

Abstract
Thioredoxin (Trx) is an intracellular redox protein with extracellular cytokine-like and chemokine-like activities. We show here that, although plasma Trx levels are unrelated to survival of HIV-infected individuals with CD4 cell counts above 200/μl blood, survival is significantly impaired (P = 0.003) when plasma Trx is chronically elevated in HIV-infected subjects with CD4 T cell counts below this level (i.e., with Centers for Disease Control (CDC)-defined AIDS). Relevant to the mechanism potentially underlying this finding, we also present data from experimental studies in mice showing that elevated plasma Trx efficiently blocks lipopolysaccharide (LPS)-induced chemotaxis, an innate immune mechanism that is particularly crucial when adaptive immunity is compromised. Thus, we propose that elevated plasma Trx in HIV-infected individuals with low CD4 T cell counts directly impairs survival by blocking pathogen-induced chemotaxis, effectively eliminating the last (innate) barrier against establishment of opportunistic and other infections in these immunodeficient individuals.
Thioredoxin (Trx), a small (12 kDa) well-characterized protein with known three-dimensional structure and a highly conserved active site (-Cys-Gly-Pro-Cys-), plays a variety of redox-related roles in organisms ranging from Escherichia coli to human (1, 2). It was originally identified in E. coli as an electron donor for ribonucleotide reductase, and has been shown to catalyze several protein disulfide reductions in combination with thioredoxin reductase and NADPH in eukaryotic organisms (1, 2). In addition, under the initial names ADF (adult T cell leukemia derived factor) and 3B6-IL-1 (3, 34), it has been shown to be released by mammalian cells, to trigger release of cytokines and growth factors (3–9), and to itself have growth-factor and cytokine-like activities (4, 10, 11). Finally, it has been identified as one of the array of acute phase proteins that are produced in vivo in response to LPS stimulation (12).
Trx also regulates aspects of cell-growth and immune responses and influences the DNA-binding of certain transcription factors (6, 8, 11, 13, 14). It is induced by oxidative stress (8, 11), is translocated into the nucleus in stressed cells (8, 11), is chemotactic in vitro for eosinophils (7), and effectively blocks ischemic injury (15–18). In addition, our recent studies show that Trx has potent in vitro and in vivo chemoattractant activity for murine neutrophils, monocytes, and lymphocytes (9).
Studies presented here introduce a remarkable association between elevated plasma Trx and decreased survival in HIV-infected subjects whose CD4 T cell counts have fallen below 200/μl blood and thus are classified by the CDC as having AIDS. We have shown previously that plasma Trx tends to be elevated in HIV-infected subjects (19), both before and after progression to AIDS. We show here that, although all subjects with AIDS who entered the study were relatively healthy when baseline plasma Trx was measured, those who also had elevated plasma Trx (>30 ng/ml) were 2- to 3-fold more likely to die within 18 months than those whose plasma Trx levels fell within normal range.
The mechanism underlying this association between elevated plasma Trx and decreased survival in subjects with AIDS traces to a Trx-mediated block in innate defenses against pathogen invasion. Previous studies have shown that neutrophil function is impaired in advanced HIV disease (20, 21), and other studies have shown that elevated chemokine levels in circulation block neutrophil chemotaxis (22, 23). Here, we demonstrate in a standard mouse chemotaxis model that raising circulating Trx to levels comparable to those seen in HIV infection blocks almost all neutrophil migration in response to locally introduced LPS. Because the adaptive immune system that normally provides the major defense against disease is largely disrupted in AIDS patients, even a partial impairment of neutrophil migration in these patients can predispose toward the development of opportunistic and other infections that hasten death. Thus, we propose that the connection we have observed between elevated plasma Trx levels and decreased survival in HIV-infection is causal rather than merely associative.
Methods
Human Subjects.
Peripheral blood samples were drawn with informed consent from HIV-infected subjects with the following characteristics: CD4 T cell counts below 500/μl blood; no currently active opportunistic infection (OI); Karnofsky scores ≥60; no current debilitating illness or condition that would prevent walking up 2 steep flights of stairs (to the study office); no current consumption of large amounts of drugs known to deplete glutathione (GSH) (e.g., alcohol and acetaminophen); and no consumption of drugs that potentially replete GSH (e.g., high dose vitamins C or E). Roughly half of the subjects were taking 3′-azido-3′-deoxythymidine (AZT) or other anti-retroviral drugs. Protease inhibitors [(highly active anti-retroviral therapy (HAART)] were not introduced until some time after the study was complete and all survival times had been ascertained.
Fluorescence-activated cell sorter (FACS) and clinical and local laboratory analyses were performed to establish baseline values. Survival status was ascertained 2–3 years later by direct contact for subjects who were still alive or from official death records or reliable reports for those who died. Data are shown for all subjects for whom we were able to acquire survival data. Among subjects with CD4 T cell counts below 200/μl blood (the focus of the survival study), data for seven subjects were missing. No attempt was made to assure that composition of the study group reflects the composition of the community from which it was drawn.
N-acetylcysteine (NAC) was administered to some subjects. These were participants in a clinical trial aimed at determining whether oral administration of NAC increases GSH levels (24). Inclusion in this trial required clinical laboratory values within or near normal range and low T cell GSH levels (see below).
FACS Assay for T Cell Surface Markers and Intracellular GSH.
Peripheral blood mononuclear cells (PBMC) isolated from heparinized whole blood by Ficoll-Hypaque density centrifugation (Ficoll-Paque; Amersham Pharmacia) were stained, as previously described (25), with fluorochrome-coupled monoclonal antibodies detecting subset-defining T cell surface markers and with monochlorobimane to detect intracellular GSH as the glutathione S-bimane (GSB) conjugate. Cell-associated fluorescence was measured with a FACS. GSB in T cell subsets is reported as median fluorescence normalized to a frozen PBMC standard run at the same time.
Plasma Trx.
Heparinized blood samples were centrifuged to remove cells and stored at −70°C. Trx levels in plasma samples were measured, as previously described (19), by a sandwich ELISA and corrected to exclude Trx contribution from lysed erythrocytes, because erythrocytes contain roughly 1000 times as much Trx. Thus, Trx and hemoglobin levels (Sigma Diagnostics, procedure no. 527) were obtained for each plasma sample, and the Trx contribution from lysed erythrocytes was estimated from the plasma hemoglobin level. If plasma hemoglobin exceeded 65 mg/dl, the sample was discarded (about 10 samples). Otherwise, the estimated value for erythrocyte-derived plasma Trx was subtracted from the total plasma Trx to yield the corrected Trx values reported here.
Statistics.
Kaplan-Meier and Cox proportional hazards analyses were used to analyze survival data and estimate risk ratios. All statistical evaluations were performed with jmp version 4 software produced by the SAS Institute (Cary, NC; ref. 26).
Results
Plasma Trx Levels Are Elevated in HIV-Infected Individuals.
Because one third of the HIV-infected subjects (57/173) that we examined had plasma Trx above 30 ng/ml while less than five percent (2/46) of uninfected control subjects had Trx levels above this level (Fig. 1), we use 30 ng/ml as the categorical threshold value to distinguish subjects with elevated Trx (≥30 ng/ml plasma) from those with Trx levels in normal range. However, even among HIV-infected subjects with plasma Trx levels in normal range (<30 ng/ml), the median Trx level was still significantly higher than in control subjects (17.8 vs 11.3; P < 0.0001).
Figure 1.

HIV-infected subjects have elevated plasma thioredoxin levels. Estimated distributions (based on histograms) showing relative numbers of subjects as a function of plasma thioredoxin levels: HIV-negative controls (Top, n = 46); HIV-infected individuals with CD4 cell counts (Middle, n = 102); HIV-infected individuals with CD4 cell counts <200l (Bottom, n = 71). The vertical thin solid line on each histogram shows the median plasma Trx for the indicated group. The dotted vertical line, which is drawn at the 95th percentile for plasma Trx uninfected control subjects (30 ng/ml), is the threshold used to separate normal and elevated Trx levels in this study.
The increased Trx levels in HIV-infected subjects do not reflect severe or debilitating HIV disease because subjects in this study were prescreened to eliminate those with active opportunistic infections, excessive weight loss, or other indications of imminent health crisis(27). Furthermore, elevated Trx levels occur in both subjects with CD4 T cell counts below 200/μl blood and those with counts above this AIDS-defining threshold (Fig. 1). Nevertheless, the proportion of subjects with elevated Trx levels was higher among AIDS subjects than among subjects with higher T cell counts, i.e., 39% (28/71) vs 28% (29/102), and, among subjects with AIDS, differences in baseline measurements for several variables approached significance for subjects with elevated versus normal Trx (Table 1).
Table 1.
Baseline data for subjects with CD4 T cell counts <200/μl blood
| Variables | Subjects grouped by plasma
Trx
|
P* | |
|---|---|---|---|
| <30 ng/ml | ≥30 ng/ml | ||
| Subjects in group | n = 43 | n = 43 | |
| Anti-retroviral monotherapy (AZT, ddI, etc.) users† | 44% | 50% | NS |
| Group medians for variables | |||
| Thioredoxin (Trx), ng/ml plasma | 21 | 45 | |
| CD4 T cells/μl blood | 79 | 73 | NS |
| AST, IU/liter plasma | 32 | 45 | 0.03 |
| ALT, IU/liter plasma | 34 | 46 | 0.06 |
| Hematocrit, percent | 39 | 37 | 0.12 |
| Hemoglobin, g/dl | 13 | 12.3 | 0.08 |
| RBC, 106/μl | 4.1 | 3.8 | 0.08 |
| B cell GSB, FACS units/cell‡ | 0.59 | 0.73 | 0.02 |
| CD4 T cell GSB, FACS units/cell‡ | 0.73 | 0.79 | NS |
| β2-Microglobulin (n = 33), μg/ml plasma | 2.1 | 2.6 | 0.04 |
| HIV copies/ml plasma (n = 55) | 96 × 103 | 106 × 103 | NS |
NS, not significant (P > 0.2). The following were also NS: CD8 T cell, monocyte, and NK GSB; Karnofsky score.
Percentage of subjects taking AZT or other nucleoside analogs at baseline; data for usage throughout the observation period are not available. Since HAART therapy was not introduced until after the end of the observation period, none of the subjects had access to HAART treatment until the study was complete and all observations were recorded.
GSB (glutathione-S-bimane) is a FACS measure of intracellular GSH. Median fluorescence for the indicated subset, normalized to values for a frozen standard run at the same time, were determined for each subset in each subject. The median of these median values is shown in the table (see Methods).
High Plasma Trx Is Associated with Impaired Survival in Advanced HIV Disease.
The recruitment criteria for this study excluded all subjects whose health history or general physical condition, other than CD4 T cell count, indicated a likelihood of serious illness within 1–2 years of baseline. Thus, although the study was completed before the current HAART drugs were introduced, there were only two deaths recorded for HIV-infected subjects with CD4 T cell counts above 200/μl blood during the 2–3 year observation period of the study (Table 1). Subjects with CD4 T cell counts below this AIDS-defining threshold fared less well. Nearly half failed to survive this period. The studies that follow focus exclusively on this group of 64 subjects with AIDS and show that, within this group, elevated plasma Trx levels are associated with decreased survival measured either as the frequency of deaths (Table 2) or as survival time relative to baseline Trx measurement (Fig. 2).
Table 2.
Impaired survival of subjects with elevated plasma Trx and low CD4 T cell counts: 400-day survival of HIV-infected subjects
| Plasma Trx | CD4 T cells/μl blood
|
|
|---|---|---|
| <200 | ≥200 | |
| <30 ng/ml | 4/38 (11%)* | 1/62 (2%) |
| ≥30 ng/ml | 15/26 (58%) | 0/23 |
| Total | 19/64 (30%) | 1/85 (1%) |
Number of deaths within 400 days/total in indicated group (% death). By 800 days, there was one additional death in the ≥200 T cell group; nine additional deaths in the groups with <200 T cells and <30 ng/ml Trx; and two additional deaths in the group with low T cell counts and elevated plasma Trx levels.
Figure 2.
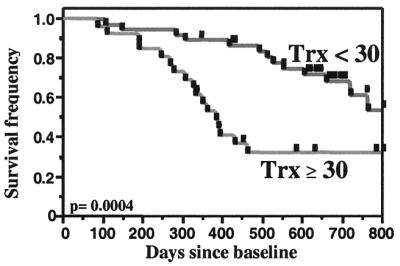
Survival is impaired in HIV-infected subjects with low CD4 T cell counts and elevated plasma Trx. Kaplan-Meier survival analysis for HIV-infected individuals with CD4 cell counts less than 200/μl (n = 71). Log rank P value is shown in the figure; Wilcoxon P value = 0.002. Data are shown for all subjects in the study. When six apparently healthy subjects who had clinical laboratory values outside the acceptable range for the NAC clinical trial were excluded, Log rank P = 0.003 and Wilcoxon P = 0.01.
Kaplan-Meier survival analysis (Fig. 2) dramatically reveals the importance of elevated Trx levels to 2–3 year (800 day) survival among subjects with AIDS (P = 0.0004, Fig. 2). The 30 ng/ml plasma Trx threshold used to divide subjects in this analysis is both the 95th percentile for control Trx levels (Fig. 1) and the receiver operating characteristic (ROC) (28) threshold for survival determined for this group of subjects. Thus, the Trx value that we use to distinguish subjects with elevated plasma Trx is rooted both in standard statistical methodology and in solid data distinguishing normal and elevated Trx levels.
Importantly, the majority of the deaths among AIDS subjects with elevated Trx levels occurred within the first 400 days of the study (Fig. 2 and Table 2). Nearly 90% (15/17) of the deaths recorded among the 26 subjects in this group occurred within this period. In contrast, deaths during this period account for only 30% (4/13) of the deaths ultimately recorded among the 38 subjects with baseline Trx levels in normal range.
Variables Known to Be Associated with Impaired Survival Do Not Explain the Trx Effect on Survival.
In a Cox proportional hazards survival analysis (Table 3), elevated plasma Trx levels remain significant when adjusted for covariates already known to be associated either with impaired survival (low CD4 T cell counts and low intracellular GSH) or with increased survival (administration of NAC; ref. 24). Thus, although Trx levels tend to be higher when CD4 T cell counts and CD4 T cell GSB are low (19), these variables do not individually or collectively explain the effect of Trx on survival. Addition of other covariates known to be of importance in HIV infection (e.g., anti-retroviral therapy, viral load, and CD38 on T cells) to the model altered the significance values of the covariates shown in Table 2 to some extent; however, elevated Trx levels always remain significantly associated with impaired survival (P = 0.0004–0.02).‡‡
Table 3.
Elevated plasma Trx is associated with impaired survival
| Variable† | Survival*
(subjects with CD4 T cell counts below 200/μl blood)
|
|||
|---|---|---|---|---|
| 0–400 days
|
0–800 days
|
|||
| P | Relative risk of dying‡ | P | Relative risk of dying | |
| Elevated plasma Trx (>30 ng/ml) | 0.0001 | 2.7 (1.6–5.2) | 0.0002 | 2.2 (1.4–3.3) |
| NAC treated§ | 0.0004 | 0.3 (0.06–0.6) | 0.002 | 0.5 (0.3–0.8) |
| CD4 T cell GSH (FACS GSB)¶ | 0.04 | 0.1 (0.02–0.9) | 0.01 | 0.1 (0.02–0.6) |
| CD4 T cell count (/μl blood) | 0.3 | NS | 0.005 | 0.8 (0.7–0.9) |
Survival analysis (proportional hazard) computed from baseline until the indicated time. Whole model fit, P < 0.0001; χ2 = 32 and 33 for 400 and 800 days, respectively. The model is computed on the basis of a total of 64 subjects, with 38 subjects in the group with <30 ng/ml and 26 subjects in the group with ≥30 ng/ml. None of the other variables listed in Table 1 showed significant effects. Reverse transcriptase inhibitor usage was similar for subjects in all groups and did not show a significant effect on survival when entered as a covariate in the above model. Since protease inhibitors were introduced after the end of the monitoring period, none of the subjects received protease inhibitors during the observation period shown here.
Baseline values representing the average of 2–3 measurements taken within 1 month for each subject. CD4 T cell counts and CD4 T cell GSH levels were entered into the model as continuous variables; plasma Trx and NAC treatment were entered as categorical variables.
Relative risk of dying (risk ratio); 95% confidence interval is shown in parentheses. Risk ratios shown for CD4 T cells are computed per increase in 20 T cells/μl blood.
Twenty-two of 64 subjects in this study were treated with NAC during the 8-week placebo-controlled and/or the 6-month open-label segments of the NAC trial. Among the subjects not treated with NAC (n = 42), 8 were in the placebo trial arm and did not elect open label NAC, 34 were screened for trial entry but either entered and rapidly withdrew (n = 3) or did not enter. Only 6/31 subjects in this latter No-NAC group were refused trial entry for medical cause, i.e., because they had protocol-defined exclusions such as elevated liver enzymes or hematologic deficiencies. We include these 6 subjects in data reported in the table. There was very little difference in the values obtained when significance and other table entries were computed with these 6 subjects excluded.
B cell GSB was significant at the 0.04 level at 400 days and not significant at 800 days.
These analyses are based on data for all of the subjects with CD4 T cell counts below 200/μl blood from whom we obtained blood samples for Trx analysis. At the time the samples were acquired, Karnofsky scores, face-to-face interviews, and the ability to climb two flights of steep stairs indicated that all subjects were relatively healthy. Clinical laboratory analyses later identified six subjects with one or more laboratory test values that were out of typical range for HIV-infected subjects at this stage of disease. Four of these subjects had Trx values above 30 ng/ml and died within 400 days of baseline measurement. The remaining two, who had Trx levels below 30 ng/ml, both survived past 400 days (one was still alive at the end of the study). The significance value for the Kaplan-Meier analysis (Fig. 2) changes from P = 0.0004 to P = 0.003 when these six subjects are excluded.
Note that survival times for all individuals in this 2–3 year study were determined before the introduction of HAART therapies. Therefore, study subjects were either untreated with anti-retroviral drugs or were receiving nucleoside analogs only. Neither the specific treatment nor the lack of treatment with these anti-retroviral drugs showed a significant survival effect in the model (P > 0.3).
NAC Blocks Trx Release in Vitro (and Possibly in Vivo).
In vitro studies demonstrate that NAC inhibits Trx release by ATL-2, a long-term cultured human T-lymphotrophic virus type I (HTLV-I)-infected human T cell leukemia line that has been used to characterize ADF and to demonstrate ADF identity to Trx (5). ATL-2 releases sizable amounts of Trx into the culture medium. However, addition of NAC at levels that are not toxic to ATL-2 decreases Trx release in a dose-dependent manner (Fig. 3). Because NAC is a source of cysteine, the amino acid that limits GSH synthesis, and because NAC can also act directly to relieve oxidative stress, these findings indicate that increasing intracellular GSH and/or decreasing oxidative stress in vitro decreases Trx release. This finding suggests that the release of Trx into plasma in HIV-infected subjects may be related to the low GSH levels that occur in many of these subjects (29) and hence that NAC treatment, which raises GSH in such subjects (27, 30), might decrease plasma Trx.
Figure 3.

NAC blocks thioredoxin release in vitro. The HTLV-I-transformed T cell line, ATL-2, was cultured in RPMI 1640 + 10% FCS in the presence of the indicated amount of NAC for 24 hr. Trx levels in the supernatants were measured by ELISA. Ten millimolar NAC is not toxic to these cells. Data shown are representative of three similar experiments.
We test this hypothesis here preliminarily by retrospectively examining baseline and exit plasma Trx levels in the subgroup of HIV-infected subjects studied here who were participating in an 8-week randomized, double-blind, placebo-controlled trial testing oral administration of NAC for GSH replenishment (Table 4). In this trial, which we completed several years ago (24, 27), NAC successfully replenished GSH in GSH-deficient HIV-infected subjects. Lowering plasma Trx was not an endpoint of this study, and collection of baseline and exit plasma samples useful for Trx analysis was begun only some time after the trial started. Furthermore, a number of samples were lost because of preservation problems. Nevertheless, with the available samples, we were able to do a least squares model fit analysis testing the effect of trial arm (NAC vs placebo) on plasma Trx levels in 21 subjects with CD4 T cell counts below 200/μl blood.
Table 4.
NAC treatment may decrease plasma Trx
| Variables | Standard least squares model fit for
plasma Trx at the end of the 8-week trial* (subjects
with CD4 T cell counts below 200/μl)
|
||||
|---|---|---|---|---|---|
| P (variables) | Adjusted mean plasma
Trx
|
Adjusted R2 | P (model) | ||
| NAC | Placebo | ||||
| Trial arm | 0.05† | 12 | 17 | 0.3 | 0.05 |
| Baseline Trx | 0.01 | ||||
| CD4 T cell count‡ | 0.5 | ||||
Subjects were treated with NAC (approx 7 g/day) or placebo during an 8-week double-blind trial. The complete Trx and GSH measurements (at 0 and 8 weeks) required for this analysis were available for a total of 24 trial subjects. Two outliers (1 from each arm) were excluded from this analysis on the basis of standard statistical criteria; one questionable admission to the trial was also removed, leaving a total of 21 subjects.
Power analysis indicates that trial arm would be significant at the 0.01 level if data for 36 comparable subjects, rather than the 21 studied here, were available.
Above or below 100/μl blood.
Data from this analysis, which includes baseline Trx as a covariate, reveals a trend (P = 0.05; Table 4) suggesting that NAC administration would significantly decrease Trx levels if tested in an appropriately designed prospective trial with enough subjects. Power analysis indicates that 36 subjects (rather than the 21 tested here) would be required to achieve significance at the 0.01 level.
Circulating Trx Impairs Chemotactic Responses.
The recent demonstration that Trx has chemokine-like activity introduces a rationale for the association of high plasma Trx with impaired survival in HIV-infected subjects with AIDS. Because circulating chemokines are known to decrease local chemotaxis (22, 23), circulating Trx can be expected to prevent neutrophil localization to infection sites and thereby cripple what little protection remains in these severely immunodeficient individuals. Preliminary studies in vitro demonstrated that Trx inhibits chemotaxis for human cells (R. Bertini, S. Pagliei, and Dompe' Research Center, L'Aquilla, Italy, personal communication). To test for inhibition in vivo, we elevated plasma Trx in mice by i.v. injection of human recombinant Trx and determined whether the elevated plasma Trx interferes with chemotaxis in a classic in vivo assay for chemotactic activity (ref. 9; Fig. 4).
Figure 4.

Circulating thioredoxin blocks local chemotaxis in mice. Recombinant (human) thioredoxin (40 μg) or saline was injected intravenously into mice prepared for the air pouch chemotaxis (9). Within 10 min, LPS (1 μg) was injected into the air pouch on all animals, and leukocyte infiltration into the pouch was measured 4 hr later. Although the ratio between neutrophils and lymphocytes can vary, this experiment has been repeated several times with similar results (decreased recruitment of total cells into the air pouch and decreased ratio of neutrophils to lymphocytes when Trx is injected intravenously).
In this model, lipopolysaccharide (LPS) injected into a preformed s.c. “air pouch” on the animal's back rapidly recruits blood cells into the air pouch. Four hours after LPS injection, the contents of the pouch are harvested and the cells it contained are counted and analyzed by FACS. In untreated animals, as in saline-treated animals, roughly 3 × 106 cells are recruited into the pouch during the 4-hour assay, and neutrophils dominate the recruited population. However, when human recombinant Trx is injected intravenously (i.v.) just before injection of LPS into the air pouch, the number of recruited cells decreases nearly 30-fold. Furthermore, within the small population of cells (approximately 105) that are recruited in the presence of Trx, lymphocytes usually outnumber neutrophils (e.g., Fig. 4). Results were similar in studies with Trx-transgenic mice (data not shown).
This Trx-mediated block in chemotactic activity occurs at Trx levels that are physiological in man. In essence, the Trx half-life is approximately 30 min measured over the 4-hour assay, and there is an immediate loss of more than half the injected Trx. Thus, the Trx dose used in this study (40 μg) brings the average plasma Trx level in the injected mice into the same range during the 4 h assay as plasma Trx levels (30–90 ng/ml) that are associated with markedly impaired survival in immunodeficient HIV-infected subjects.
Discussion
We have shown that chronically elevated plasma Trx in HIV infection is associated with impaired short-term (18-month) survival, but only in immunodeficient HIV-infected subjects with CD4 T cell counts below 200/μl blood, i.e., with AIDS. Although we were not able to specifically ascertain the cause of death in these subjects, their rapid demise despite having been in apparent good health roughly a year earlier (at the time plasma Trx was measured) implicates infection as the likely cause. Because, as we have shown, elevating plasma Trx in mice blocks LPS-stimulated chemotaxis of neutrophils, we propose that the chronic elevation of plasma Trx in subjects with AIDS interferes with the attraction of neutrophils to sites of opportunistic or other serious infection and thus blocks innate immune responses necessary for the survival of these subjects.
The link we have demonstrated between elevated plasma Trx and survival in AIDS is particularly dramatic because all subjects with active opportunistic infections or other illnesses associated with advanced HIV disease were screened from the study before collection of baseline measurements (24). Furthermore, among subjects included in the study, there were only minimal differences in baseline serologic and other values between those with elevated Trx and those with Trx levels in normal range. Nevertheless, among subjects with AIDS, 58% of those who had elevated plasma Trx died within 400 days of baseline data collection whereas only 11% of subjects with Trx levels in normal range died during this period.
The link that we have demonstrated between elevated plasma Trx and inhibition of chemotaxis introduces a mechanism to explain these survival findings. Previous studies have established that circulating chemokines block chemotactic activity (22, 31) and that locally introduced Trx is chemotactic for neutrophils, lymphocytes, and monocytes (9). Consistent with these findings, we have shown here that elevation of plasma Trx in mice results in essentially complete prevention of neutrophil chemotaxis to the local site of inflammation (LPS stimulation). Furthermore, we have shown (i) that Trx functions in a dose-dependent manner to decrease neutrophil recruitment in the air pouch model; (ii) that the active site on Trx is necessary to block recruitment; and (iii) that oxidized Trx is more effective than reduced Trx for this purpose (unpublished data). These findings collectively demonstrate that elevated plasma Trx directly interferes with recruitment of the neutrophils and lymphocytes that provide the first defense against invading pathogens.
Failure of this innate response may have little impact in subjects whose immune system is intact. However, in subjects with AIDS, it can mean the difference between life and death. Our data suggest that elevated plasma Trx does indeed suppress neutrophil recruitment to infection sites in these subjects, because the levels of circulating Trx that suppress chemotaxis in mice are comparable to the elevated plasma Trx in the subjects with AIDS who died within 400 days of Trx measurement. Thus, we propose that elevated plasma Trx levels were directly involved in the increased mortality in these subjects.
The evidence presented here from studies with NAC-treated subjects is consistent with this argument. We have shown here that NAC treatment is a significant covariate in the Trx survival model and have presented evidence suggesting that NAC decreases plasma Trx in a group of subjects to whom NAC was administered in a previously reported (24, 27) placebo-controlled trial. Because decrease in Trx was not an endpoint of this previous trial and because the number of NAC-treated subjects for whom Trx data were acquired is small, these data basically recommend a larger study specifically designed to test NAC and Trx interaction in survival of HIV-infected subjects. Nevertheless, the trend evident in these findings, coupled with data showing that NAC decreases Trx release in vitro, suggests that decreasing Trx levels may be important for survival, potentially by decreasing Trx.
Paradoxically, although chronic elevation of circulating Trx is likely to be harmful in HIV infection, acute elevation of circulating Trx has been shown to be helpful in clinical situations where avoidance of sudden influx of neutrophils and other inflammatory cells is important. Several studies, for example, have already reported that injection of a bolus of Trx substantially prevents ischemic reperfusion injury (15–18). These findings were previously ascribed to the reducing power of Trx and its ability to remove locally generated reactive oxygen intermediates. However, they are more likely explained by findings presented here demonstrating that Trx interferes with the recruitment of neutrophils and other inflammatory cells that worsen the injury at the ischemic site.
Explorations of this unique clinically viable approach to preventing ischemic and other injury because of intensive neutrophil infiltration in ischemia-reperfusion, including acute myocardial infarction, impending acute respiratory distress syndrome, and/or interstitial pneumonia, make it important to emphasize that even chronically elevated Trx levels are not necessarily dangerous. As we have shown, elevated Trx levels in HIV-infected subjects whose disease has not progressed to AIDS (i.e., <200 CD4 T cells) are not associated with impaired survival. Although there were 23 subjects with elevated Trx levels in this group, none died during the first 400 days of the observation period and only two died by 800 days. Furthermore, in several studies in which elevated Trx levels were found in subjects with diseases or conditions other than AIDS (32, 33), there was no evidence to indicate that the elevated Trx levels were associated with impaired survival. Thus, the survival impairment associated with chronically elevated Trx levels in subjects with AIDS occurs in a unique setting where normal immune defenses against invading pathogens have been severely disrupted.
In sum, we have shown here that Trx blocks chemotactic recruitment of neutrophils. Further, we have shown that elevated plasma Trx levels are associated with impaired short-term survival in HIV-infected individuals with CD4 T cell counts below 200/μl blood. On the strength of this evidence, we propose that chronically high levels of circulating Trx block migration of protective cells to sites of pathogen invasion and suggest that this block in innate immunity, coupled with the immunodeficiency in AIDS, permits development of opportunistic and other infections that contribute to rapid demise of HIV-infected individuals with insufficient T cell function.
Acknowledgments
The authors thank all members of the NAC Clinical Trial Working Group in the Herzenberg laboratory at Stanford. Dr. Mario Roederer, in particular, played a key-organizing role in this project and the trial from which the samples were drawn. The studies with human subjects were done at Stanford. The studies with mice were done in Kyoto. We thank Dr. E. Winger, Immunodiagnostic Laboratories, San Leandro, CA for enabling sample collection. Finally, we remember Dr. J. Gregson Dubs, who helped initiate this project and ran the project office and who died recently because of an AIDS-related neoplasm. This work was supported by grants from the Foundation of Human Frontier Science Program in Japan, Research for the Future from the Japan Society for the Promotion of Science, the Swedish Cancer Society (961 and 3505-B93-01VAA), and the California University-wide AIDS Research Program (F97-ST-044) and by National Institutes of Health Grants CA-42059, AI-07290, and LM-04836.
Abbreviations
- HAART
highly active anti-retroviral therapy
- LPS
lipopolysaccharide
- Trx
thioredoxin
- GSH
glutathione
- GSB
glutathione S-bimane
- NAC
N-acetylcysteine
- AZT
3′-azido-3′-deoxythymidine
Footnotes
In a subgroup of subjects for whom β2-microglobulin analyses were done (n = 33), β2-microglobulin was significantly correlated with Trx and hence decreased the Trx significance in the Cox proportional hazards analysis.
References
- 1.Holmgren A. Annu Rev Biochem. 1985;54:237–271. doi: 10.1146/annurev.bi.54.070185.001321. [DOI] [PubMed] [Google Scholar]
- 2.Holmgren A. Structure. 1995;3:239–243. doi: 10.1016/s0969-2126(01)00153-8. [DOI] [PubMed] [Google Scholar]
- 3.Wakasugi N, Tagaya Y, Wakasugi H, Mitsui A, Maeda M, Yodoi J, Tursz T. Proc Natl Acad Sci USA. 1990;87:8282–8286. doi: 10.1073/pnas.87.21.8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tagaya Y, Maeda Y, Mitsui A, Kondo N, Matsui H, Hamuro J, Brown N, Arai K, Yokota T, Wakasugi H, Yodoi J. EMBO J. 1989;8:757–764. doi: 10.1002/j.1460-2075.1989.tb03436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tagaya Y, Wakasugi H, Masutani H, Nakamura H, Iwata S, Mitsui A, Fujii S, Wakasugi N, Tursz T, Yodoi J. Mol Immunol. 1990;27:1279–1289. doi: 10.1016/0161-5890(90)90032-u. [DOI] [PubMed] [Google Scholar]
- 6.Okamoto T, Ogiwara H, Hayashi T, Mitsui A, Kawabe T, Yodoi J. Int Immunol. 1992;4:811–819. doi: 10.1093/intimm/4.7.811. [DOI] [PubMed] [Google Scholar]
- 7.Hori K, Hirashima M, Ueno M, Matsuda M, Waga S, Tsurufuji S, Yodoi J. J Immunol. 1993;151:5624–5630. [PubMed] [Google Scholar]
- 8.Hirota K, Matsui M, Iwata S, Nishiyama A, Mori K, Yodoi J. Proc Natl Acad Sci USA. 1997;94:3633–3638. doi: 10.1073/pnas.94.8.3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bertini R, Howard OM, Dong H F, Oppenheim J J, Bizzarri C, Sergi R, Caselli G, Pagliei S, Romines B, Wilshire J A, et al. J Exp Med. 1999;189:1783–1789. doi: 10.1084/jem.189.11.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yodoi J, Uchiyama T. Immunol Today. 1992;13:405–411. doi: 10.1016/0167-5699(92)90091-K. [DOI] [PubMed] [Google Scholar]
- 11.Nakamura H, Nakamura K, Yodoi J. Annu Rev Immunol. 1997;15:351–369. doi: 10.1146/annurev.immunol.15.1.351. [DOI] [PubMed] [Google Scholar]
- 12.Buetler T M. Hepatology. 1998;28:1551–1560. doi: 10.1002/hep.510280615. [DOI] [PubMed] [Google Scholar]
- 13.Matthews J R, Wakasugi N, Virelizier J L, Yodoi J, Hay R T. Nucleic Acids Res. 1992;20:3821–3830. doi: 10.1093/nar/20.15.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Silberstein D S, McDonough S, Minkoff M S, Balcewicz-Sablinska M K. J Biol Chem. 1993;268:9138–9142. [PubMed] [Google Scholar]
- 15.Aota M, Matsuda K, Isowa N, Wada H, Yodoi J, Ban T. J Cardiovasc Pharmacol. 1996;27:727–732. doi: 10.1097/00005344-199605000-00016. [DOI] [PubMed] [Google Scholar]
- 16.Isowa N, Yoshimura T, Kosaka S, Liu M, Hitomi S, Yodoi J, Wada H. J Cell Physiol. 2000;182:33–40. doi: 10.1002/(SICI)1097-4652(200001)182:1<33::AID-JCP4>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 17.Okubo K, Kosaka S, Isowa N, Hirata T, Hitomi S, Yodoi J, Nakano M, Wada H. J Thorac Cardiovasc Surg. 1997;113:1–9. doi: 10.1016/S0022-5223(97)70393-3. [DOI] [PubMed] [Google Scholar]
- 18.Takagi Y, Tokime T, Nozaki K, Gon Y, Kikuchi H, Yodoi J. J Cereb Blood Flow Metab. 1998;18:206–214. doi: 10.1097/00004647-199802000-00012. [DOI] [PubMed] [Google Scholar]
- 19.Nakamura H, De Rosa S, Roederer M, Anderson M T, Dubs J G, Yodoi J, Holmgren A, Herzenberg L A. Int Immunol. 1996;8:603–611. doi: 10.1093/intimm/8.4.603. [DOI] [PubMed] [Google Scholar]
- 20.Kuritzkes D R. Clin Infect Dis. 2000;30:256–260. doi: 10.1086/313642. [DOI] [PubMed] [Google Scholar]
- 21.Pitrak D L. Am J Health Syst Pharm. 1999;56, Suppl. 5:S9–S16. doi: 10.1093/ajhp/56.suppl_5.S9. [DOI] [PubMed] [Google Scholar]
- 22.Hechtman D H, Cybulsky M I, Fuchs H J, Baker J B, Gimbrone M A., Jr J Immunol. 1991;147:883–892. [PubMed] [Google Scholar]
- 23.Rutledge B J, Rayburn H, Rosenberg R, North R J, Gladue R P, Corless C L, Rollins B J. J Immunol. 1995;155:4838–4843. [PubMed] [Google Scholar]
- 24.Herzenberg L A, De Rosa S C, Dubs J G, Roederer M, Anderson M T, Ela S W, Deresinski S C. Proc Natl Acad Sci USA. 1997;94:1967–1972. doi: 10.1073/pnas.94.5.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roederer M, Herzenberg L A. Int Immunol. 1996;8:1–11. doi: 10.1093/intimm/8.1.1. [DOI] [PubMed] [Google Scholar]
- 26.jmp Development Group (SAS) jmp Statistics and Graphics Guide. Cary, NC: SAS Inst.; 1995. , Version 3.1. [Google Scholar]
- 27.De Rosa S C, Zaretsky M D, Dubs J G, Roederer M, Anderson M, Green A, Mitra D, Watanabe N, Nakamura H, Tjioe I, Deresinski S C, et al. Eur J Clin Invest. 2000;30:915–929. doi: 10.1046/j.1365-2362.2000.00736.x. [DOI] [PubMed] [Google Scholar]
- 28.Metz C E. Semin Nucl Med. 1978;8:283–298. doi: 10.1016/s0001-2998(78)80014-2. [DOI] [PubMed] [Google Scholar]
- 29.Staal F J, Ela S W, Roederer M, Anderson M T, Herzenberg L A, Herzenberg L A. Lancet. 1992;339:909–912. doi: 10.1016/0140-6736(92)90939-z. [DOI] [PubMed] [Google Scholar]
- 30.Breitkreutz R, Pittack N, Nebe C T, Schuster D, Brust J, Beichert M, Hack V, Daniel V, Edler L, Droge W. J Mol Med. 2000;78:55–62. doi: 10.1007/s001099900073. [DOI] [PubMed] [Google Scholar]
- 31.Simonet W S, Hughes T M, Nguyen H Q, Trebasky L D, Danilenko D M, Medlock E S. J Clin Invest. 1994;94:1310–1319. doi: 10.1172/JCI117450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakamura H, Vaage J, Valen G, Padilla C A, Bjornstedt M, Holmgren A. Free Radical Biol Med. 1998;24:1176–1186. doi: 10.1016/s0891-5849(97)00429-2. [DOI] [PubMed] [Google Scholar]
- 33.Sumida Y, Nakashima T, Yoh T, Nakajima Y, Ishikawa H, Mitsuyoshi H, Sakamoto Y, Okanoue T, Kashima K, Nakamura H, Yodoi J. J Hepatol. 2000;33:616–622. doi: 10.1034/j.1600-0641.2000.033004616.x. [DOI] [PubMed] [Google Scholar]
- 34.Rosen A, Lundman P, Carlsson M, Bhavani K, Srinivasa B R, Kjellstrom G, Nilsson K, Holmgren A. Int Immunol. 1995;7:625–633. doi: 10.1093/intimm/7.4.625. [DOI] [PubMed] [Google Scholar]