

Abstract
The classical nuclear factor kappa B (NF-κB) signaling pathway is an important regulator of inflammation and innate immunity that is activated by a wide variety of stimuli, including virus infection, tumor necrosis factor alpha (TNF-α), and interleukin 1β (IL-1β). Poxviruses, including vaccinia virus (VV) and ectromelia virus, encode multiple proteins that function in immune evasion. Recently, a growing number of genes encoded by poxviruses have been shown to target and disrupt the NF-κB signaling pathway. To determine if additional gene products that interfere with NF-κB signaling existed, we used a vaccinia virus deletion mutant, VV811, which is missing 55 open reading frames lacking all known inhibitors of TNF-α-induced NF-κB activation. Immunofluorescence analysis of HeLa cells treated with TNF-α and IL-1β revealed that NF-κB translocation to the nucleus was inhibited in VV811-infected cells. This was further confirmed through Western blotting of cytoplasmic and nuclear extracts for NF-κB. Additionally, VV811 infection inhibited TNF-α-induced IκBα degradation. In contrast to vaccinia virus strain Copenhagen (VVCop)-infected cells, VV811 infection resulted in the dramatic accumulation of phosphorylated IκBα. Correspondingly, coimmunoprecipitation assays demonstrated that the NF-κB-inhibitory IκBα-p65-p50 complex was intact in VV811-infected cells. Significantly, cells treated with 1-β-d-arabinofuranosylcytosine, an inhibitor of poxvirus late gene expression, demonstrated that an additional vaccinia virus late gene was involved in the stabilization of IκBα. Overall, this work indicates that unidentified inhibitors of NF-κB exist in vaccinia virus. The complex inhibition of NF-κB by vaccinia virus illustrates the importance of NF-κB activation in the antiviral response.
The nuclear factor kappa B (NF-κB) family of proteins function as transcription factors that regulate a wide range of genes involved in inflammation, innate immunity, and apoptosis (17, 63). The canonical NF-κB pathway is activated by a variety of stimuli, including virus infection, lipopolysaccharide, and proinflammatory cytokines, such as tumor necrosis factor alpha (TNF-α) and interleukin 1β (IL-1β) (25, 63). In unstimulated cells, the NF-κB dimer, composed of p65 and p50, is found as an inactive form bound to one of the inhibitors of NF-κB (IκB) proteins in the cytoplasm, most commonly IκBα (2, 25, 63). Upon stimulation of the TNF receptor (TNFR) or Toll-like receptor/IL-1 receptor (TLR/IL-1R), signaling cascades are activated which converge at the phosphorylation and activation of components of the inhibitor of NF-κB kinase (IKK) complex, most importantly, IKKβ (25, 34). IKKβ phosphorylates IκBα, which is subsequently polyubiquitinated by the ubiquitin ligase Skp1-cullin-1-F-box SCFβTrCP complex and degraded by the 26S proteasome (24, 60, 67). The degradation of IκB releases the NF-κB p65-p50 dimer, which translocates to the nucleus, binds κB sites on DNA, and regulates transcriptional activation of target genes (25, 63).
Many viruses manipulate the NF-κB pathway in order to regulate the diverse immune responses initiated by the pathway (27, 28, 49). For example, the enhancer region of human immunodeficiency virus type 1 (HIV-1) contains NF-κB binding sites required for activation of viral transcription (39). Alternatively, viruses such as Epstein-Barr virus and human T-cell leukemia virus activate constitutive NF-κB signaling to inhibit apoptosis and support viral transcription (32, 58). Other viruses balance NF-κB activation and inhibition. Upon infection, glycoprotein D and UL37 of herpes simplex virus type 1 (HSV-1) rapidly induce NF-κB activation to promote viral replication and inhibit apoptosis (33, 53). However, the infected cell protein 0 (ICP0) protein of HSV-1 redirects the deubiquitinating enzyme, ubiquitin-specific peptidase 7, to deubiquitinate TNF receptor-associated factor 6 (TRAF6) and IKKγ and prevent activation of NF-κB (13). Additionally, African swine fever virus encodes an IκB-like protein, A238L, that binds and inhibits the NF-κB heterodimer (46, 47). Viruses have also developed mechanisms to degrade certain proteins in the NF-κΒ pathway. For example, the poliovirus 3C protease cleaves p65, and coxsackievirus B3 protease cleaves IκBα, resulting in nuclear translocation of a fragment of IκBα and inhibition of NF-κB (40, 71). The regulation of NF-κB by viruses is a common strategy for evading the innate immune response.
Poxviruses are a large family of double-stranded DNA viruses that encode an array of proteins that interfere with signaling cascades and antiviral responses (38, 54). Variola virus, the causative agent of smallpox, is the most well-known member of the family, and mass vaccination campaigns used vaccinia virus, a closely related poxvirus, to globally eradicate smallpox (37). Vaccinia virus (VV), the prototypic member of the poxvirus family, contains approximately 200 open reading frames, including inhibitors of the NF-κB pathway (35).
Recently, a growing list of NF-κB inhibitors has been identified in vaccinia virus (7, 9, 16, 20, 52, 55). The TLR/IL-1R pathway of NF-κB activation is inhibited by A46R, A52R, and K7R (7, 52). A46R contains a Toll/IL-1 receptor (TIR) domain and interacts with other TIR-containing adaptor proteins at the receptor complex (7, 56). A52R and K7R interact with TRAF6 and interleukin 1 receptor-associated kinase-like 2 (IRAK2) to inhibit downstream NF-κB signaling (7, 23, 52). The vaccinia virus proteins B14R and N1L interact with different components of the IKK complex to inhibit the NF-κB pathway (9, 16). Specifically, B14R prevents the phosphorylation and activation of IKKβ, leading to the inhibition of IκBα phosphorylation, while N1L associates with members of the IKK complex to inhibit NF-κB signaling (9, 16). Finally, K1L suppresses modified vaccinia virus Ankara strain (MVA)-induced protein kinase R (PKR) phosphorylation and IκBα degradation, whereas the endoplasmic reticulum (ER)-localized protein M2L prevents MVA-induced ERK2 phosphorylation (20, 55, 66). Notably, A52R, K7R, B14R, and N1L display structural similarity to the Bcl-2 family of proteins; however, only N1L shows evidence of antiapoptotic activity (11, 22, 29). Overall, the presence of multiple NF-κB inhibitors in vaccinia virus underlines the importance of inhibiting the antiviral response during poxvirus infection.
A powerful approach to identify viral proteins that manipulate complex signaling pathways is through the use of large-deletion viruses such as MVA or VV811 (45, 69). VV811 is missing 55 open reading frames of the parental vaccinia virus strain Copenhagen (VVCop) genome (45). Importantly, VV811 lacks the known inhibitors of TNF-α-induced NF-κB activation but contains A46R and A52R, the inhibitors of TLR-mediated and IL-1β-induced NF-κB activation (35, 45). As such, VV811 is an attractive tool to identify previously unknown inhibitors of NF-κB (45). Given the importance of poxviral inhibition of the NF-κB pathway and the growing number of poxvirus-encoded NF-κB inhibitors, we sought to determine if other NF-κB inhibitors existed in the vaccinia virus genome by utilizing the large-deletion virus VV811, which is devoid of the known inhibitors that regulate TNF-α-induced NF-κB activation. We report here that vaccinia virus encodes one or more previously unknown inhibitors of the NF-κB pathway. Infection with VV811 inhibited TNF-α-induced p65 translocation to the nucleus and IκBα degradation, even though VV811 lacks the known inhibitors of TNF-α-induced NF-κB activation. Additionally, cells infected with VV811 and treated with TNF-α demonstrated an accumulation of phosphorylated IκBα. Significantly, VV811 required late gene expression for inhibition of IκBα degradation. Overall, our results indicate that vaccinia virus requires late protein synthesis that regulates IκBα degradation to effectively inhibit NF-κB signaling during infection.
MATERIALS AND METHODS
Cells and viruses.
HeLa cells were obtained from the American Type Culture Collection (ATCC) and maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 50 U of penicillin per ml, 50 μg of streptomycin per ml, and 200 μM glutamine (Invitrogen). Baby green monkey kidney (BGMK) cells were also obtained from ATCC and were maintained in DMEM supplemented with 10% newborn calf serum (NCS), 50 U of penicillin per ml, 50 μg of streptomycin per ml, and 200 μM glutamine (Invitrogen). VVCop was provided by G. McFadden (University of Florida). The deletion mutant VV811 was generated as previously described and was provided by E. Paoletti (45). VVCop and VV811 were propagated in BGMK cells (57). UV-inactivated VVCop and UV-inactivated VV811 were generated by treating VVCop or VV811 with 200 mJ/cm2 UV light in a Stratalinker UV cross-linker (Stratagene). Uninfected and infected HeLa cells were treated with 10 ng/ml TNF-α (Roche Diagnostics), 10 ng/ml IL-1β (PeproTech Inc.), 10 μM MG132 (Sigma-Aldrich), or 80 μg/ml 1-β-d-arabinofuranosylcytosine (araC; Sigma-Aldrich).
Antibodies.
Antibodies used in the study included rabbit anti-NF-κB p65 (C-20) and rabbit anti-IκBα (C-21) (Santa Cruz Biotechnology Inc.), mouse anti-IκBα (L35A5), anti-p105/p50, and mouse anti-phospho-IκBα (Ser32/36; 5A5) (Cell Signaling Technology Inc.); mouse anti-β-tubulin (ECM Biosciences); anti-poly(ADP-ribose) polymerase (anti-PARP; BD Biosciences); rabbit anti-I5L, generated as previously described (64); rabbit anti-E9L, which was kindly provided by D. Evans (University of Alberta); and mouse anti-Mcl-1 (Rockland). Secondary antibodies for Western blots were peroxidase-conjugated antirabbit and peroxidase-conjugated antimouse (Jackson ImmunoResearch Laboratories) used at a concentration of 1:25,000. The secondary antibody used for immunofluorescence and confocal microscopy was antirabbit Alexa Fluor 546 (Invitrogen), used at a concentration of 1:400. To detect mouse-anti-IκBα (L35A5) for flow cytometry, phycoerythrin-conjugated antimouse (Jackson ImmunoResearch Laboratories Inc.) was used at a concentration of 1:1,000.
Immunofluorescence microscopy.
HeLa cells (5 × 105) were grown on 18-mm glass coverslips and infected with the indicated virus at a multiplicity of infection (MOI) of 5 for 12 h at 37°C. Infection was followed by treatment with 10 ng/ml of either TNF-α or IL-1β for 20 min. Cells were washed twice with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde in PBS for 10 min at room temperature. After 3 washes with PBS containing 1% FBS, cells were permeabilized in 0.5% NP-40 (Sigma-Aldrich) in PBS for 5 min at room temperature. Primary antibody, anti-NF-κB p65 (C-20) at a concentration of 1:250, was applied and the cells were incubated overnight at room temperature. After 3 washes with PBS containing 1% FBS, the cells were stained with Alexa Fluor 546 goat antirabbit secondary antibody (1:400) (Invitrogen) for 1 h. Coverslips were applied to microscope slides with 7 μl of 250 μg/ml 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen) diluted in 4 mg/ml n-propyl gallate (Sigma-Aldrich) in 50% glycerol and 50% PBS. Slides were visualized by immunofluorescence microscopy using a Zeiss Axiovert 200 M fluorescent microscope with an ApoTome 10 optical sectioning device (Zeiss) and Axiovision software (Zeiss). Alternatively, slides were visualized by confocal microscopy with a Zeiss LSM510 laser scanning confocal microscope and were analyzed with LSM510 imaging software (Zeiss).
Cytoplasmic and nuclear extracts.
HeLa cells (2 × 106) were infected at an MOI of 5 for the indicated times, in the presence or absence of 80 μg/ml of araC, followed by treatment with 10 ng/ml of TNF-α or IL-1β for 20 min (31, 43). Cells were resuspended in 3 volumes of cold cytoplasmic extract buffer containing 10 mM HEPES, 10 mM KCl, 0.1 mM EDTA, pH 8.0, 0.1 mM EGTA, pH 8.0, 1 mM dithiothreitol (DTT), 0.05% NP-40, and protease inhibitor cocktail (Roche Diagnostics) and incubated at 4°C for 30 min with constant agitation. Lysed cells were centrifuged at 1,000 × g for 5 min at 4°C, and supernatant (the cytoplasmic extract) was collected and stored at −80°C. The pellet was washed with 5 volumes of cytoplasmic extract buffer, centrifuged for 5 min at 1,000 × g, and resuspended in 3 volumes of nuclear extract buffer containing 20 mM HEPES, 25% glycerol, 0.4 M NaCl, 1 mM EDTA, pH 8.0, 1 mM EGTA, pH 8.0, 1 mM DTT, and protease inhibitor cocktail, followed by a 20-min incubation on ice. Extracts were vortexed for 5 s, and debris was pelleted by centrifugation at 18,000 × g for 10 min at 4°C. Supernatants from the nuclear extracts were collected and stored at −80°C.
Detection of IκBα degradation by flow cytometry.
Degradation of IκBα was analyzed by flow cytometry using a modified version of the antibody manufacturer's instructions (Cell Signaling). HeLa cells (1 × 106) were infected at an MOI of 5 for 12 h, in the presence or absence of 80 μg/ml of araC, followed by treatment with 10 ng/ml of TNF-α for 20 min. Mock-infected cells were alternatively treated with 10 μM MG132 for 1 h before treatment with TNF-α. Cells were collected by trypsinization, fixed at 37°C for 10 min in 0.5% paraformaldehyde in PBS, and permeabilized on ice for 30 min in 90% ice-cold methanol. Cells were washed twice with PBS containing 1% FBS and stained with mouse anti-IκBα (L35A5; 1:400) at room temperature for 1 h. Cells were washed twice with PBS containing 1% FBS, stained with phycoerythrin-conjugated goat antimouse antibody (1:1,000) at room temperature for 1 h, and washed twice with PBS containing 1% FBS. Fluorescence was analyzed by flow cytometry (FACScan; Becton Dickinson) by measuring through the FL-2 channel with a 585-nm filter (42-nm band-pass filter). Data from the collection of 20,000 cells were analyzed with CellQuest software. The mean fluorescence intensity for each sample from three independent experiments was calculated with CellQuest software, and averages with standard error, were displayed graphically. Whole-cell lysates were collected by centrifugation of 250 μl of the samples used for flow cytometry at 2,000 × g for 5 min, followed by resuspension of pelleted cells in 25 μl of sodium dodecyl sulfate (SDS) sample loading buffer.
Immunoprecipitation.
HeLa cells (7 × 106) were lysed in 1% NP-40 lysis buffer containing 150 mM NaCl, 1% NP-40, 50 mM Tris, pH 8.0, and protease inhibitor cocktail, followed by precipitation with mouse anti-IκBα (L35A5 and protein G-Sepharose (GE Healthcare). Immunoprecipitations were blotted with rabbit anti-NF-κB p65, mouse anti-IκBα (L35A5), anti-p105/p50, mouse anti-phospho-IκBα (Ser32/36), rabbit anti-I5L, and mouse anti-β-tubulin.
SDS-PAGE and immunoblotting.
To separate isolated proteins, 12% or 15% SDS-polyacrylamide gel electrophoresis (PAGE) was used. Proteins were transferred to nitrocellulose membranes (GE Water and Process Technologies) using a semidry transfer apparatus (Tyler Research Corp.) at 420 mA. Membranes were probed with the indicated primary antibodies and secondary antibodies, and enhanced chemiluminescence was used for visualization according to the manufacturer's instructions (GE Healthcare).
Proteasome function assay.
HeLa cells (1 × 104) were mock infected or infected for 14 h with VV811 or VVCop at an MOI of 10. Cells were untreated or treated with 10 μM MG132 at 12 h postinfection. The chymotrypsin-like activity of the proteasome was assessed using the cell-based Proteasome-Glo assay, according to the manufacturer's directions (Promega) (36). Luminescence was recorded 10 min after addition of the Proteasome-Glo reagent using an EnVision 2104 multilabel reader (Perkin-Elmer). The assay was performed in triplicate, and average relative light units were displayed.
Analysis of Mcl-1 degradation.
HeLa cells (1 × 106) were mock infected or infected with VV811 or VVCop at an MOI of 5. Cells were treated with 10 μM MG132 2 h prior to collection. Whole-cell lysates were collected at 4, 8, 12, and 24 h postinfection and subjected to Western blotting with an antibody specific for Mcl-1.
RESULTS
VV811 inhibits TNF-α-induced NF-κB p65 translocation to the nucleus.
The NF-κB pathway is a crucial regulator of inflammation and innate immunity and is tightly regulated by multiple NF-κB inhibitors encoded by vaccinia virus (35). We utilized a large-deletion vaccinia virus, VV811, in an attempt to determine if other NF-κB inhibitors were present in the vaccinia virus genome (45). A comparison of the genome of VV811 with that of the parental wild-type vaccinia virus strain, VVCop, displays the open reading frames missing in VV811 (Fig. 1) (21, 45). The vaccinia virus mutant VV811 genome lacks 55 open reading frames from the variable termini of the VVCop genome (45). VV811 possesses the known inhibitors of TLR and IL-1β-induced NF-κB activation, A46R and A52R, but lacks all the currently known inhibitors of TNF-α-induced NF-κB activation (45).
FIG. 1.

VV811 lacks the known inhibitors of TNF-α-induced NF-κB activation. A schematic representation of wild-type strain VVCop and the mutant vaccinia virus VV811 are represented. Letters correspond to the HindIII restriction digest map (15). Large deletions in VV811 representing the open reading frames C23L-F4L in the left variable region and B13R-B29R in the right variable region and locations of the known NF-κB inhibitors are indicated.
To determine if VV811 infection retained the ability to inhibit TNF-α-induced NF-κB activation, microscopy was used to visualize localization of the p65 subunit of NF-κB (Fig. 2 A). HeLa cells were mock infected, infected with VVCop, or infected with VV811 for 12 h. Nuclei were visualized by DAPI staining, and infected cells were verified by the presence of perinuclear virus factories, also visualized with DAPI staining. Staining with an antibody to the p65 subunit of NF-κB revealed that p65 was present in the cytoplasm of mock-infected cells (Fig. 2A, panels a and b), while mock-infected cells treated with TNF-α displayed an accumulation of p65 in the nucleus, as expected (Fig. 2A, panels c and d) (42, 48). Cells infected with VVCop or VV811 in the absence of TNF-α stimulation displayed cytoplasmic localization of p65, indicating that infection with both viruses did not stimulate p65 nuclear translocation (Fig. 2A, panels g, h, m, and n). VVCop- and VV811-infected cells treated with TNF-α demonstrated clear inhibition of p65 translocation into the nucleus, indicating that both viruses inhibited NF-κB activation (Fig. 2A, panels i, j, o, and p). The experiment was repeated by exposing cells to IL-1β. Mock-infected cells treated with IL-1β showed a substantial accumulation of p65 in the nucleus, as expected (Fig. 2A, panels e and f). In contrast, VVCop and VV811 inhibited p65 translocation to the nucleus upon IL-1β stimulation (Fig. 2A, panels k, l, q, and r). Overall, infection with VVCop or the deletion mutant VV811 clearly inhibited the p65 nuclear translocation induced by TNF-α or IL-1β.
FIG. 2.

VVCop- and VV811-infected cells inhibit p65 nuclear translocation. (A) HeLa cells were mock infected (a to f) or infected with VVCop (g to l) or VV811 (m to r) at an MOI of 5 for 12 h. Following infection, cells were mock treated (a, b, g, h, m, and n) or treated with 10 ng/ml of TNF-α (c, d, i, j, o, and p) or IL-1β (e, f, k, l, q, and r) for 20 min. Localization of endogenous NF-κB p65 was detected using an anti-p65 antibody, and nuclei and virus factories were detected with DAPI and visualized by confocal microscopy. (B) HeLa cells were infected with UV-inactivated VVCop (a to d) or UV-inactivated VV811 (e to h) at an MOI of 5. Twelve hours postinfection cells were mock treated (a, b, e, and f) or treated with 10 ng/ml of TNF-α for 20 min (c, d, g, and h). Endogenous p65 was detected using a p65-specific antibody, and nuclei were detected with DAPI. (C) Cells demonstrating p65 nuclear translocation were counted from three independent experiments. Error bars represent standard deviations. (D) Fourteen hours postinfection cells were mock treated or treated with TNF-α or IL-1β for 20 min and cytoplasmic and nuclear extracts were generated. Cytoplasmic (C) and nuclear (N) extracts were Western blotted with anti-p65, as well as anti-PARP and anti-β-tubulin, which served as controls for nuclear and cytoplasmic extracts.
To confirm that the inhibition of p65 translocation by VV811 was not due to effects resulting from an unproductive infection, a single-step growth curve was performed, which demonstrated that VV811 productively infected HeLa cells (data not shown). As a control, cells were infected with UV-inactivated VVCop (Fig. 2B, panels a to d) or UV-inactivated VV811 (Fig. 2B, panels e to h) and subsequently treated with TNF-α. In the absence of TNF-α treatment, infection with UV-inactivated VVCop (Fig. 2B, panels a and b) or UV-inactivated VV811 (Fig. 2B, panels e and f) retained p65 in the cytoplasm. In contrast, upon treatment with TNF-α, infection with UV-inactivated VVCop (Fig. 2B, panels c and d) or UV-inactivated VV811 (Fig. 2B, panels g and h) demonstrated p65 translocation to the nucleus, indicating the necessity for viral protein production for inhibition of p65 translocation. Nuclear translocation of p65 was quantified by counting cells. Approximately 89 and 86% of mock-infected cells treated with TNF-α or IL-1β, respectively, displayed p65 translocation to the nucleus (Fig. 2C). Overall, however, less than 5% of cells infected with VVCop or VV811 treated with TNF-α or IL-1β displayed p65 translocation to the nucleus. Strikingly, cells infected with UV-inactivated VVCop or VV811 and treated with TNF-α displayed greater than 85% p65 nuclear translocation.
To verify that VV811 inhibited TNF-α-induced p65 translocation to the nucleus, cytoplasmic and nuclear extracts were generated from HeLa cells that were mock infected or infected with VVCop or VV811. Fourteen hours postinfection, cells were mock treated or treated with TNF-α for 20 min, and cytoplasmic and nuclear extracts were Western blotted with anti-p65 (Fig. 2D). In mock-infected cells lacking TNF-α stimulation, a large amount of p65 was detected in the cytoplasmic fraction but no p65 was present in the nuclear fraction, indicative of cells that do not display NF-κB activation (Fig. 2D). Following TNF-α treatment of mock-infected cells, p65 accumulated in the nuclear fraction, demonstrating activation of the NF-κB pathway (Fig. 2D). Significantly, VVCop- and VV811-infected cells inhibited p65 translocation into the nuclear fraction following TNF-α stimulation. A similar pattern of p65 localization was observed in samples treated with IL-1β (Fig. 2E). The purity of the cytoplasmic and nuclear extracts was determined by Western blotting with anti-PARP, a nuclear protein, and anti-β-tubulin, a cytoplasmic protein. These data further confirmed that despite lacking the known vaccinia virus-encoded inhibitors of NF-κB, VV811 infection still inhibited NF-κB p65 nuclear translocation stimulated by both TNF-α and IL-1β, providing further evidence that VV811 contains at least one inhibitor of TNF-α-induced NF-κB activation.
VV811 inhibits TNF-α-induced IκBα degradation.
In unstimulated cells, IκBα retains NF-κB in the cytoplasm (2). Upon NF-κB activation, IκBα is degraded, allowing translocation of the NF-κB heterodimer to the nucleus (3, 26). To further establish how VV811 infection manipulates the NF-κB pathway, we examined IκBα degradation in mock-infected, VVCop-infected, or VV811-infected cells (Fig. 3 A). In the absence of TNF-α stimulation, IκBα was present in cytoplasmic extracts of mock-infected HeLa cells, indicative of cells lacking activated NF-κB (Fig. 3A). Upon treatment with TNF-α, mock-infected cells demonstrated a loss of IκBα due to degradation (Fig. 3A) (3, 26). In dramatic contrast, cells infected with VVCop or VV811 and stimulated with TNF-α retained IκBα expression, indicating that both VVCop and VV811 infection inhibited the degradation of IκBα (Fig. 3A). Infection with VVCop or VV811 also inhibited IL-1β-induced IκBα degradation (Fig. 3B), since both viruses contain inhibitors of the IL-1β pathway to NF-κB activation, and these inhibitors act upstream of IκBα degradation (7). The ability of VV811 to inhibit IκBα degradation suggested that inhibition of NF-κB occurred upstream or at the point of IκBα degradation.
FIG. 3.
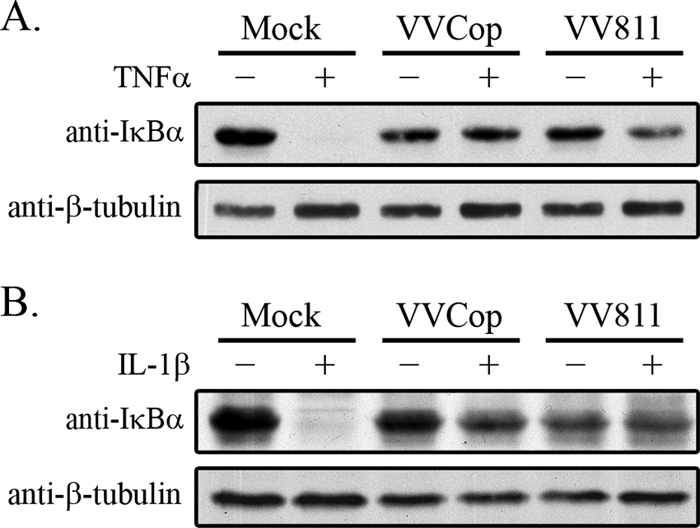
IκBα is not degraded in VV811-infected cells. HeLa cells were mock infected or infected with VVCop or VV811 at an MOI of 5. Twelve hours postinfection cells were mock treated or stimulated with TNF-α (A) or IL-1β (B) for 20 min. Cytoplasmic extracts were Western blotted with rabbit anti-IκBα and anti-β-tubulin, as a loading control.
In order to verify the ability of VVCop or VV811 to inhibit IκBα degradation, we used a flow cytometry assay to monitor IκBα levels (Fig. 4). HeLa cells were mock infected or infected with VVCop or VV811 for 12 h and subsequently mock treated or treated with TNF-α, and expression of IκBα was detected using an antibody specific for IκBα. In the absence of TNF-α, HeLa cells (labeled mock in Fig. 4) demonstrated a high level of IκBα (Fig. 4a). In HeLa cells treated with TNF-α (labeled mock + TNF-α), the level of IκBα dropped significantly due to the degradation of IκBα (Fig. 4a) (3, 26). Degradation of IκBα induced by TNF-α was rescued by treatment with the proteasome inhibitor MG132 (labeled MG132 + TNF-α), as previously demonstrated (18, 44, 62). Treatment of HeLa cells with MG132 in the absence of TNF-α (labeled MG132) had no effect on the level of IκBα (Fig. 4a). Notably, however, both VV811- and VVCop-infected cells treated with TNF-α (labeled VVCop + TNF-α and VV811 + TNF-α, respectively) displayed no loss of IκBα fluorescence (Fig. 4b and c), indicating that IκBα was not degraded. Similar results were also found when the experiment was repeated using IL-1β (Fig. 4d to f). Altogether, the data indicated that cells infected with VV811, which is lacking the known inhibitors of TNF-α-induced NF-κB activation, retained the ability to inhibit degradation of IκBα, a crucial event for the activation of the NF-κB pathway.
FIG. 4.

VV811 and VVCop inhibit IκBα degradation. HeLa cells were mock infected or infected with VVCop or VV811 at an MOI of 5. Twelve hours postinfection cells were mock treated or treated with TNF-α or IL-1β for 20 min. IκBα levels were detected by flow cytometry using anti-IκBα, followed by fluorescently labeled anti-mouse secondary antibody. The proteasome inhibitor MG132 was used as a control to inhibit IκBα degradation.
VV811 infection leads to an accumulation of phosphorylated IκBα.
Upon activation of the NF-κB pathway, IκBα is phosphorylated by IKKβ, a member of the IKK complex (34, 68, 70). Phosphorylation of IκBα at serines 32 and 36 signals for polyubiquitination, resulting in IκBα degradation by the 26S proteasome (8, 10). To further determine how VV811 infection inhibits the NF-κB pathway, phosphorylation of IκBα was examined. HeLa cells were mock infected or infected with VVCop or VV811 and subsequently mock treated or treated with TNF-α. Cytoplasmic extracts were generated and blotted with an antibody specific for phosphorylated IκBα (Fig. 5 A). Mock-infected cells treated with TNF-α displayed no accumulation of phospho-IκBα, since IκBα is quickly ubiquitinated and degraded once it is phosphorylated (Fig. 5A) (8, 10). Little phospho-IκBα was detected in VVCop-infected cells treated with TNF-α, likely due to the presence of multiple virally encoded NF-κB inhibitors, including B15R, which inhibits IKKβ function, leading to inhibition of IκBα phosphorylation (Fig. 5A) (9). Interestingly, an accumulation of phospho-IκBα was present in the VV811-infected cells treated with TNF-α (Fig. 5A). Since VV811 does not contain B15R, the activity of IKKβ could account for the observed accumulation of phospho-IκBα in VV811-infected cells (34, 70). VV811-infected cells treated with IL-1β also displayed this increase in phospho-IκBα (Fig. 5B) (9, 35). As a control, cytoplasmic extracts were blotted with an antibody to a vaccinia virus late protein, I5L, to confirm virus infection, and β-tubulin was used as a loading control. Together, this is strong evidence that VV811 expresses an inhibitor of NF-κB that inhibits the degradation of phosphorylated IκBα, leading to an accumulation of phosphorylated IκBα in infected cells.
FIG. 5.
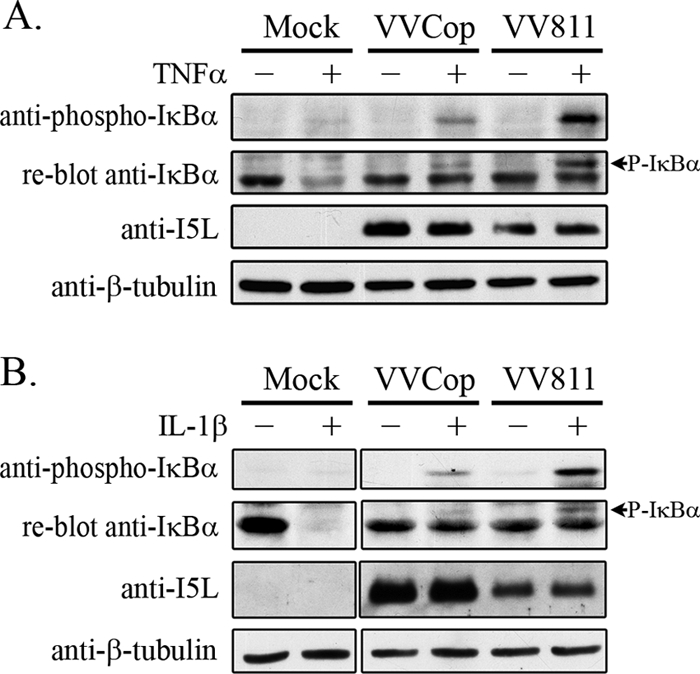
Phosphorylated IκBα accumulates in TNF-α-treated VV811-infected cells. Cells were mock infected or infected with VVCop or VV811 at an MOI of 5 for 14 h and treated with TNF-α or IL-1β for 20 min. Cytoplasmic extracts of samples treated with TNF-α (A) or IL-1β (B) were blotted with anti-phospho-IκBα. The same membranes were reblotted with anti-IκBα. Samples were Western blotted for the vaccinia virus protein I5L as a control for virus infection and β-tubulin, as a loading control.
To gain insight into the kinetics of phospho-IκBα accumulation during VV811 infection, cells were treated with TNF-α for various amounts of time. Cytoplasmic extracts were generated and blotted for phospho-IκBα (Fig. 6 A). In mock-infected cells, no phospho-IκBα was detected, as expected; however, treatment with MG132 and TNF-α induced accumulation of phosphorylated IκBα, since proteasome inhibition blocks phospho-IκBα degradation (18, 44, 62). In the absence of TNF-α, cells infected with VV811 displayed no phospho-IκBα; however, after 5 min of TNF-α stimulation a large amount of phospho-IκBα was detected, and the presence of phospho-IκBα was detected up to 90 min of TNF-α treatment (Fig. 6A). Compared to VV811, VVCop displayed minimal phospho-IκBα present after 20, 45, and 90 min of TNF-α treatment. Therefore, in contrast to VVCop-infected cells, VV811 induced accumulation of phosphorylated IκBα after TNF-α treatment, suggesting the presence of an NF-κB inhibitor that acts downstream of IκBα phosphorylation.
FIG. 6.
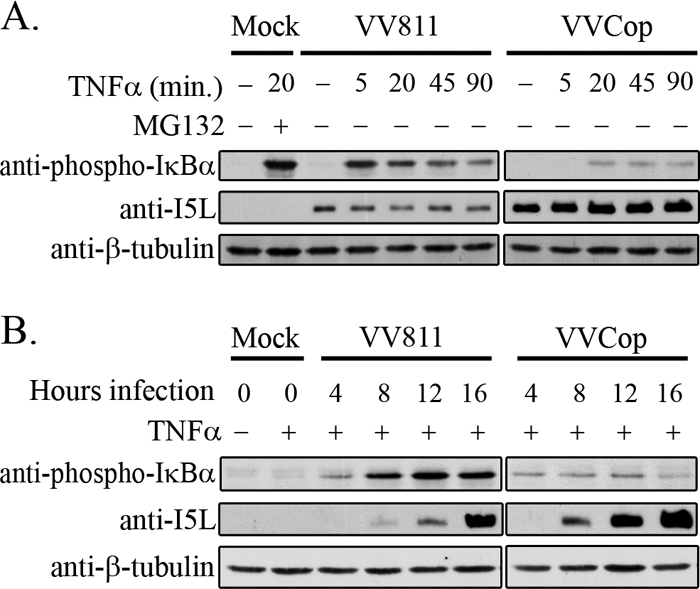
VV811 infection accumulates phospho-IκBα upon extended TNF-α treatment. (A) HeLa cells were mock infected or infected with VV811 or VVCop at an MOI of 5 for 14 h, followed by mock treatment or treatment with TNF-α for 5, 20, 45, or 90 min, and cytoplasmic extracts were collected. Mock-infected cells were treated with MG132 and TNF-α for 20 min as a control for accumulation of phospho-IκBα. Cytoplasmic extracts were blotted for phospho-IκBα. Anti-I5L was used as a control for virus infection, and anti-β-tubulin was used as a loading control. (B) HeLa cells were mock infected or infected with VV811 or VVCop at an MOI of 5. HeLa cells were treated with TNF-α for 20 min at 4, 8, 12, and 16 h postinfection. Cytoplasmic extractions were performed, and samples were blotted for phospho-IκBα. Samples were also blotted with anti-I5L to detect increasing late gene expression during infection, and anti-β-tubulin was used as a loading control.
To determine if the accumulation of phospho-IκBα occurred throughout VV811 infection, at 4, 8, 12, and 16 h postinfection, cells were treated with TNF-α for 20 min and cytoplasmic extracts were collected (Fig. 6B). Whereas no phospho-IκBα was detected in mock-infected cells after either mock treatment or treatment with TNF-α, a small amount of phospho-IκBα was detected as early as 4 h postinfection with VV811. Increasing amounts of phospho-IκBα were detected at 8, 12, and 16 h of VV811 infection (Fig. 6B). VVCop-infected cells treated with TNF-α again displayed only a minimal amount of phospho-IκBα, likely due to the presence of previously identified virus-encoded inhibitors of TNF-α-induced NF-κB (35). Infection with VV811, followed by TNF-α treatment, resulted in an accumulation of phosphorylated IκBα, suggesting that an unknown inhibitor expressed during VV811 infection inhibits the degradation of IκBα.
Since IκBα degradation was inhibited in infected cells, the possibility existed that IκBα stabilization was due to a dysfunctional proteasome. Recent reports demonstrate that proteasome activity is required for poxvirus infection (50, 61). Since proteasome inhibition affects poxvirus late protein production, we sought to determine if the proteasome was functional during vaccinia virus infection (50, 61). The chymotrypsin-like activity of the proteasome was analyzed during vaccinia virus infection using a cell-based luciferase assay (36). Cells were mock infected or infected with VVCop or VV811 for 14 h, and MG132 was added at 12 h postinfection. The amount of luminescence of each sample correlated to the amount of chymotrypsin-like proteasomal activity, and the recorded luminescence of each sample was displayed graphically as relative light units (Fig. 7 A) (36). Mock-infected cells demonstrated high luminescence, corresponding to functional proteasome activity. Compared to untreated cells, the addition of MG132 resulted in a significant decrease in luminescence. In contrast, cells infected with VV811 or VVCop demonstrated considerable proteasome activity; however, treatment with MG132 resulted in less than 1% proteasome activity (Fig. 7A). Therefore, although some decrease in proteasome function was evident late during vaccinia virus infection, the chymotrypsin-like activity of the proteasome remained functional. To further confirm that the proteasome was functional during infection, Mcl-1, an antiapoptotic cellular protein known to be polyubiquitinated and degraded by the proteasome, was also examined (12, 41). Cells were mock infected or infected with VVCop or VV811, and whole-cell lysates were collected at various time points during infection (Fig. 7B). Western blotting with an antibody to Mcl-1 revealed that levels of Mcl-1 were low during infection, whereas Mcl-1 levels increased substantially after treatment with MG132. Lysates were also blotted with anti-E9L for virus infection and anti-β-tubulin as a loading control. These data demonstrated that the proteasome is functional during VVCop and VV811 infection and that overall proteasome dysfunction is not responsible for inhibition of IκBα degradation during VV811 infection.
FIG. 7.

The proteasome is functional during vaccinia virus infection. (A) HeLa cells were mock infected or infected with VVCop or VV811 at an MOI of 10 for 14 h. MG132 (10 μM) was added at 12 h postinfection. A luciferase-based proteasome assay was utilized to examine the chymotrypsin-like activity of the proteasome, displayed as relative light units. The assay was completed in triplicate, and error bars represent standard deviations. (B) HeLa cells were mock infected or infected with VVCop or VV811 at an MOI of 5, and whole-cell lysates were collected at the indicated times. Cells were also mock treated or treated with 10 μM MG132 for 2 h prior to collection of lysates. Whole-cell lysates were blotted with antibodies to Mcl-1, E9L, and β-tubulin.
IκBα remains associated with p65 and p50 in VV811-infected cells.
Upon phosphorylation and degradation of IκBα, the p65-p50 NF-κB heterodimer is released and translocates to the nucleus due to the presence of an exposed nuclear localization signal in p65 (4, 19). Given that IκBα was phosphorylated but not degraded in VV811-infected cells stimulated with TNF-α, we sought to determine if IκBα remained bound to the NF-κB heterodimer in the inactive IκBα-p65-p50 complex. HeLa cells were infected with VVCop or VV811, and at 12 h postinfection cells were mock treated or treated with TNF-α and immunoprecipitated with an antibody specific for IκBα (Fig. 8 A). As expected, immunoprecipitation of IκBα demonstrated binding to p65 and p50 in unstimulated mock-infected cells (Fig. 8A). Immunoprecipitated samples blotted for IκBα confirmed that IκBα was immunoprecipitated in all the samples except for mock-infected cells treated with TNF-α, in which IκBα was degraded (Fig. 8A) (3, 26). Importantly, in cells infected with VVCop or VV811 and immunoprecipitated with IκBα, IκBα also associated with p65 and p50 (Fig. 8A). A significant amount of phospho-IκBα was immunoprecipitated in mock-infected cells treated with MG132 and TNF-α, indicating that inhibition of the proteasome maintained the levels of phospho-IκBα. A small amount of phospho-IκBα was detected in VVCop-infected cells treated with TNF-α, and conversely, phospho-IκBα was clearly present in VV811-infected cells treated with TNF-α, confirming that a portion of the IκBα associated with p65 and p50 in VV811-infected cells treated with TNF-α was phosphorylated (Fig. 8A). Cell lysates demonstrated expression of p65, p50, and IκBα in the mock-infected and virus-infected cells (Fig. 8B). Blotting with anti-I5L and anti-β-tubulin confirmed virus infection and ensured equal amounts in all samples. The interaction observed between IκBα, p65, and p50 is indicative of the NF-κB-inhibitory IκBα-p65-p50 complex. Therefore, in both VVCop- and VV811-infected cells stimulated with TNF-α, the inactive IκBα-p65-p50 complex remained intact. Furthermore, a portion of the IκBα in VV811-infected cells stimulated with TNF-α was phosphorylated. This observation indicates that VV811 expresses a previously unidentified inhibitor of the NF-κB pathway, resulting in inhibition of IκBα degradation after phosphorylation of IκBα.
FIG. 8.
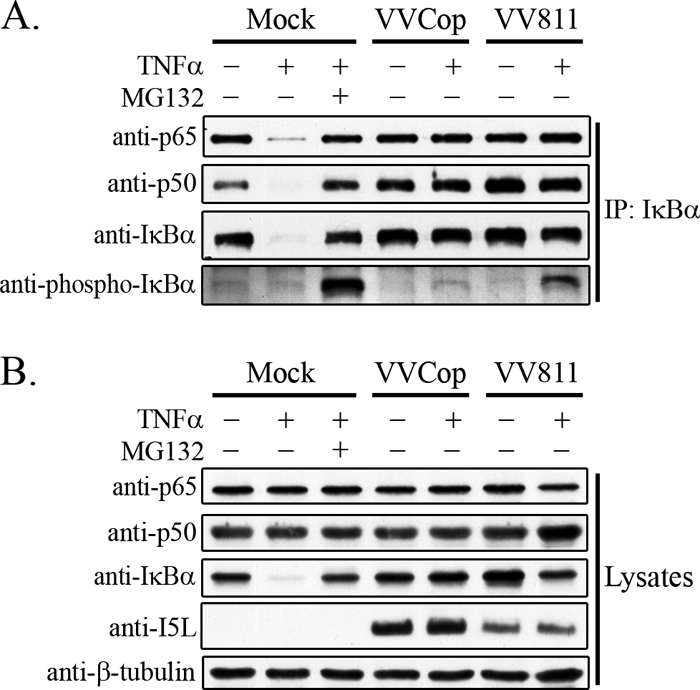
NF-κB p65-p50 remains associated with IκBα after VV811 infection. HeLa cells were mock infected or infected with VVCop or VV811 at an MOI of 5 for 12 h and were mock treated or treated with TNF-α. (A) Cells were lysed in NP-40 lysis buffer and immunoprecipitated with anti-IκBα. Western blotting was performed with antibodies to p65, p50, IκBα, and phospho-IκBα. (B) Cell lysates were Western blotted for expression of endogenous p65, p50, IκBα, the vaccinia virus protein I5L, and β-tubulin, as a loading control.
Late protein expression inhibits IκBα degradation.
To effectively inhibit the antiviral immune response, many vaccinia virus immune evasion proteins are expressed early during infection (37, 54). To determine if late protein expression contributed to inhibition of TNF-α-induced IκBα degradation, HeLa cells were infected with VVCop or VV811 and IκBα fluorescence was examined by flow cytometry in the presence and absence of araC, an inhibitor of late viral gene expression (Fig. 9 A) (1, 14). As expected, mock-infected cells treated with TNF-α (labeled mock + TNF-α in Fig. 9) resulted in IκBα degradation (Fig. 9A, panels a, b, c, and d). As previously shown in Fig. 4, IκBα degradation was inhibited in infected cells treated with TNF-α (Fig. 9A, panels a and b). In contrast, cells infected with VVCop or VV811 in the presence of TNF-α and araC resulted in loss of IκBα (Fig. 9A, panels c and d). The loss of IκBα in VV811-infected cells was more prominent than that in cells infected with VVCop (Fig. 9A, panels c and d). The results of the flow cytometry analysis were also quantified, and mean fluorescence intensities of samples were displayed graphically (Fig. 9B).
FIG. 9.

A late vaccinia virus protein regulates IκBα degradation. (A) HeLa cells were infected with VVCop or VV811 at an MOI of 5 for 12 h, and cells were either mock treated or treated with TNF-α for 20 min. Cells were treated with 80 μg/ml of araC to inhibit late poxvirus gene expression. Mock-infected cells treated with MG132 and TNF-α were used as a positive control for inhibition of IκBα degradation. IκBα levels were detected by flow cytometry by staining with mouse anti-IκBα, followed by fluorescently tagged anti-mouse secondary antibody, and flow cytometry analysis was performed to examine IκBα fluorescence. (B) Quantification of IκBα fluorescence was determined from the mean fluorescence intensities represented by the average from three independent experiments. Error bars represent standard deviations. (C) HeLa cells were mock infected or infected with VVCop or VV811 for 12 h at an MOI of 5 and either mock treated or treated with TNF-α for 20 min. Additionally, cells infected with VVCop or VV811 were treated with 80 μg/ml araC, to inhibit late genes at 12 h postinfection. MG132 combined with TNF-α treatment was used as a positive control for inhibition of IκBα degradation. Cytoplasmic extracts were generated and Western blotted with antibodies to IκBα, phospho-IκBα, E9L, I5L, and β-tubulin, as a loading control.
To further verify the role of late protein synthesis in the inhibition of the NF-κB pathway, cells were treated with araC and Western blotted for IκBα (Fig. 9C). After 12 h of infection with VVCop or VV811, IκBα was maintained in all cytoplasmic extracts in the absence and presence of TNF-α stimulation (Fig. 9C, lanes 4 to 7). Upon TNF-α stimulation and treatment with araC, a loss of IκBα was observed in VVCop-infected cells (Fig. 9C, lane 9). However, infection with VV811 in the presence of araC and TNF-α resulted in a loss of IκBα that was significantly greater than that observed with VVCop infection (Fig. 9C, lane 11). Levels of IκBα were maintained in VVCop- or VV811-infected cells treated with only araC (Fig. 9C, lanes 8 and 10). Along with the flow cytometry data, these results reinforce the notion that late protein synthesis is required to inhibit IκBα degradation. To examine the effect of araC treatment on phospho-IκBα accumulation during virus infection, cytoplasmic extracts were also blotted with anti-phospho-IκBα. As expected, phospho-IκBα accumulated in TNF-α-treated cells infected with VV811, and, notably, phospho-IκBα was maintained even in the presence of araC (Fig. 9C, lanes 7 and 11). Interestingly, an increase in phospho-IκBα was detected in VVCop cells treated with araC and TNF-α (Fig. 9C; compare lane 5 to lane 9), suggesting that additional late gene products regulate accumulation of phospho-IκBα during infection. To verify that late proteins were not expressed upon treatment with araC, cytoplasmic extracts were blotted for a late vaccinia virus protein, I5L, and an early vaccinia protein, E9L, and the presence of β-tubulin confirmed equal loading. The data indicate that a late vaccinia protein(s) is responsible for the observed inhibition of IκBα degradation in VVCop- and VV811-infected cells.
DISCUSSION
This study was initiated by the intriguing possibility that a large-deletion vaccinia virus could be utilized to identify additional inhibitors of the NF-κB pathway (45). Given the importance of the NF-κB pathway during poxvirus infection (35), we postulated that unidentified NF-κB inhibitors might be present in the vaccinia virus genome. Our initial observation that infection with VV811 inhibited TNF-α-induced p65 translocation to the nucleus, even though VV811 is devoid of the known inhibitors of TNF-α-induced NF-κB activation, indicated that additional NF-κB inhibitors exist in vaccinia virus. Upon infection with VV811, we have determined that at least one previously unidentified inhibitor of TNF-α-induced NF-κB activation is present in the genome of VV811 and that late viral gene expression is required to inhibit IκBα degradation.
Comparison of VV811 with the parental strain, VVCop, revealed that VV811 possesses vaccinia virus inhibitors of IL-1β-induced NF-κB activation, A46R and A52R, but lacks the NF-κB inhibitors that suppress MVA-induced NF-κB activation, K1L and M2L, as well as those shown to block TNF-α-induced NF-κB signaling (Fig. 1) (7, 35, 45). The observation that VV811 infection inhibited p65 translocation to the nucleus after TNF-α treatment was the first evidence that an additional inhibitor of NF-κB activation in vaccinia virus existed (Fig. 2). Upon further characterization, we determined that VV811 infection inhibited IκBα degradation induced by TNF-α stimulation (Fig. 3, 4). The degradation of IκBα occurs after the convergence point of the TNFR and the TLR/IL-1R pathways, making it an attractive target for viral inhibition of NF-κB signaling induced by a wide variety of stimuli (8, 10). For example, infected cell protein 27 (ICP27), expressed by HSV-1, interacts with IκBα to inhibit IκBα phosphorylation and subsequent ubiquitination (30). In addition, the coxsackievirus B3 protease cleaves IκBα, producing an inhibitory fragment of IκBα that prevents DNA binding by the NF-κB transcription factor (51, 71).
Since IκBα levels were stabilized during infection, we examined the possibility that the proteasome was dysfunctional during vaccinia virus infection. By demonstrating that the proteasome retained a significant level of chymotrypsin-like activity (Fig. 7A) and the ability to degrade cellular proteins during infection (Fig. 7B), we ruled out the possibility that proteasome dysfunction was responsible for the inhibition of IκBα degradation. We detected only a minor decrease in proteasome function during infection, which could minimally contribute to the inhibition of IκBα degradation (Fig. 7); however, it is likely that cell growth suppression during virus infection may be responsible for the smaller amount of luminescence compared to that of uninfected samples (36).
The regulation of IκBα has been studied intensely since the discovery of its vital role in NF-κB signaling (25, 63). Activation of the IKK complex results in phosphorylation of the IKKβ kinase (34, 68, 70). IKKβ phosphorylates IκBα at serines 32 and 36, and this phosphorylation marks IκBα for polyubiquitination by the SCFβTrCP ubiquitin ligase, resulting in the release of the NF-κB transcription factor and nuclear translocation of NF-κB (8, 10). In this study, we demonstrated that VV811 infection resulted in an accumulation of phosphorylated IκBα after TNF-α stimulation, suggesting that IκBα was phosphorylated but not degraded (Fig. 5 and 6). We were unable to detect phospho-IκBα ubiquitination in VV811-infected cells (data not shown). However, given that phospho-IκBα accumulated in VV811-infected cells, this strongly suggests that phospho-IκBα was not subjected to ubiquitination and subsequent degradation (Fig. 5 and 6). In contrast to VV811 infection, only a small amount of phosphorylated IκBα was present in VVCop-infected cells treated with TNF-α (Fig. 5 and 6). This is likely due to the presence of the known inhibitors of TNF-α-induced NF-κB activation that act upstream of IκBα phosphorylation in VVCop (35). Notably, B15R, also known as B14R in VV strain Western Reserve, prevents the phosphorylation and activation of IKKβ, resulting in inhibition of IκBα phosphorylation (9). However, VV811 lacks B15R, leading us to propose that a previously uncharacterized inhibitor of the NF-κB pathway functions to prevent degradation of phosphorylated IκBα (9). Surprisingly, accumulation of phosphorylated IκBα was also observed in VV811-infected cells treated with IL-1β, suggesting that expression of A46R and A52R may not be adequately inhibiting IL-1β-mediated NF-κB activation (Fig. 5) (7, 45). A46R and A52R were originally characterized in vaccinia virus strain Western Reserve; however, the A46R gene in vaccinia virus strain VVCop is truncated by 26 amino acids at the C terminus, but the consequences on protein function are unknown (7, 21). Additionally, the A52R gene of VVCop contains a phenylalanine-to-serine substitution at amino acid 57, which may have consequences for the function of A52R (7, 21). Overall, the evidence overwhelmingly suggests that a previously uncharacterized protein(s) expressed by vaccinia virus inhibits NF-κB following IκBα phosphorylation.
Our results indicated that infection with VV811 inhibited IκBα degradation (Fig. 3 and 4). However, it was possible that a vaccinia virus protein prevented NF-κB translocation to the nucleus by displacing IκBα from the NF-κB dimer. Notably, A238L, encoded by African swine fever virus, replaces IκBα, sequestering the NF-κB transcription factor in the cytoplasm (46, 47, 59). To examine the possibility that VV811 prevented NF-κB translocation to the nucleus by displacing IκBα from the NF-κB dimer, we immunoprecipitated IκBα from VV811- and VVCop-infected cells (Fig. 8). Association of IκBα with both p65 and p50 was detected, and a portion of immunoprecipitated IκBα was phosphorylated (Fig. 8). Therefore, the inhibitory complex of IκBα-p65-p50 was not disrupted during virus infection, confirming that phosphorylated IκBα was still bound to the NF-κB dimer, providing evidence that inhibition of IκBα degradation by VV811 occurs prior to dissociation of IκBα from the p65-p50 heterodimer. Using flow cytometry, in conjunction with cytoplasmic extracts, we determined that late protein expression was required for suppression of IκBα degradation during VV811 infection (Fig. 9). Significantly, late protein expression was also required for inhibition of IκBα degradation in VVCop-infected cells (Fig. 9). Additionally, treatment with araC, which blocks DNA replication and, subsequently, viral late protein synthesis, resulted in IκBα degradation after 12 h of VV811 infection followed by TNF-α treatment (Fig. 9B). Vaccinia virus expresses proteins in a temporal manner (37). Late proteins are typically structural components of the progeny virions (37); therefore, it was somewhat surprising that a late protein was essential for the inhibition of NF-κB, given that all of the known inhibitors of NF-κB signaling in vaccinia virus are expressed early during infection to block the rapid immune response elicited after detection of virus infection (35). Interestingly, inhibition of late protein synthesis during VVCop infection resulted in an increase of phospho-IκBα upon TNF-α treatment (Fig. 9B), suggesting that a late protein contributes to reduction of phospho-IκBα in VVCop-infected cells late during infection. Therefore, multiple proteins expressed by vaccinia virus contribute to the inhibition of NF-κB during infection, underlining the complexity of NF-κB regulation by vaccinia virus. Overall, the data indicate that an additional inhibitor of NF-κB activation is required late in infection to aid in the persistent suppression of NF-κB signaling.
The existence of a vaccinia virus protein that inhibits IκBα degradation directly has not been reported, although IκBα is clearly a vital component of the NF-κB pathway. To test this, we immunoprecipitated IκBα from VV811-infected cells and identified interacting partners by mass spectrometry (K. Fagan-Garcia and M. Barry, unpublished data). Both p65 and c-Rel coprecipitated with IκBα, suggesting that further examination of the effects of vaccinia virus infection on other NF-κB dimers is also warranted; however, we failed to identify any viral proteins interacting with IκBα. Additionally, the possibility exists that the inhibition of IκBα degradation by VV811 is an indirect effect of viral exploitation. The best example of this is the role of Vpu in HIV-1 infection that is responsible for directing degradation of CD4 during HIV-1 infection via the SCFβTrCP (5, 6). Vpu also affects other substrates of SCFβTrCP, since Vpu competitively binds βTrCP, significantly reducing the degradation of cellular proteins normally targeted by SCFβTrCP, including β-catenin and phospho-IκBα (5, 6). Since our data indicate an accumulation of phospho-IκBα in VV811-infected cells after TNF-α stimulation, it is possible that a vaccinia virus protein may be sequestering or redirecting the SCFβTrCP ubiquitin ligase during vaccinia virus infection, in a manner similar to that of Vpu, thereby preventing the ubiquitination of phosphorylated IκBα and subsequent degradation of IκBα. We compared the open reading frames present in VV811 and absent in MVA, which is known to induce NF-κB activation (43). Using this approach we identified 18 open reading frames; 6 of these genes were late genes, which we will test for IκBα degradation. However, it is possible that multiple open reading frames contribute to the observed effects, and future examination of other proteins expressed during VV811 and VVCop infection is warranted.
NF-κB signaling is a pivotal regulator of the immune response to infection, and poxviruses must therefore suppress NF-κB signaling through the use of multiple poxvirus-encoded inhibitors of NF-κB to promote a successful infection (35). Homologs to the NF-κB transcription factor and IκBα are found in the horseshoe crab, indicating that the NF-κB pathway has been an important player in the immune response for millions of years (65). Overall, the vital importance of NF-κB signaling in the immune response, combined with millions of years of coevolution, has led to the development of multiple strategies utilized by poxviruses to effectively suppress NF-κB signaling. Including those identified in this study, eight or more NF-κB inhibitors have been identified in vaccinia virus, and it is possible that this list will continue to grow, given the overall interest in poxvirus regulation of NF-κB.
Acknowledgments
We thank Logan Banadyga, Stephanie Campbell, and Kelly Mottet for critical evaluation of the manuscript.
This research was supported by a grant from the Canadian Institutes of Health Research. K.F.-G. was supported by funds from the National Sciences and Engineering Council of Canada and a Queen Elizabeth II Scholarship. M.B. is a senior scholar of the Alberta Heritage Foundation for Medical Research, a Tier I Canada Research Chair, and a Howard Hughes Institute Scholar in Infection and Parasitology.
Footnotes
Published ahead of print on 27 October 2010.
REFERENCES
- 1.Babiuk, L. A., B. Meldrum, V. S. Gupta, and B. T. Rouse. 1975. Comparison of the antiviral effects of 5-methoxymethyl-deoxyuridine with 5-iododeoxyuridine, cytosine arabinoside, and adenine arabinoside. Antimicrob. Agents Chemother. 8:643-650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baeuerle, P. A., and D. Baltimore. 1988. I kappa B: a specific inhibitor of the NF-kappa B transcription factor. Science 242:540-546. [DOI] [PubMed] [Google Scholar]
- 3.Beg, A. A., T. S. Finco, P. V. Nantermet, and A. S. Baldwin, Jr. 1993. Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of I kappa B alpha: a mechanism for NF-kappa B activation. Mol. Cell. Biol. 13:3301-3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beg, A. A., S. M. Ruben, R. I. Scheinman, S. Haskill, C. A. Rosen, and A. S. Baldwin, Jr. 1992. I kappa B interacts with the nuclear localization sequences of the subunits of NF-kappa B: a mechanism for cytoplasmic retention. Genes Dev. 6:1899-1913. [DOI] [PubMed] [Google Scholar]
- 5.Besnard-Guerin, C., N. Belaidouni, I. Lassot, E. Segeral, A. Jobart, C. Marchal, and R. Benarous. 2004. HIV-1 Vpu sequesters beta-transducin repeat-containing protein (betaTrCP) in the cytoplasm and provokes the accumulation of beta-catenin and other SCFbetaTrCP substrates. J. Biol. Chem. 279:788-795. [DOI] [PubMed] [Google Scholar]
- 6.Bour, S., C. Perrin, H. Akari, and K. Strebel. 2001. The human immunodeficiency virus type 1 Vpu protein inhibits NF-kappa B activation by interfering with beta TrCP-mediated degradation of Ikappa B. J. Biol. Chem. 276:15920-15928. [DOI] [PubMed] [Google Scholar]
- 7.Bowie, A., E. Kiss-Toth, J. A. Symons, G. L. Smith, S. K. Dower, and L. A. O'Neill. 2000. A46R and A52R from vaccinia virus are antagonists of host IL-1 and Toll-like receptor signaling. Proc. Natl. Acad. Sci. U. S. A. 97:10162-10167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown, K., S. Gerstberger, L. Carlson, G. Franzoso, and U. Siebenlist. 1995. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science 267:1485-1488. [DOI] [PubMed] [Google Scholar]
- 9.Chen, R. A., G. Ryzhakov, S. Cooray, F. Randow, and G. L. Smith. 2008. Inhibition of IkappaB kinase by vaccinia virus virulence factor B14. PLoS Pathog. 4:e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen, Z., J. Hagler, V. J. Palombella, F. Melandri, D. Scherer, D. Ballard, and T. Maniatis. 1995. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 9:1586-1597. [DOI] [PubMed] [Google Scholar]
- 11.Cooray, S., M. W. Bahar, N. G. Abrescia, C. E. McVey, N. W. Bartlett, R. A. Chen, D. I. Stuart, J. M. Grimes, and G. L. Smith. 2007. Functional and structural studies of the vaccinia virus virulence factor N1 reveal a Bcl-2-like anti-apoptotic protein. J. Gen. Virol. 88:1656-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cuconati, A., C. Mukherjee, D. Perez, and E. White. 2003. DNA damage response and MCL-1 destruction initiate apoptosis in adenovirus-infected cells. Genes Dev. 17:2922-2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daubeuf, S., D. Singh, Y. Tan, H. Liu, H. J. Federoff, W. J. Bowers, and K. Tolba. 2009. HSV ICP0 recruits USP7 to modulate TLR-mediated innate response. Blood 113:3264-3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Clercq, E., E. Darzynkiewicz, and D. Shugar. 1975. Antiviral activity of O′-alkylated derivatives of cytosine arabinoside. Biochem. Pharmacol. 24:523-527. [DOI] [PubMed] [Google Scholar]
- 15.DeFilippes, F. M. 1982. Restriction enzyme mapping of vaccinia virus DNA. J. Virol. 43:136-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DiPerna, G., J. Stack, A. G. Bowie, A. Boyd, G. Kotwal, Z. Zhang, S. Arvikar, E. Latz, K. A. Fitzgerald, and W. L. Marshall. 2004. Poxvirus protein N1L targets the I-kappaB kinase complex, inhibits signaling to NF-kappaB by the tumor necrosis factor superfamily of receptors, and inhibits NF-kappaB and IRF3 signaling by Toll-like receptors. J. Biol. Chem. 279:36570-36578. [DOI] [PubMed] [Google Scholar]
- 17.Dong, J., E. Jimi, H. Zhong, M. S. Hayden, and S. Ghosh. 2008. Repression of gene expression by unphosphorylated NF-kappaB p65 through epigenetic mechanisms. Genes Dev. 22:1159-1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fiedler, M. A., K. Wernke-Dollries, and J. M. Stark. 1998. Inhibition of TNF-alpha-induced NF-kappaB activation and IL-8 release in A549 cells with the proteasome inhibitor MG-132. Am. J. Respir. Cell Mol. Biol. 19:259-268. [DOI] [PubMed] [Google Scholar]
- 19.Ganchi, P. A., S. C. Sun, W. C. Greene, and D. W. Ballard. 1992. I kappa B/MAD-3 masks the nuclear localization signal of NF-kappa B p65 and requires the transactivation domain to inhibit NF-kappa B p65 DNA binding. Mol. Biol. Cell 3:1339-1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gedey, R., X. L. Jin, O. Hinthong, and J. L. Shisler. 2006. Poxviral regulation of the host NF-kappaB response: the vaccinia virus M2L protein inhibits induction of NF-kappaB activation via an ERK2 pathway in virus-infected human embryonic kidney cells. J. Virol. 80:8676-8685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goebel, S. J., G. P. Johnson, M. E. Perkus, S. W. Davis, J. P. Winslow, and E. Paoletti. 1990. The complete DNA sequence of vaccinia virus. Virology 179:247-266, 517-563. [DOI] [PubMed] [Google Scholar]
- 22.Graham, S. C., M. W. Bahar, S. Cooray, R. A. Chen, D. M. Whalen, N. G. Abrescia, D. Alderton, R. J. Owens, D. I. Stuart, G. L. Smith, and J. M. Grimes. 2008. Vaccinia virus proteins A52 and B14 share a Bcl-2-like fold but have evolved to inhibit NF-kappaB rather than apoptosis. PLoS Pathog. 4:e1000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harte, M. T., I. R. Haga, G. Maloney, P. Gray, P. C. Reading, N. W. Bartlett, G. L. Smith, A. Bowie, and L. A. O'Neill. 2003. The poxvirus protein A52R targets Toll-like receptor signaling complexes to suppress host defense. J. Exp. Med. 197:343-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hatakeyama, S., M. Kitagawa, K. Nakayama, M. Shirane, M. Matsumoto, K. Hattori, H. Higashi, H. Nakano, K. Okumura, K. Onoe, and R. A. Good. 1999. Ubiquitin-dependent degradation of IkappaBalpha is mediated by a ubiquitin ligase Skp1/Cul 1/F-box protein FWD1. Proc. Natl. Acad. Sci. U. S. A. 96:3859-3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hayden, M. S., and S. Ghosh. 2008. Shared principles in NF-kappaB signaling. Cell 132:344-362. [DOI] [PubMed] [Google Scholar]
- 26.Henkel, T., T. Machleidt, I. Alkalay, M. Kronke, Y. Ben-Neriah, and P. A. Baeuerle. 1993. Rapid proteolysis of I kappa B-alpha is necessary for activation of transcription factor NF-kappa B. Nature 365:182-185. [DOI] [PubMed] [Google Scholar]
- 27.Hiscott, J., H. Kwon, and P. Genin. 2001. Hostile takeovers: viral appropriation of the NF-kappaB pathway. J. Clin. Invest. 107:143-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hiscott, J., T. L. Nguyen, M. Arguello, P. Nakhaei, and S. Paz. 2006. Manipulation of the nuclear factor-kappaB pathway and the innate immune response by viruses. Oncogene 25:6844-6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalverda, A. P., G. S. Thompson, A. Vogel, M. Schroder, A. G. Bowie, A. R. Khan, and S. W. Homans. 2009. Poxvirus K7 protein adopts a Bcl-2 fold: biochemical mapping of its interactions with human DEAD box RNA helicase DDX3. J. Mol. Biol. 385:843-853. [DOI] [PubMed] [Google Scholar]
- 30.Kim, J. C., S. Y. Lee, S. Y. Kim, J. K. Kim, H. J. Kim, H. M. Lee, M. S. Choi, J. S. Min, M. J. Kim, H. S. Choi, and J. K. Ahn. 2008. HSV-1 ICP27 suppresses NF-kappaB activity by stabilizing IkappaBalpha. FEBS Lett. 582:2371-2376. [DOI] [PubMed] [Google Scholar]
- 31.Laegreid, A., A. Medvedev, U. Nonstad, M. P. Bombara, G. Ranges, A. Sundan, and T. Espevik. 1994. Tumor necrosis factor receptor p75 mediates cell-specific activation of nuclear factor kappa B and induction of human cytomegalovirus enhancer. J. Biol. Chem. 269:7785-7791. [PubMed] [Google Scholar]
- 32.Laherty, C. D., H. M. Hu, A. W. Opipari, F. Wang, and V. M. Dixit. 1992. The Epstein-Barr virus LMP1 gene product induces A20 zinc finger protein expression by activating nuclear factor kappa B. J. Biol. Chem. 267:24157-24160. [PubMed] [Google Scholar]
- 33.Liu, X., K. Fitzgerald, E. Kurt-Jones, R. Finberg, and D. M. Knipe. 2008. Herpesvirus tegument protein activates NF-kappaB signaling through the TRAF6 adaptor protein. Proc. Natl. Acad. Sci. U. S. A. 105:11335-11339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mercurio, F., H. Zhu, B. W. Murray, A. Shevchenko, B. L. Bennett, J. Li, D. B. Young, M. Barbosa, M. Mann, A. Manning, and A. Rao. 1997. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science 278:860-866. [DOI] [PubMed] [Google Scholar]
- 35.Mohamed, M. R., and G. McFadden. 2009. NFkB inhibitors: strategies from poxviruses. Cell Cycle 8:3125-3132. [DOI] [PubMed] [Google Scholar]
- 36.Moravec, R. A., M. A. O'Brien, W. J. Daily, M. A. Scurria, L. Bernad, and T. L. Riss. 2009. Cell-based bioluminescent assays for all three proteasome activities in a homogeneous format. Anal. Biochem. 387:294-302. [DOI] [PubMed] [Google Scholar]
- 37.Moss, B. 1996. Poxviridae: the viruses and their replication, p. 2637-2671. In B. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology. Lippincott-Raven Publishers, Philadelphia, PA.
- 38.Moss, B. 2001. Poxviridae: the viruses and their replication, p. 2849-2883. In Fields virology, vol. 2, 4th ed. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 39.Nabel, G., and D. Baltimore. 1987. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 326:711-713. [DOI] [PubMed] [Google Scholar]
- 40.Neznanov, N., K. M. Chumakov, L. Neznanova, A. Almasan, A. K. Banerjee, and A. V. Gudkov. 2005. Proteolytic cleavage of the p65-RelA subunit of NF-kappaB during poliovirus infection. J. Biol. Chem. 280:24153-24158. [DOI] [PubMed] [Google Scholar]
- 41.Nijhawan, D., M. Fang, E. Traer, Q. Zhong, W. Gao, F. Du, and X. Wang. 2003. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 17:1475-1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nolan, G. P., S. Ghosh, H. C. Liou, P. Tempst, and D. Baltimore. 1991. DNA binding and I kappa B inhibition of the cloned p65 subunit of NF-kappa B, a rel-related polypeptide. Cell 64:961-969. [DOI] [PubMed] [Google Scholar]
- 43.Oie, K. L., and D. J. Pickup. 2001. Cowpox virus and other members of the orthopoxvirus genus interfere with the regulation of NF-kappaB activation. Virology 288:175-187. [DOI] [PubMed] [Google Scholar]
- 44.Palombella, V. J., O. J. Rando, A. L. Goldberg, and T. Maniatis. 1994. The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell 78:773-785. [DOI] [PubMed] [Google Scholar]
- 45.Perkus, M. E., S. J. Goebel, S. W. Davis, G. P. Johnson, E. K. Norton, and E. Paoletti. 1991. Deletion of 55 open reading frames from the termini of vaccinia virus. Virology 180:406-410. [DOI] [PubMed] [Google Scholar]
- 46.Powell, P. P., L. K. Dixon, and R. M. Parkhouse. 1996. An IkappaB homolog encoded by African swine fever virus provides a novel mechanism for downregulation of proinflammatory cytokine responses in host macrophages. J. Virol. 70:8527-8533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Revilla, Y., M. Callejo, J. M. Rodriguez, E. Culebras, M. L. Nogal, M. L. Salas, E. Vinuela, and M. Fresno. 1998. Inhibition of nuclear factor kappaB activation by a virus-encoded IkappaB-like protein. J. Biol. Chem. 273:5405-5411. [DOI] [PubMed] [Google Scholar]
- 48.Ruben, S. M., P. J. Dillon, R. Schreck, T. Henkel, C. H. Chen, M. Maher, P. A. Baeuerle, and C. A. Rosen. 1991. Isolation of a rel-related human cDNA that potentially encodes the 65-kD subunit of NF-kappa B. Science 254:11. [DOI] [PubMed] [Google Scholar]
- 49.Santoro, M. G., A. Rossi, and C. Amici. 2003. NF-kappaB and virus infection: who controls whom? EMBO J. 22:2552-2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Satheshkumar, P. S., L. C. Anton, P. Sanz, and B. Moss. 2009. Inhibition of the ubiquitin-proteasome system prevents vaccinia virus DNA replication and expression of intermediate and late genes. J. Virol. 83:2469-2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saura, M., T. R. Lizarbe, C. Rama-Pacheco, C. J. Lowenstein, and C. Zaragoza. 2007. Inhibitor of NF kappa B alpha is a host sensor of coxsackievirus infection. Cell Cycle 6:503-506. [DOI] [PubMed] [Google Scholar]
- 52.Schroder, M., M. Baran, and A. G. Bowie. 2008. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKepsilon-mediated IRF activation. EMBO J. 27:2147-2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sciortino, M. T., M. A. Medici, F. Marino-Merlo, D. Zaccaria, M. Giuffre-Cuculletto, A. Venuti, S. Grelli, and A. Mastino. 2008. Involvement of HVEM receptor in activation of nuclear factor kappaB by herpes simplex virus 1 glycoprotein D. Cell. Microbiol. 10:2297-2311. [DOI] [PubMed] [Google Scholar]
- 54.Seet, B. T., J. B. Johnston, C. R. Brunetti, J. W. Barrett, H. Everett, C. Cameron, J. Sypula, S. H. Nazarian, A. Lucas, and G. McFadden. 2003. Poxviruses and immune evasion. Annu. Rev. Immunol. 21:377-423. [DOI] [PubMed] [Google Scholar]
- 55.Shisler, J. L., and X. L. Jin. 2004. The vaccinia virus K1L gene product inhibits host NF-kappaB activation by preventing IkappaBalpha degradation. J. Virol. 78:3553-3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stack, J., I. R. Haga, M. Schroder, N. W. Bartlett, G. Maloney, P. C. Reading, K. A. Fitzgerald, G. L. Smith, and A. G. Bowie. 2005. Vaccinia virus protein A46R targets multiple Toll-like-interleukin-1 receptor adaptors and contributes to virulence. J. Exp. Med. 201:1007-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stuart, D., K. Graham, M. Schreiber, C. Macaulay, and G. McFadden. 1991. The target DNA sequence for resolution of poxvirus replicative intermediates is an active late promoter. J. Virol. 65:61-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun, S. C., and D. W. Ballard. 1999. Persistent activation of NF-kappaB by the tax transforming protein of HTLV-1: hijacking cellular IkappaB kinases. Oncogene 18:6948-6958. [DOI] [PubMed] [Google Scholar]
- 59.Tait, S. W., E. B. Reid, D. R. Greaves, T. E. Wileman, and P. P. Powell. 2000. Mechanism of inactivation of NF-kappa B by a viral homologue of I kappa b alpha. Signal-induced release of i kappa b alpha results in binding of the viral homologue to NF-kappa B. J. Biol. Chem. 275:34656-34664. [DOI] [PubMed] [Google Scholar]
- 60.Tan, P., S. Y. Fuchs, A. Chen, K. Wu, C. Gomez, Z. Ronai, and Z. Q. Pan. 1999. Recruitment of a ROC1-CUL1 ubiquitin ligase by Skp1 and HOS to catalyze the ubiquitination of I kappa B alpha. Mol. Cell 3:527-533. [DOI] [PubMed] [Google Scholar]
- 61.Teale, A., S. Campbell, N. Van Buuren, W. C. Magee, K. Watmough, B. Couturier, R. Shipclark, and M. Barry. 2009. Orthopoxviruses require a functional ubiquitin-proteasome system for productive replication. J. Virol. 83:2099-2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Traenckner, E. B., S. Wilk, and P. A. Baeuerle. 1994. A proteasome inhibitor prevents activation of NF-kappa B and stabilizes a newly phosphorylated form of I kappa B-alpha that is still bound to NF-kappa B. EMBO J. 13:5433-5441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vallabhapurapu, S., and M. Karin. 2009. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 27:693-733. [DOI] [PubMed] [Google Scholar]
- 64.van Buuren, N., B. Couturier, Y. Xiong, and M. Barry. 2008. Ectromelia virus encodes a novel family of F-box proteins that interact with the SCF complex. J. Virol. 82:9917-9927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang, X. W., N. S. Tan, B. Ho, and J. L. Ding. 2006. Evidence for the ancient origin of the NF-kappaB/IkappaB cascade: its archaic role in pathogen infection and immunity. Proc. Natl. Acad. Sci. U. S. A. 103:4204-4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Willis, K. L., S. Patel, Y. Xiang, and J. L. Shisler. 2009. The effect of the vaccinia K1 protein on the PKR-eIF2alpha pathway in RK13 and HeLa cells. Virology 394:73-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Winston, J. T., P. Strack, P. Beer-Romero, C. Y. Chu, S. J. Elledge, and J. W. Harper. 1999. The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkappaBalpha and beta-catenin and stimulates IkappaBalpha ubiquitination in vitro. Genes Dev. 13:270-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Woronicz, J. D., X. Gao, Z. Cao, M. Rothe, and D. V. Goeddel. 1997. IkappaB kinase-beta: NF-kappaB activation and complex formation with IkappaB kinase-alpha and NIK. Science 278:866-869. [DOI] [PubMed] [Google Scholar]
- 69.Wyatt, L. S., P. L. Earl, L. A. Eller, and B. Moss. 2004. Highly attenuated smallpox vaccine protects mice with and without immune deficiencies against pathogenic vaccinia virus challenge. Proc. Natl. Acad. Sci. U. S. A. 101:4590-4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zandi, E., D. M. Rothwarf, M. Delhase, M. Hayakawa, and M. Karin. 1997. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell 91:243-252. [DOI] [PubMed] [Google Scholar]
- 71.Zaragoza, C., M. Saura, E. Y. Padalko, E. Lopez-Rivera, T. R. Lizarbe, S. Lamas, and C. J. Lowenstein. 2006. Viral protease cleavage of inhibitor of kappaBalpha triggers host cell apoptosis. Proc. Natl. Acad. Sci. U. S. A. 103:19051-19056. [DOI] [PMC free article] [PubMed] [Google Scholar]