

Abstract
The large RNA polymerase (L) protein of human parainfluenza virus type 2 (hPIV2) binds the nucleocapsid, phosphoprotein, and V protein, as well as itself, and these interactions are essential for transcription and replication of the viral RNA genome. Although all of these interactions were found to be mediated through the domains within the N terminus of L, the C terminus of the L protein was also required for minigenome reporter gene expression. We have identified a highly conserved rubulavirus domain near the C terminus of the L protein that is required for mRNA synthesis but not for genome replication. Remarkably, this region of L shares homology with a conserved region of cellular capping enzymes that binds GTP and forms a lysyl-GMP enzyme intermediate, the first step in the cellular capping reaction. We propose that this conserved region of L also binds GTP (or GDP) to carry out the second step of the unconventional nonsegmented negative-strand virus capping reaction.
Human parainfluenza virus type 2 (hPIV2) is a major human respiratory pathogen and a member of the Rubulavirus genus of the family Paramyxoviridae. Its negative-stranded RNA genome is 15,654 nucleotides (nt) long and encodes seven viral proteins from six genes: the nucleocapsid (NP), phosphoprotein (P) and V, matrix (M), hemagglutinin-neuraminidase (HN), fusion (F), and large RNA polymerase (L) proteins (9). The NP, P, and L proteins are required for transcription and replication of the viral RNA genome. The V mRNA is a faithful transcript of the V/P gene, whereas the P mRNA is synthesized through a cotranscriptional RNA editing process in which two pseudotemplated G residues are inserted into the mRNA during transcription. Therefore, the N-terminal 164 amino acids (aa) of the V and P proteins are common, while their C termini are unique (21). The C terminus of the V protein contains seven invariant cysteine residues capable of binding two atoms of zinc, and these C termini are approximately 50% identical among all paramyxovirus V proteins (9, 23).
The multifunctional L protein is the major polymerase subunit, required for RNA synthesis, mRNA capping, methylation, and polyadenylation. Although the structure of L has not been determined, alignment of the L proteins of the order Mononegavirales shows six regions of relatively high conservation (conserved region I [CRI] to CRVI), which are proposed to specify the essential activities common to all L proteins (24, 26). The L proteins of Sendai virus (SeV), measles virus (MeV), and PIV3 are present as oligomers, and these L-L interactions have been mapped to the N-terminal 200, 408, and 1,305 aa of the L protein, respectively (2, 3, 27). L-P complex formation is required to stabilize the L protein intracellularly (28), and the sites on L required for binding to the P protein have been mapped to the N-terminal 360 aa (SeV [3, 7]), 380 aa (rinderpest virus [RPV] [5]), 408 aa (MeV [2]), 1,305 aa (PIV3 [27]), and 1,247 aa (PIV5, formerly known as simian virus 5 [22]).
We previously identified regions on the PIV2 NP protein that are required for binding to the P, V, or L protein and for its assembly as nucleocapsids (13, 14, 16). We also identified regions on the P protein that are required for binding to NP or L protein and for its oligomerization (14, 17, 18). Moreover, the C-terminal region of the V protein was found to be required for binding to the L or NP proteins and for its oligomerization (12, 13). However, it was not clear which region(s) on the PIV2 L protein is required for binding to the NP, P, or V protein, and this question is addressed in the first part of this paper. We also identified a domain near the C terminus of the L protein that is essential for minigenome reporter gene expression, outside conserved domains I to VI. This region of L bears homology to a region of cellular capping enzymes that forms a phosphoramidate linkage to GMP, the first step of the cellular capping reaction. Mutational analysis showed that this L region, like that of the conserved HR domain in the middle of L, is required for mRNA synthesis but not for genome replication, consistent with its role in mRNA capping.
MATERIALS AND METHODS
Cells and antibodies.
BSR T7/5 (1) cells were cultured in Eagle's minimal essential medium supplemented with 10% fetal calf serum and 1 mg/ml G418 (Geneticin; Gibco). Monoclonal antibodies (MAbs) against hPIV2 P/V protein (315-1), hPIV2 NP protein (306-1), and hPIV2 L protein (L70-6) were as described previously (14, 16, 17). Anti-Flag polyclonal antibody and polyclonal antibody to green fluorescent protein (GFP) (sc-8334) were purchased from Sigma or Santa Cruz Biotechnology.
Construction of expression plasmids.
Various C-terminally truncated L genes were introduced by PCR amplification of the wild-type (wt) L gene using synthetic oligonucleotides corresponding to nucleotides of the L mRNA, including an in-frame stop codon. Various L genes, the NP gene, the P gene, and the V gene of hPIV2 cloned into pTM1, which contains a T7 promoter and an encephalomyocarditis virus (EMCV) internal ribosome entry site (IRES) (B. Moss, National Institutes of Health), were as described previously (15). Plasmid pPIV2-GFP was as described previously (12). Plasmid pPIV2, containing the full-length cDNA, was as described previously (15). pPIV2-LΔC plasmids carrying various C-terminally truncated L genes were constructed similarly to what was described previously (15). All of these constructs were confirmed by DNA sequencing.
Immunoprecipitation analysis.
BSR T7/5 cells in six-well plates were transfected with 1 μg pTM1-L, pTM1-L mutants, pTM1-P, pTM1-V, pTM1-NP, or pTM1-FlagL and 5 μl of FuGENE 6 (Roche) according to the manufacturer's instructions. At 42 h posttransfection (hpt), cells were lysed in lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM.NaCl, 0.6% NP-40, and 4 mM phenylmethylsulfonyl fluoride). The supernatants obtained by centrifugation were incubated with MAbs or anti-Flag and protein A-Sepharose for 6 h as described previously (18). Polypeptides were analyzed by a Western blotting technique. Cell lysates were also subjected directly to Western blotting with MAbs or anti-Flag to confirm expression of the proteins.
Transient expression analysis.
Analysis of transient minigenome-carried gene expression was performed in BSR T7/5 cells cultured in six-well plates. pPIV2-GFP, pTM1-P, pTM1-NP, and various pTM1-L plasmids were transfected into the cells by using FuGENE 6. The amounts of plasmids per well were as follows: pPIV2-GFP, 1 μg; pTM1-NP, 0.75 μg; pTM1-P, 0.4 μg; and pTM1-L or mutants, 0.75 μg. After 2 days, the transfected cells were lysed in lysis buffer. The cell extracts were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and analyzed by a Western blot technique.
Viral growth kinetics.
Monolayers of Vero cells were infected with several viruses at a multiplicity of infection (MOI) of about 0.01 and incubated at 37°C in minimal essential medium (MEM) with 2% fetal calf serum. Supernatants were harvested at 48 or 72 h postinfection, and virus titers were determined by plaque assay on Vero cells as described previously (15).
RNA isolation, RT-PCR, and primer extension analysis.
Total RNAs were isolated from transfected cells by use of Isogen (Nippongene) according to the manufacturer's protocol. After treatment with DNase I (Invitrogen) to eliminate possible DNA contamination, the first-strand cDNA synthesis was carried out by use of the SuperScript first-strand synthesis system for reverse transcription-PCR (RT-PCR) (Invitrogen) with 2 μg of total RNA and oligo(dT)12-18 for mRNA or the specific primer for the GFP gene (5′-CAAGTCCGGACTCAGATCT-3′) for antigenome. For PCRs, 5 μl of RT reaction product was used in a reaction volume of 50 μl. GFP gene-specific primers were 5′-ATGGTGAGCAAGGGCGAG-3′ and 5′-TTAAGATCTGAGTCCGGACTTG-3′. The actin control primers were 5′-TTGTCACCAACTGGGACGATATGG-3′ and 5′-GATCTTGATCTTCATGGTGCTAG-3′. Ten-microliter samples from each PCR were analyzed in 1.5% agarose gels.
The same RNAs (10 μg total) were analyzed with 0.25 pmol of 32P-5′-CTCCTCGCCCTTGCTCACC-3′, representing complementary positions 178 to 160 from the 5′ end of the antigenome, using Moloney murine leukemia virus (MMLV) reverse transcriptase (RTase) (Promega). The extensions were examined on a 6% sequencing gel.
RESULTS
The C-terminal region of the L protein is not required for its interaction with other viral proteins.
To identify the essential regions for hPIV2 L-P, L-V, and L-NP complex formation, a series of C-terminally truncated PIV2 L proteins were constructed and expressed together with the P, V, or NP proteins (Fig. 1A, rows 2 to 7). At 48 h posttransfection, cell lysates were immunoprecipitated with anti-P/V monoclonal antibody (MAb) (Fig. 1B and C) or anti-NP MAb (Fig. 1D). As shown in Fig. 1B, the complexes of P/L-575 and P/L-374 were not detected (lanes 6 and 7). The N-terminal 747 aa of the L protein are thus necessary for L interaction with P. As shown in Fig. 1C and D, truncated L-374, in which 1,889 residues are removed from the C terminus of the full-length 2,263-residue L protein, still formed L-V and L-NP complexes. Thus, the N-terminal 374 aa on the L protein are sufficient for interaction with the V or NP proteins.
FIG. 1.

Analysis of interactions between the C-terminally truncated L and P, V, NP, or L proteins by immunoprecipitation (IP) and Western blot (WB) assay. (A) Schematic diagram of the C-terminally truncated L proteins. Binding to P, V, NP, or L protein is summarized at right. (B to E) The proteins from transfected BSR T7/5 cell lysates were analyzed by Western blotting for anti-P/V MAb (B and C), anti-NP MAb (D), or anti-Flag polyclonal antibody (E) (top panels) and anti-L MAb (middle panels). Immunoprecipitates with anti-P/V MAb (B and C), anti-NP MAb (D), or anti-Flag antibody (E) were probed by anti-L MAb in a Western blot assay (bottom panels). Numbers on the bottom correspond to the L proteins depicted in panel A. Sizes of molecular mass markers (in kDa) are shown. The asterisk at the left indicates the immunoglobulin heavy chain.
To test whether the hPIV2 L protein forms an L-L complex, pTM1-FlagL (L fused to an N-terminal Flag epitope tag) was constructed. Flag-tagged hPIV2 L and a series of C-terminal truncations were expressed in BSR T7/5 cells and immunoprecipitated by anti-Flag polyclonal antibody (Fig. 1E). Flag-tagged L protein could not coprecipitate L-575 and L-374 (Fig. 1E, lanes 6 and 7). Thus, the N-terminal 747 aa of L are necessary for its oligomerization, as well as its interaction with P. These data are summarized in Fig. 1A.
The N-terminal region of the L protein is essential for its interaction with other viral proteins.
To further study L complex formation, we also constructed a series of N-terminal truncations as illustrated in Fig. 2A. The Flag tag is cloned in frame with the N-terminal end of the L-Stu protein that is necessary for L interactions with other viral proteins. Flag-L/Stu could coprecipitate the P, V, NP, and L proteins by anti-Flag polyclonal antibody (Fig. 2B, C, D, and E, lanes 2). However, deletion of the N-terminal 23 aa ablated its interaction with the P, V, NP, and L proteins (Fig. 2B, C, D, and E, lanes 3). These data are summarized in Fig. 2A. From the results of Fig. 1 and 2, amino acids within the N-terminal 374 aa of the L protein are essential for L interaction with the V and NP proteins and aa 1 to 747 are essential for L interaction with the P protein and L oligomerization.
FIG. 2.

Analysis of interactions between the N- and C-terminally truncated L and P, V, NP, or L proteins by immunoprecipitation and Western blot assay. (A) Schematic diagram of the N- and C-terminally truncated L proteins. Binding to P, V, NP, or L protein is summarized at right. (B to E) The proteins from transfected BSR T7/5 cell lysates were analyzed by Western blotting for anti-P/V MAb (B and C), anti-NP MAb (D), or anti-L MAb (E) (top panels) and for anti-Flag polyclonal antibody (middle panels). Immunoprecipitates with anti-P/V MAb (B and C), anti-NP MAb (D), or anti-Flag polyclonal antibody (E) were probed by anti-Flag polyclonal antibody or anti-L MAb in Western blot assays (bottom panels). Numbers on the bottom correspond to the L proteins depicted in panel A. Sizes of molecular mass markers (in kDa) are shown.
A region of the L protein essential for minigenome reporter gene expression.
We recently established a vaccinia virus-free minigenome system for hPIV2, in which viral RNA synthesis can be studied in the absence of virus infection (Fig. 3A) (12). BSR T7/5 cells are transfected with a minigenome plasmid (pPIV2-GFP), containing the hPIV2 leader and trailer and a reporter gene (GFP), together with the plasmids encoding NP, P, and L proteins (pTM1-NP, pTM1-P, and pTM1-L). When all three support proteins are expressed, the minigenome template is assembled (and replicated) and transcribed into the reporter gene mRNA, resulting in GFP expression. The N-terminal 747 L protein residues are important for L interaction with the other polymerase subunits and are thus essential for its function. To assess the importance of the C-terminal region of L, a set of C-terminally truncated L proteins was tested. As shown in Fig. 3B, whereas removal of the C-terminal 30 aa had little effect on GFP expression, removal of the C-terminal 40 aa eliminated all activity. We also attempted hPIV2 recovery using these C-terminally truncated L proteins. Plasmid pPIV2 or pPIV2-LΔC carrying various C-terminally truncated L genes at 10 μg together with pTM1-NP at 2 μg, pTM1-P at 1 μg, and C-terminally truncated mutants of pTM1-L at 2 μg was transfected into BSR T7/5 cells. After 2 days, the supernatants of the transfected cells were used to infect Vero cells. We rescued infectious viruses from a set of plasmids including L-CΔ10, L-CΔ20, or L-CΔ30 but not L-CΔ40 or L-CΔ50 ( Table 1). These results confirmed that a region located 30 to 40 residues from the C terminus of L is essential for minigenome reporter gene expression and virus recovery.
FIG. 3.

GFP expression from the minigenome system by C-terminally truncated L proteins. (A) Schematic diagram of the minigenome system. Plasmids pTM1-NP, pTM1-P, and pTM1-L were used to express NP, P, and L proteins in BSR T7/5 cells, a cell line that constitutively expresses T7 RNA polymerase (RNAP). The negative-strand minigenome can be generated from transcription of primary T7 transcript in BSR T7/5 cells. This template is assembled and transcribed into the reporter gene mRNA, resulting in expression of GFP as well as being used as a template for replication by a complex of NP, P, and L proteins. (B) The plasmids pPIV2-GFP (1 μg), pTM1-NP (0.75 μg), pTM1-P (0.4 μg), and pTM1-L or mutants (0.75 μg) were transfected into BSR T7/5 cells. At 48 h posttransfection, cells were lysed and assayed by Western blotting using antibodies against NP, P, and L proteins and GFP.
TABLE 1.
Growth kinetics of recombinant PIV2s carrying C-terminally truncated L proteins in tissue culturea
| L protein | Virus growth at h p.i.: |
|
|---|---|---|
| 48 | 72 | |
| wt | 7.0 × 106 | 2.5 × 107 |
| C-terminally truncated | ||
| CΔ10 | 5.0 × 106 | 1.8 × 107 |
| CΔ20 | 5.2 × 106 | 2.0 × 107 |
| CΔ30 | 8.2 × 105 | 1.0 × 107 |
| CΔ40 | 0 | 0 |
| CΔ50 | 0 | 0 |
Monolayers of Vero cells were infected with the various recombinant PIV2s at an MOI of ca. 0.01. Samples of supernatants were harvested at 48 or 72 h postinfection (p.i.), and their titers (PFU/ml) were determined by plaque formation on Vero monolayers (Materials and Methods).
Identification of amino acids essential for L protein activity.
Figure 4A shows the sequence located between 30 and 50 residues from the C terminus of L aligned with those of the other rubulavirus L genes sequenced to date: simian virus 41 (SV41), PIV5, mumps virus (MuV), PIV4A, PIV4B, Mapuera virus (MPRV), porcine rubulavirus (PoRV), Menangle virus (MenV), and Tioman virus (TiV). The sequence of −49 to −38 (from the C terminus) is highly conserved among rubulaviruses, henipaviruses, and Newcastle disease virus (NDV). When the conserved residues of the 2214R (P/S) X Q K (Q/R) (I/V) W K X (L/I/V) G2225 consensus sequence (amino acids in bold are conserved among rubulaviruses) were individually mutated to Ala, none of these mutations by themselves eliminated GFP expression (Fig. 4C). Moreover, the mutation of both lysines (K2218 and K2222) or all 3 basic residues (R2214, K2218, and K2222) to alanine had no effect on GFP expression. However, when both lysines plus G2225, Q2217 plus W2221, or W2221 plus G2225 was mutated to Ala, GFP expression was eliminated. As G2225 appeared to be particularly important, we also mutated this residue to Q and M to introduce a much bulkier side chain at this position (Fig. 5A, rows 3 and 4). Both of these point mutations also eliminated GFP expression (Fig. 5B, lanes 3 and 4). Thus, a domain near the C terminus of the rubulavirus L proteins is clearly important for minigenome reporter gene expression. It was recently reported that the HR motif of CRV is essential for mRNA capping (10, 19). We mutated these residues of the hPIV2 HR motif to Ala (Fig. 5A, rows 5 to 7). These mutants also eliminated GFP expression (Fig. 5B, lanes 5 to 7).
FIG. 4.

GFP expression from the minigenome system by L proteins that have mutations in the amino acids conserved among paramyxoviruses. (A) Comparison of sequences at the C termini of paramyxovirus L proteins. hPIV2, human parainfluenza virus type 2 (GenBank accession number AB176531); SV41, simian virus 41 (NC 006428); PIV5, parainfluenza virus type 5 (NC 006430); MuV, mumps virus (D10575); PIV4A or -4B, parainfluenza virus type 4A (AB543336) or 4B (AB543337); MPRV, Mapuera virus (EF095490); PoRV, porcine rubulavirus (NC 009640); MenV, Menangle virus (AF326114); TiV, Tioman virus (AF298895); NDV, Newcastle disease virus; HeV, Hendra virus; NiV, Nipah virus; MeV, measles virus; RPV, rinderpest virus; SeV, Sendai virus; PIV3, parainfluenza virus type 3. The amino acids are numbered from the amino terminus of each L protein. The asterisks indicate amino acids conserved among rubulaviruses. (B) Schematic diagram of L proteins that have mutations in the conserved motif. (C) GFP expression from the minigenome system by the L mutants as shown in Fig. 3. Numbers on the bottom correspond to each L mutant protein listed in panel B.
FIG. 5.

GFP expression from the minigenome system by L proteins that have mutations in the conserved amino acids. (A) Schematic diagram of the L mutants. (B) GFP expression from minigenome system by the L mutants as shown in Fig. 3. Numbers on the bottom correspond to each L mutant protein listed in panel A.
A C-terminal L protein domain required for mRNA capping.
When the Swiss-Prot database is scanned for this conserved sequence (R [P/S] X Q K [Q/R] [I/V] W K X [L/I/V] G), a conserved region of cellular capping enzymes is identified (Fig. 6). This conserved cellular sequence can also be aligned (albeit to a lesser extent) with the same region of all paramyxovirus L proteins, including those of the pneumoviruses. However, this conserved sequence cannot be aligned with any of the rhabdovirus L proteins. This sequence conservation between paramyxovirus L proteins and cellular capping enzymes was indeed surprising, because nonsegmented negative-strand viruses (NNSV) cap their mRNAs differently than do cellular and other eucaryotic capping enzymes. Cellular or conventional capping enzymes are guanylyltransferases, in which a conserved lysine acts as a nucleophile to form a phosphoramidate linkage to GMP (5′ pG), which is then transferred to 5′ ppRNA to form a GpppRNA cap (25). Rhabdovirus (and presumably all NNSV) capping enzymes, in contrast, are polyribonucleotidyltransferases, in which a histidine of the conserved HR motif (at least for rhabdoviruses and probably for all NNSV) forms a phosphoramidate linkage to 5′ pRNA, which is then transferred to 5′ ppG to form the same GpppRNA cap (20).
FIG. 6.

Alignment of negative-stranded RNA virus polymerase domains displaying homology with the catalytic site of eukaryotic mRNA guanylyltransferases. The catalytic lysine is marked in bold. SeV, Sendai virus (UniProtKB accession number Q9DUD8); PIV3, parainfluenza virus type 3 (P12577); MeV, measles virus (P12576); RPV, rinderpest virus (P41357); CDV, canine distemper virus (P24658); HRSVA, human respiratory syncytial virus A (strain A2) (P28887); HRSVL, human respiratory syncytial virus A (long strain) (Q9IWW8); HRSVB, human respiratory syncytial virus B (strain B1) (O36635); BRSV, bovine respiratory syncytial virus (O91940); MPV, murine pneumonia virus (Q50EW2); NDV, Newcastle disease virus (Q9DLD3); HeV, Hendra virus (O89344); NiV, Nipah virus (Q997F0); hPIV2, human parainfluenza virus type 2 (P26676); SV41, simian virus 41 (P35341); PIV5, parainfluenza virus type 5 (Q03396); MuV, mumps virus (P30929); mouse (O55236); rat (B5DFA8); bovine (Q2KHX7); human (O60942); Xenopus laevis (Q6NWX1); zebrafish (Q6NY98); tick (B7PBC2); Drosophila melanogaster (Q9VY44); yeast, Saccharomyces cerevisiae (Q01159); PBCV, Paramecium bursaria Chlorella virus (Q84424); VARV, variola major virus (P33057). ScanProsite software (http://www.expasy.org/tools/scanprosite/) was used to scan the experimentally defined C-terminal motif against the UniProt/Swiss-Prot database. The alignment of paramyxovirus polymerases was made by the Muscle method; cellular motifs were added manually.
Capping of NNSV mRNAs is thought to occur shortly after their synthesis is initiated (when the nascent mRNA is 40 to 50 nucleotides [nt] long), and nascent mRNAs which are not capped are thought not to be elongated further (an mRNA quality control step appears to operate at this point) (29). We reasoned that a capping-deficient L protein should specifically ablate GFP mRNA synthesis in our minigenome system, as minigenome replication depends only on the products of the N, P, and L support plasmids, whose mRNAs are translated due to the presence of an IRES and need not be capped by the L protein. To determine whether mutations in this conserved sequence that ablated GFP expression specifically affected mRNA synthesis, mRNA and antigenome levels were quantified by RT-PCR and primer extension. As shown in Fig. 7A and B, L proteins in which the C-terminal 50 aa were deleted (lanes 4); in which K2218, K2222, and G2225 were mutated to Ala (lanes 5); in which G2225 was mutated to methionine (lanes 6); or in which H1298 and R1299 were mutated to Ala (lanes 7) all eliminated GFP mRNA accumulation [in Fig. 7A, whose RTase reaction was primed by oligo(dT)12-18], whereas these mutations had little or no effect on antigenome accumulation (in Fig. 7A, whose RTase reaction was primed by the GFP reverse primer). Thus, mutations in either the HR motif of CRV or the conserved C-terminal domain appear to specifically target reporter gene mRNA synthesis, consistent with the function of both regions in mRNA capping.
FIG. 7.
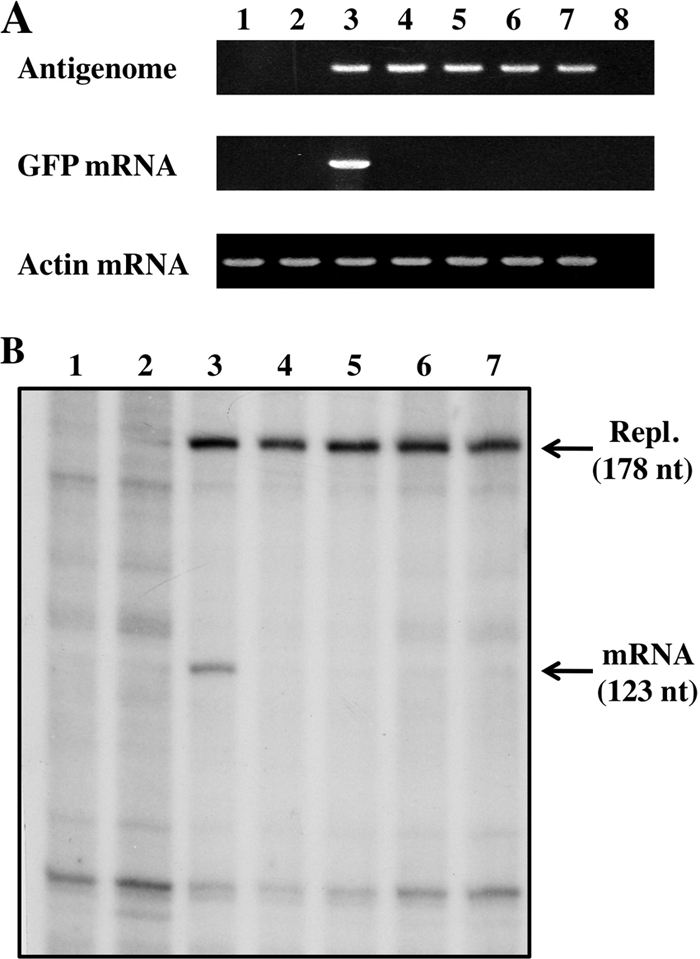
Minigenome RNA expression and L mutants. (A) RT-PCR. Total RNAs were purified from BSR T7/5 cells transfected with pPIV2-GFP, pTM1-P, pTM1-NP, and pTM1-L or mutants. RT-PCRs were carried out as described in Materials and Methods. Ethidium bromide staining of products from the PCRs is shown. Lane 1, untransfected; lane 2, without pTM1-L; lane 3, PIV2 wt; lane 4, C-terminal-50-aa-truncated L (Fig. 3B, CΔ50); lane 5, K2218/2222/G2225A (Fig. 4B, row 4); lane 6, G2225M (Fig. 5A, row 4); lane 7, H1298/R1299A (Fig. 5A, row 7); lane 8, without the RT reaction. (B) Primer extension. The same RNAs were analyzed by primer extension, using a 32P-oligonucleotide representing positions 178 to 160 (Materials and Methods). Lane 1, untransfected; lane 2, without pTM1-L; lane 3, PIV2 wt; lane 4, C-terminal-50-aa-truncated L; lane 5, K2218/2222/G2225A; lane 6, G2225M; lane 7, H1298/R1299A.
DISCUSSION
Paramyxoviruses are similar in genome structure and replication strategy. A common theme in their replication is the use of a multifunctional L protein to catalyze many of the steps in RNA synthesis and processing. These functions are carried out by a polymerase complex, not only the L protein (9). Previous work with SeV, PIV3, MeV, and PIV5 L proteins showed that their N-terminal regions were required for binding to their respective P proteins or oligomerization (2, 3, 7, 22, 27, 28). In this study, we first identified the region(s) on the hPIV2 L protein that is essential for binding other viral proteins. All these regions mapped within the N-terminal third of hPIV2 L, similar to other paramyxoviruses. Likewise, an N-terminal PIV2 L protein site also forms oligomers, where L-L complex formation is independent of other viral proteins. While the L proteins of a number of NNSV appear to form oligomers, the degree of the L oligomerization and its function are unknown. Surprisingly, hPIV2 L missing only the N-terminal 23 amino acids had lost its interaction with other viral proteins. Recently, Cevik et al. (4) showed that SeV L with deletion of the first 25 aa had lost its biological activity but that this deletion did not affect P-L or L-L interactions.
NNSV L proteins contain 6 conserved regions (I to VI) that reside within aa 230 to 1857 of the hPIV2 L protein. Other highly conserved L protein domains are located in the N- and C-terminal regions: aa 7 to 19 and aa 1987 to 1993 on the hPIV2 L protein (8, 24, 26). By testing C-terminally truncated L proteins in a GFP-expressing minigenome system, we found that the removal of the C-terminal 40 aa abolished its activity. As shown in Fig. 4A, the sequence of −49 to −38 (from the C terminus) is highly conserved among rubulaviruses, henipaviruses, and NDV, whereas the remainder of the C-terminal 250 residues is poorly conserved. This island of strong conservation (−49R-[P/S]-x-Q-K-[Q/R]-[I/V]-W-K-x-[L/I/V]-G−38) contains 3 invariant basic residues (and no acidic residues) and one invariant aromatic residue, in a hydrophobic background. Mutational analysis of this C-terminal rubulavirus sequence clearly demonstrated its importance for minigenome reporter gene expression and specifically for mRNA synthesis. This C-terminal domain is not required for genome replication.
It is indeed surprising that paramyxovirus L proteins have conserved part of the motif that characterizes cellular capping enzymes (25). Significantly, this cellular motif includes the conserved lysine that acts as a nucleophile to form the enzyme-5′ pG intermediate, the first step of the conventional capping reaction. In the case of hPIV2, neither this lysine nor the two other basic residues of this conserved C-terminal domain are essential for capping, as their mutation to alanine has no effect on GFP expression (Fig. 4). In contrast, mutation of either residue of the hPIV2 HR motif to alanine eliminates GFP expression without affecting genome replication, i.e., it appears to specifically target mRNA synthesis (Fig. 5 and 7). The HR motif is part of a larger GxxT[n]HR motif of CRV of NNSV L proteins (where n is ∼80 residues in length) (10). Mutation of these conserved residues to Ala eliminates capping activity in a reconstituted vesicular stomatitis virus (VSV) capping reaction. More recently, the histidine of the HR motif was shown to act as the nucleophile that forms the VSV L-5′ pRNA intermediate (19), the first step of the VSV capping reaction.
Liuzzi et al. (11) have studied small molecule inhibitors of mRNA capping by the L protein of human respiratory syncytial virus (RSV), a pneumovirus that contains the conserved C-terminal domain (Fig. 6). They found that mutant viruses resistant to this inhibition carry L proteins with amino acid substitutions near the HR motif. Thus, if as seems increasingly likely, the histidine of the hPIV2 HR motif also acts as the nucleophile that forms the L-5′ pRNA intermediate, why should paramyxovirus L proteins have conserved a domain of cellular capping enzymes? In contrast to NNSV L proteins, for which structure-function analysis is in its infancy, a great deal is known about conventional capping enzymes, and a possible answer to this conservation may be gleaned by examining how guanylyltransferases carry out their reaction. Conventional capping enzymes are part of a superfamily of nucleotidyltransferases, including ATP- and NAD-dependent DNA ligases and ATP-dependent RNA ligases (25). This superfamily is defined by five peptide motifs that line the nucleotide binding pocket and contribute amino acid side chains essential for catalysis. The region of cellular capping enzymes conserved in rubulavirus L proteins includes the WKADG sequence (part of the most N-terminal peptide motif I, which includes the nucleophilic lysine [bold]; WKALG in rubulaviruses), which is separated from the presumably catalytic HR motif by almost 1,000 aa. Besides forming a histidyl-5′ pRNA intermediate, NNSV L proteins must also bind GTP, remove its gamma-PO4, and then present 5′ ppG as a nucleophile that attacks the histidyl-5′ pRNA intermediate, to effect the transfer of pRNA to 5′ ppG. We presume that motif I has been conserved in rubulavirus L proteins because it continues to act as a GTP (and/or GDP) binding site, in this case for the second step of the unconventional NNSV capping reaction.
For cellular capping enzymes, substrate binding and the chemical steps of the reaction pathway are coordinated with large rearrangements of the component protein domains (25). During the catalytic cycle, for example, rearrangements of domain I which close and reopen the active site twice occur, entailing movement of this domain by 80 Å (6). If the conserved rubulavirus C-terminal domain functions to present GDP to histidyl-pRNA at the catalytic HR domain, these two domains must directly interact with each other for the second step of the transfer reaction to occur, possibly also over large distances. Given that the NNSV capping reaction is so intimately connected to the mechanism of viral mRNA synthesis, e.g., occurring only when the nascent mRNA chain is presumably just long enough to interact with the HR domain, and given the further elongation of the mRNA being dependent on its successful capping at this stage, a choreography of large domain rearrangements may also be a hallmark of NNSV L protein's multiple catalytic properties.
Finally, although this C-terminal L protein domain is present in all paramyxoviruses and avipaviruses, this sequence is absent in rhabdoviruses and filoviruses. It is likely that another sequence of these other NNSV L proteins, possibly in the same relative location, functions in its stead, e.g., to present 5′ ppG to the histidyl-pRNA intermediate. In that case, it would appear that paramyxoviruses and avipaviruses may have been the first among NNSV to have evolved from a primordial ancestor whose mRNA capping activity was derived from a cellular gene. The other NNSV L proteins may then have evolved another sequence to fill this function.
Acknowledgments
This study was supported in part by a grant-in-aid for scientific research from the Ministry of Education, Science, Culture, Sports and Technology of Japan and by the Swiss National Fund.
Footnotes
Published ahead of print on 10 November 2010.
REFERENCES
- 1.Buchholz, U. J., S. Finke, and K. K. Conzelmann. 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 73:251-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cevik, B., D. E. Holmes, E. Vrotsos, J. A. Feller, A. Smallwood, and S. A. Moyer. 2004. The phosphoprotein (P) and L binding sites reside in the N-terminus of the L subunit of the measles virus RNA polymerase. Virology 327:297-306. [DOI] [PubMed] [Google Scholar]
- 3.Cevik, B., S. Smallwood, and S. A. Moyer. 2003. The L-L oligomerization domain resides at the very N-terminus of the Sendai virus L RNA polymerase protein. Virology 313:525-536. [DOI] [PubMed] [Google Scholar]
- 4.Cevik, B., S. Smallwood, and S. A. Moyer. 2007. Two N-terminal regions of the Sendai virus L RNA polymerase protein participate in oligomerization. Virology 363:189-197. [DOI] [PubMed] [Google Scholar]
- 5.Chattopadhyay, A., and M. S. Shaila. 2004. Rinderpest virus RNA polymerase subunits: mapping of mutual interacting domains on the large protein L and phosphoprotein P. Virus Genes 28:352-363. [DOI] [PubMed] [Google Scholar]
- 6.Håkansson, K., A. J. Doherty, S. Shuman, and D. B. Wigley. 1997. X-ray crystallography reveals a large conformational change during guanyl transfer by mRNA capping enzymes. Cell 89:545-553. [DOI] [PubMed] [Google Scholar]
- 7.Holmes, D. E., and S. A. Moyer. 2002. The phosphoprotein (P) binding site resides in the N terminus of the L polymerase subunit of Sendai virus. J. Virol. 76:3078-3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawano, M., K. Okamoto, H. Bando, K. Kondo, M. Tsurudome, H. Komada, M. Nishio, and Y. Ito. 1991. Characterizations of the human parainfluenza type 2 virus gene encoding the L protein and the intergenic sequences. Nucleic Acids Res. 19:2739-2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lamb, R. A., and G. D. Parks. 2007. Paramyxoviridae: the viruses and their replication, p. 1449-1490. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 10.Li, J., A. Rahmeh, M. Morelli, and S. P. Whelan. 2008. A conserved motif in region V of the large polymerase proteins of nonsegmented negative-sense RNA viruses that is essential for mRNA capping. J. Virol. 82:775-784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liuzzi, M., S. W. Mason, M. Cartier, C. Lawetz, R. S. McCollum, N. Dansereau, G. Bolger, N. Lapeyré, Y. Gaudette, L. Lagace, M. J. Massariol, F. Dô, P. Whitehead, L. Lamarre, E. Scouten, J. Bordeleau, S. Landry, J. Rancourt, G. Fazal, and B. Simoneau. 2005. Inhibitors of respiratory syncytial virus replication target cotranscriptional mRNA guanylylation by viral RNA-dependent RNA polymerase. J. Virol. 79:13105-13115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nishio, M., J. Ohtsuka, M. Tsurudome, T. Nosaka, and D. Kolakofsky. 2008. Human parainfluenza virus type 2 V protein inhibits genome replication by binding to the L protein; possible role in promoting viral fitness. J. Virol. 82:6130-6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishio, M., M. Tsurudome, H. Ishihara, M. Ito, and Y. Ito. 2007. The conserved carboxyl terminus of human parainfluenza virus type 2 V protein plays an important role in virus growth. Virology 362:85-98. [DOI] [PubMed] [Google Scholar]
- 14.Nishio, M., M. Tsurudome, M. Ito, and Y. Ito. 2000. Mapping of domains on the human parainfluenza type 2 virus P and NP proteins that are involved in the interaction with the L protein. Virology 273:241-247. [DOI] [PubMed] [Google Scholar]
- 15.Nishio, M., M. Tsurudome, M. Ito, D. Garcin, D. Kolakofsky, and Y. Ito. 2005. Identification of paramyxovirus V protein residues essential for STAT protein degradation and promotion of virus replication. J. Virol. 79:8591-8601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishio, M., M. Tsurudome, M. Ito, M. Kawano, S. Kusagawa, H. Komada, and Y. Ito. 1999. Mapping of domains on the human parainfluenza virus type 2 nucleocapsid protein (NP) required for NP-phosphoprotein or NP-NP interaction. J. Gen. Virol. 80:2017-2022. [DOI] [PubMed] [Google Scholar]
- 17.Nishio, M., M. Tsurudome, M. Ito, N. Watanabe, M. Kawano, H. Komada, and Y. Ito. 1997. Human parainfluenza virus type 2 phosphoprotein: mapping of monoclonal antibody epitopes and location of the multimerization domain. J. Gen. Virol. 78:1303-1308. [DOI] [PubMed] [Google Scholar]
- 18.Nishio, M., M. Tsurudome, M. Kawano, N. Watanabe, S. Ohgimoto, M. Ito, H. Komada, and Y. Ito. 1996. Interaction between nucleocapsid protein (NP) and phosphoprotein (P) of human parainfluenza virus type 2: one of the two NP binding sites on P is essential for granule formation. J. Gen. Virol. 77:2457-2463. [DOI] [PubMed] [Google Scholar]
- 19.Ogino, T., S. P. Yadav, and A. K. Banerjee. 2010. Histidine-mediated RNA transfer to GDP for unique mRNA capping by vesicular stomatitis virus RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 107:3463-3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ogino, T., and A. K. Banerjee. 2007. Unconventional mechanism of mRNA capping by the RNA-dependent RNA polymerase of vesicular stomatitis virus. Mol. Cell 25:85-97. [DOI] [PubMed] [Google Scholar]
- 21.Ohgimoto, S., H. Bando, M. Kawano, K. Okamoto, K. Kondo, M. Tsurudome, M. Nishio, and Y. Ito. 1990. Sequence analysis of P gene of human parainfluenza type 2 virus: P and cysteine-rich proteins are translated by two mRNAs that differ by two nontemplated G residues. Virology 177:116-123. [DOI] [PubMed] [Google Scholar]
- 22.Parks, G. 1994. Mapping of a region of the paramyxovirus L protein required for the formation of a stable complex with the viral phosphoprotein P. J. Virol. 68:4862-4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paterson, R. G., M. G. Leser, P. A. Shaughnessy, and R. A. Lamb. 1995. The paramyxovirus SV5 V protein binds two atoms of zinc and is a structural component of virions. Virology 208:121-131. [DOI] [PubMed] [Google Scholar]
- 24.Poch, O., B. M. Blumberg, L. Bougueleeret, and N. Tordo. 1990. Sequence comparison of five polymerases (L proteins) of unsegmented negative-strand RNA viruses: theoretical assignment of functional domains. J. Gen. Virol. 71:1153-1162. [DOI] [PubMed] [Google Scholar]
- 25.Shuman, S., and C. D. Lima. 2004. The polynucleotide ligase and RNA capping enzyme superfamily of covalent nucleotidyltransferases. Curr. Opin. Struct. Biol. 14:757-764. [DOI] [PubMed] [Google Scholar]
- 26.Sidhu, M. S., J. P. Menonna, S. D. Cook, and S. A. Udem. 1993. Canine distemper virus L gene; sequence and comparison with related viruses. Virology 193:50-65. [DOI] [PubMed] [Google Scholar]
- 27.Smallwood, S., and S. A. Moyer. 2004. The L polymerase protein of parainfluenza virus 3 forms an oligomer and can interact with the heterologous Sendai virus L, P and C proteins. Virology 318:439-450. [DOI] [PubMed] [Google Scholar]
- 28.Smallwood, S., K. W. Ryan, and S. A. Moyer. 1994. Deletion analysis defines a carboxyl-proximal region of Sendai virus P protein that binds to the polymerase L protein. Virology 202:154-163. [DOI] [PubMed] [Google Scholar]
- 29.Stillman, E. A., and M. A. Whitt. 1999. Transcript initiation and 5′-end modifications are separable events during vesicular stomatitis virus transcription. J. Virol. 73:7199-71209. [DOI] [PMC free article] [PubMed] [Google Scholar]