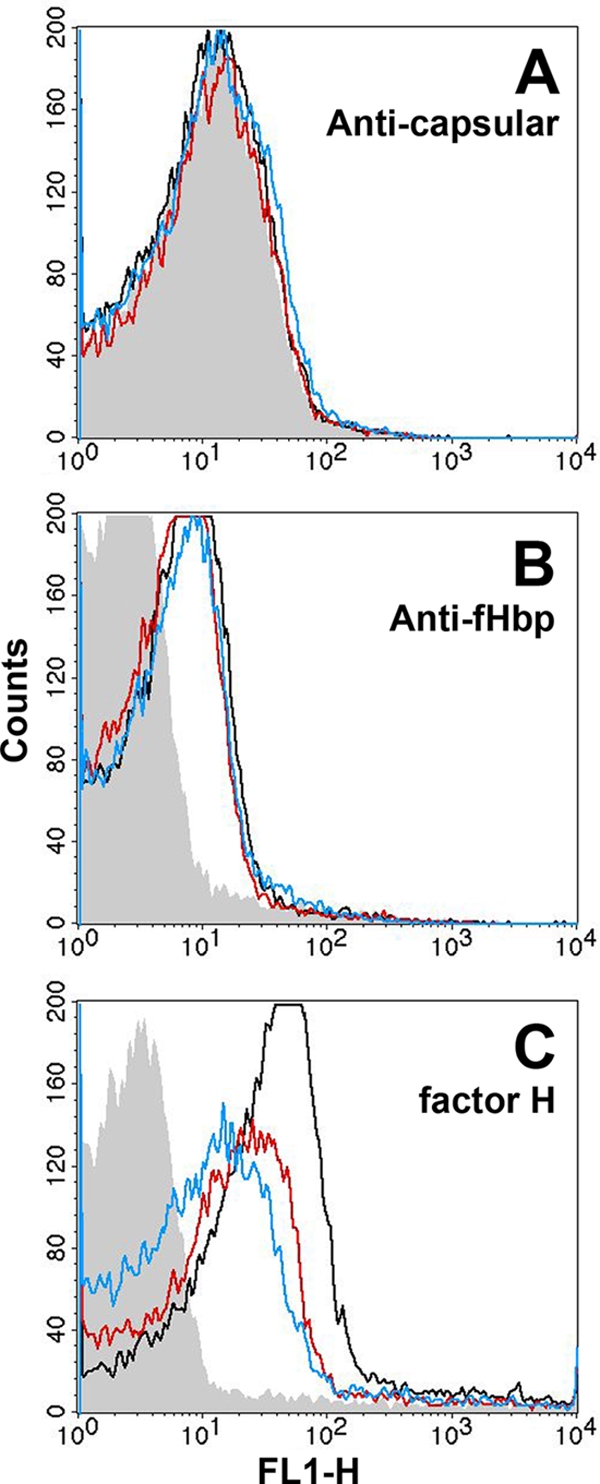
fHbp surface accessibility and binding of human fH to the surface of live bacteria from mutants of strain H44/76 as measured by indirect fluorescence flow cytometry. (A) Binding of control anti-capsular polysaccharide MAb SEAM 12 (5 μg/ml) (gray, fHbp knockout; black, complemented ID 1; red, ID 14; blue, ID 15). (B) Binding of a combination of mouse anti-fHbp mouse MAbs JAR 4 and JAR 5 (25 μg/ml each). (C) Binding of fH (10 μg/ml). Color designations in panels B and C are the same as in panel A. The respective results in each panel were replicated in two or three independent experiments.
The H44/76 fHbp knockout mutant, which showed growth in broth that was similar to that of the mutants with fHbp expression (Fig. 5 A and B), was rapidly killed by blood or plasma from both donors (panels C to G). Despite the lower fH binding of the mutants with fHbp ID 14 or 15, all three mutants reached similar cell densities (CFU/ml) at 4 h when incubated in nonimmune plasma from both donors (Fig. 5E, F, and G). There were no differences in the growth rate of the mutant with ID 14 and that of the mutant with ID 1 in blood or plasma of donor 1. The growth rate of the mutant with fHbp ID 15 was lower than that of the mutant with ID 1 or ID 14 in blood and/or plasma of donors 1 and 2 (P < 0.01; see also the legend to Fig. 5). The respective results for both donors were confirmed in independent experiments.
FIG. 5.

Survival of mutants of strain H44/76 in nonimmune human blood or plasma. The bacteria were incubated in Mueller-Hinton broth (A and B), whole blood (C and D), or plasma (E, F, and G). The error bars representing the respective ranges for triplicate values are too small to be evident for some data points. In plasma of donor 1, the mutant with fHbp ID 15 had a lower mean CFU/ml at 2 h than the mutant with fHbp ID 14 (P = 0.0004). In blood or plasma of donor 2, the mutant with fHbp ID 15 had a lower mean CFU/ml at the 2-h time point than the mutant with ID 1 (P = 0.0042 for blood and P = 0.0004 for plasma) and a lower CFU/ml at the 4-h time point for blood (P = 0.0097). Representative data from independent experiments are shown for donor 1 (panels A, C, and E compare growth of mutants with fHbp ID 1 and 14 in one experiment, whereas panel G compares mutants with fHbp ID 14 and 15 in another experiment).
DISCUSSION
Binding of fH to the surface of encapsulated N. meningitidis strains is an important mechanism for evasion of innate host defenses (9, 16, 20, 22). An important fH ligand is the meningococcal vaccine candidate fHbp (16). A conundrum, therefore, is why previous studies found wide variability in fHbp expression among isolates from patients with invasive meningococcal disease (19), and why in the present study fH binding by different recombinant fHbp amino acid sequence variants varied over a 50-fold range (i.e., up to 5-fold higher fH binding by fHbp ID 45 than by fHbp ID 1 and 10-fold lower binding by fHbp IDs 14 and 15 than by fHbp ID 1; Table 1).
To determine the effect of increasing fHbp ID 14 expression in a naturally low-fHbp-expressing strain on survival in human blood, we prepared a mutant of strain NZ98/254 that overexpressed its native fHbp sequence, ID 14. The increased fH binding by the mutant, however, did not translate into increased survival or growth in nonimmune human plasma or blood. As previously observed (26), the control NZ98/254 fHbp KO mutant, which had little or no detectable fH on its surface by flow cytometry, also grew well in human blood or plasma. Flow cytometry is a relatively insensitive method to detect binding of ligands with relatively low affinity, but it may have functional importance. It is possible that strain NZ98/254 expressed an alternative fH-binding ligand such as the recently described neisserial surface protein A (14), which allowed sufficient fH binding to the fHbp KO mutant for survival in human blood. Alternatively, the ability of N. meningitidis to bind C4bp would promote inactivation of C4b by factor I and help some strains with low fH binding to escape classical pathway complement activation (11).
To determine the effect of expression of fHbp variants with relatively low or high fH affinity on the survival of a strain that had an absolute requirement for fHbp expression for survival in human blood or plasma, we created mutants of H44/76 that expressed genes encoding fHbp ID 1, 14, or 15. We hypothesized that expression of genes encoding fHbp ID 14 or 15 with lower fH binding would result in decreased fitness and poorer survival of the mutants in blood or plasma. There was, however, no or minimal observable effects of expression of fHbp variants with lower fH affinity on survival of the mutants in human blood or plasma.
By surface plasmon resonance, the principal kinetic difference between the two fHbp variants with lower fH binding by ELISA was up to 10-fold higher fH dissociation rates than those of fHbp ID 1 (P ≤ 0.01; Table 1), which reflected less stable fH-fHbp ID 14 or 15 complexes. These kinetic studies were performed under conditions where the amount of fH was limiting. When bacteria invade the bloodstream, the high serum fH concentrations present (∼250 μg/ml) (10) would be expected to saturate binding of fH to fHbp on the surface of the organism, even for strains with fHbp variants that have increased dissociation rates. Finally, the dissociation constant (KD) between fH and fHbp ID 1 measured in the present study (45 nM) was approximately 10-fold higher than that reported by Schneider et al. (4 nM) (21). These different results may have reflected measurement of binding with a full-length fH molecule in the present study and a fragment of fH containing short consensus repeat (SCR) domains 6 and 7 in the previous study (21).
Recent data indicated that immune selection played a role in generating amino acid sequence diversity on certain surface-exposed portions of the fHbp molecule (5). Given the importance of fH binding on the ability of the bacteria to resist complement-mediated killing (16, 20), we hypothesized that preserving this fH binding function also could be important in limiting polymorphisms involving fHbp residues that affected fH binding. Our data, however, suggested that naturally low fH-binding sequence variants can be expressed by strains causing epidemic and endemic disease (Table 1), which is indirect evidence of their fHbp fitness (19). Further, mutants expressing fHbp variants with higher or lower fH binding had similar respective growth rates in nonimmune human blood or plasma. Collectively, our data suggest that fHbp mutations resulting in up to 10-fold decreased fH binding are likely to be well tolerated with respect to the ability of the organism to cause invasive disease. Thus, preserving high fH binding may not be a prerequisite for fHbp fitness during immune selection.
Supplementary Material
Acknowledgments
This work was supported, in part, by Public Health Service grants R01 AI 046464 and AI 082263 (to D.M.G.) and AI 070955 (to P.T.B.) from the National Institute of Allergy and Infectious Diseases, NIH. The work at Children's Hospital Oakland Research Institute was performed in a facility funded by Research Facilities Improvement Program grant number C06 RR 016226 from the National Center for Research Resources, NIH.
We are grateful to Paolo Costantino and Francesco Berti, Novartis Vaccines, Siena, for review of our data and helpful discussions.
Editor: F. C. Fang
Footnotes
Published ahead of print on 1 November 2010.
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1.Baker, M. G., D. R. Martin, C. E. Kieft, and D. Lennon. 2001. A 10-year serogroup B meningococcal disease epidemic in New Zealand: descriptive epidemiology, 1991-2000. J. Paediatr. Child Health 37:S13-S19. [DOI] [PubMed] [Google Scholar]
- 2.Beernink, P. T., and D. M. Granoff. 2008. Bactericidal antibody responses induced by meningococcal recombinant chimeric factor H-binding protein vaccines. Infect. Immun. 76:2568-2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beernink, P. T., and D. M. Granoff. 2009. The modular architecture of meningococcal factor H-binding protein. Microbiology 155:2873-2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beernink, P. T., J. A. Welsch, M. Bar-Lev, O. Koeberling, M. Comanducci, and D. M. Granoff. 2008. Fine antigenic specificity and cooperative bactericidal activity of monoclonal antibodies directed at the meningococcal vaccine candidate factor H-binding protein. Infect. Immun. 76:4232-4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brehony, C., D. J. Wilson, and M. C. Maiden. 2009. Variation of the factor H-binding protein of Neisseria meningitidis. Microbiology 155:4155-4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dereeper, A., V. Guignon, G. Blanc, S. Audic, S. Buffet, F. Chevenet, J. F. Dufayard, S. Guindon, V. Lefort, M. Lescot, J. M. Claverie, and O. Gascuel. 2008. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36:W465-W469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fletcher, L. D., L. Bernfield, V. Barniak, J. E. Farley, A. Howell, M. Knauf, P. Ooi, R. P. Smith, P. Weise, M. Wetherell, X. Xie, R. Zagursky, Y. Zhang, and G. W. Zlotnick. 2004. Vaccine potential of the Neisseria meningitidis 2086 lipoprotein. Infect. Immun. 72:2088-2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Granoff, D. M., A. Bartoloni, S. Ricci, E. Gallo, D. Rosa, N. Ravenscroft, V. Guarnieri, R. C. Seid, A. Shan, W. R. Usinger, S. Tan, Y. E. McHugh, and G. R. Moe. 1998. Bactericidal monoclonal antibodies that define unique meningococcal B polysaccharide epitopes that do not cross-react with human polysialic acid. J. Immunol. 160:5028-5036. [PubMed] [Google Scholar]
- 9.Granoff, D. M., J. A. Welsch, and S. Ram. 2009. Binding of complement factor H (fH) to Neisseria meningitidis is specific for human fH and inhibits complement activation by rat and rabbit sera. Infect. Immun. 77:764-769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hakobyan, S., C. L. Harris, A. Tortajada, E. Goicochea de Jorge, A. Garcia-Layana, P. Fernandez-Robredo, S. Rodriguez de Cordoba, and B. P. Morgan. 2008. Measurement of factor H variants in plasma using variant-specific monoclonal antibodies: application to assessing risk of age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 49:1983-1990. [DOI] [PubMed] [Google Scholar]
- 11.Jarva, H., S. Ram, U. Vogel, A. M. Blom, and S. Meri. 2005. Binding of the complement inhibitor C4bp to serogroup B Neisseria meningitidis. J. Immunol. 174:6299-6307. [DOI] [PubMed] [Google Scholar]
- 12.Join-Lambert, O., P. C. Morand, E. Carbonnelle, M. Coureuil, E. Bille, S. Bourdoulous, and X. Nassif. 2010. Mechanisms of meningeal invasion by a bacterial extracellular pathogen, the example of Neisseria meningitidis. Prog. Neurobiol. 91:130-139. [DOI] [PubMed] [Google Scholar]
- 13.Koeberling, O., S. Giuntini, A. Seubert, and D. M. Granoff. 2009. Meningococcal outer membrane vesicle vaccines derived from mutant strains engineered to express factor H binding proteins from antigenic variant groups 1 and 2. Clin. Vaccine Immunol. 16:156-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lewis, L. A., J. Ngampasutadol, R. Wallace, J. E. Reid, U. Vogel, and S. Ram. 2010. The meningococcal vaccine candidate neisserial surface protein A (NspA) binds to factor H and enhances meningococcal resistance to complement. PLoS Pathog. 6:e1001027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lo, H., C. M. Tang, and R. M. Exley. 2009. Mechanisms of avoidance of host immunity by Neisseria meningitidis and its effect on vaccine development. Lancet Infect. Dis. 9:418-427. [DOI] [PubMed] [Google Scholar]
- 16.Madico, G., J. A. Welsch, L. A. Lewis, A. McNaughton, D. H. Perlman, C. E. Costello, J. Ngampasutadol, U. Vogel, D. M. Granoff, and S. Ram. 2006. The meningococcal vaccine candidate GNA1870 binds the complement regulatory protein factor H and enhances serum resistance. J. Immunol. 177:501-510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masignani, V., M. Comanducci, M. M. Giuliani, S. Bambini, J. Adu-Bobie, B. Arico, B. Brunelli, A. Pieri, L. Santini, S. Savino, D. Serruto, D. Litt, S. Kroll, J. A. Welsch, D. M. Granoff, R. Rappuoli, and M. Pizza. 2003. Vaccination against Neisseria meningitidis using three variants of the lipoprotein GNA1870. J. Exp. Med. 197:789-799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murphy, E., L. Andrew, K. L. Lee, D. A. Dilts, L. Nunez, P. S. Fink, K. Ambrose, R. Borrow, J. Findlow, M. K. Taha, A. E. Deghmane, P. Kriz, M. Musilek, J. Kalmusova, D. A. Caugant, T. Alvestad, L. W. Mayer, C. T. Sacchi, X. Wang, D. Martin, A. von Gottberg, M. du Plessis, K. P. Klugman, A. S. Anderson, K. U. Jansen, G. W. Zlotnick, and S. K. Hoiseth. 2009. Sequence diversity of the factor H binding protein vaccine candidate in epidemiologically relevant strains of serogroup B Neisseria meningitidis. J. Infect. Dis. 200:379-389. [DOI] [PubMed] [Google Scholar]
- 19.Pajon, R., P. T. Beernink, L. H. Harrison, and D. M. Granoff. 2010. Frequency of factor H-binding protein modular groups and susceptibility to cross-reactive bactericidal activity in invasive meningococcal isolates. Vaccine 28:2122-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19a.Plested, J. S., J. A. Welsch, and D. M. Granoff. 2009. Ex vivo model of meningococcal bacteremia using human blood for measuring vaccine-induced serum passive protective activity. Clin. Vaccine Immunol. 16:785-791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schneider, M. C., R. M. Exley, H. Chan, I. Feavers, Y. H. Kang, R. B. Sim, and C. M. Tang. 2006. Functional significance of factor H binding to Neisseria meningitidis. J. Immunol. 176:7566-7575. [DOI] [PubMed] [Google Scholar]
- 21.Schneider, M. C., B. E. Prosser, J. J. Caesar, E. Kugelberg, S. Li, Q. Zhang, S. Quoraishi, J. E. Lovett, J. E. Deane, R. B. Sim, P. Roversi, S. Johnson, C. M. Tang, and S. M. Lea. 2009. Neisseria meningitidis recruits factor H using protein mimicry of host carbohydrates. Nature 458:890-893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seib, K. L., D. Serruto, F. Oriente, I. Delany, J. Adu-Bobie, D. Veggi, B. Arico, R. Rappuoli, and M. Pizza. 2009. Factor H-binding protein is important for meningococcal survival in human whole blood and serum and in the presence of the antimicrobial peptide LL-37. Infect. Immun. 77:292-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sprong, T., P. Brandtzaeg, M. Fung, A. M. Pharo, E. A. Hoiby, T. E. Michaelsen, A. Aase, J. W. van der Meer, M. van Deuren, and T. E. Mollnes. 2003. Inhibition of C5a-induced inflammation with preserved C5b-9-mediated bactericidal activity in a human whole blood model of meningococcal sepsis. Blood 102:3702-3710. [DOI] [PubMed] [Google Scholar]
- 24.Stephens, D. S., B. Greenwood, and P. Brandtzaeg. 2007. Epidemic meningitis, meningococcaemia, and Neisseria meningitidis. Lancet 369:2196-2210. [DOI] [PubMed] [Google Scholar]
- 25.Welsch, J. A., and D. Granoff. 2007. Immunity to Neisseria meningitidis group B in adults despite lack of serum bactericidal antibody. Clin. Vaccine Immunol. 14:1596-1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Welsch, J. A., S. Ram, O. Koeberling, and D. M. Granoff. 2008. Complement-dependent synergistic bactericidal activity of antibodies against factor H-binding protein, a sparsely distributed meningococcal vaccine antigen. J. Infect. Dis. 197:1053-1061. [DOI] [PubMed] [Google Scholar]
- 27.Welsch, J. A., R. Rossi, M. Comanducci, and D. M. Granoff. 2004. Protective activity of monoclonal antibodies to genome-derived neisserial antigen 1870, a Neisseria meningitidis candidate vaccine. J. Immunol. 172:5606-5615. [DOI] [PubMed] [Google Scholar]
- 28.Zhu, D., Y. Zhang, V. Barniak, L. Bernfield, A. Howell, and G. Zlotnick. 2005. Evaluation of recombinant lipidated P2086 protein as a vaccine candidate for group B Neisseria meningitidis in a murine nasal challenge model. Infect. Immun. 73:6838-6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.