

Abstract
The hemagglutinin (HA) surface glycoprotein promotes influenza virus entry and is the key protective antigen in natural immunity and vaccines. The HA protein is a trimeric envelope glycoprotein consisting of a globular receptor-binding domain (HA-RBD) that is inserted into a membrane fusion-mediating stalk domain. Similar to other class I viral fusion proteins, the fusogenic stalk domain spontaneously refolds into its postfusion conformation when expressed in isolation, consistent with this domain being trapped in a metastable conformation. Using X-ray crystallography, we show that the influenza virus HA-RBD refolds spontaneously into its native, immunogenic structure even when expressed in an unglycosylated form in Escherichia coli. In the 2.10-Å structure of the HA-RBD, the receptor-binding pocket is intact and its conformational epitopes are preserved. Recombinant HA-RBD is immunogenic and protective in ferrets, and the protein also binds with specificity to sera from influenza virus-infected humans. Overall, the data provide a structural basis for the rapid production of influenza vaccines in E. coli. From an evolutionary standpoint, the ability of the HA-RBD to refold spontaneously into its native conformation suggests that influenza virus acquired this domain as an insertion into an ancestral membrane-fusion domain. The insertion of independently folding domains into fusogenic stalk domains may be a common feature of class I viral fusion proteins.
The genetic drift of seasonal influenza viruses and the occasional emergence of pandemic strains represent a continuing and serious burden on human health. Pandemic influenza viruses arise at irregular intervals, can infect up to 50% or more of the population, and vary in disease severity. Most notably, the H1N1 Spanish influenza pandemic of 1918 killed an estimated 20 to 50 million people worldwide, and the 1957 H2N2 Asian flu and 1968 H3N2 Hong Kong flu pandemics killed between 0.5 and 1 million people in the United States alone (30). The ongoing danger of influenza was recently emphasized by the emergence of the novel H1N1 pandemic virus from Mexico in April of 2009. The urgent need to speed up vaccine production was highlighted by this outbreak because over 340,000 confirmed cases and 4,100 deaths had occurred worldwide during the 6 months that were necessary to produce a vaccine using current procedures (39).
As the major surface antigen of influenza A viruses, the hemagglutinin (HA) envelope glycoprotein is the primary source of natural immunity and the key target in vaccination. However, changes in the antigenic sites of the HA protein due to antigenic drift result in lost or diminished immunity acquired from previous infection or vaccination (35). This necessitates the production of new vaccines against seasonal influenza viruses each year. The HA protein also plays a central role in the emergence of human pandemic influenza viruses. There are 16 known antigenic subtypes of HA proteins in influenza A viruses (H1 through H16), and a pandemic occurs when an influenza virus that has an HA protein to which most of the population lacks immunity acquires the ability to be efficiently transmitted from person to person.
The HA protein has multiple roles in the virus life cycle, notably receptor binding and membrane fusion. The protein is synthesized as a single precursor protein, HA0, that trimerizes and becomes glycosylated in the endoplasmic reticulum as it traffics to the cell surface (33). The HA protein contains multiple disulfide bonds and is cleaved into a mature form consisting of two subunits, HA1 and HA2 (9, 18). HA2 and the N- and C-terminal portions of HA1 form a membrane-proximal stalk that mediates membrane fusion during viral entry (40). A receptor-binding domain (HA-RBD) forms the distal head of the molecule and is inserted into the HA1 subunit. During virus entry, the HA-RBD engages sialic acid-containing receptors on the surface of the host cell, and the virion is subsequently internalized by endocytosis (33). Structurally and functionally, the HA-RBD is a member of the lectin superfamily, and the specificity of the binding pocket contributes to the host range of influenza viruses. For example, α(2,6)-containing sialosides are typically preferred by the HA protein from human viruses and α(2,3) sialosides by the HA proteins from avian viruses (13, 28). Upon triggering by the low-pH environment of endosomes, the HA protein undergoes an irreversible conformational change (6, 40) during which the intact HA-RBDs dissociate from the stalk of the trimer (3, 14, 19, 21). This observation, together with the manner in which the lectin-like domain is inserted as a folded module into the full-length HA protein, led us to hypothesize that the HA-RBD is able to adopt its native structure in isolation. Proper folding of the isolated HA-RBD into its native immunogenic structure has important therapeutic implications because the domain contains all of the known HA antigenic epitopes responsible for antibody recognition (5), and producing a protein-based influenza vaccine composed of isolated HA-RBD would dramatically speed up vaccine development during the early stages of a pandemic.
In a recently published report, a construct of the 2009 pandemic H1N1 HA protein that encompasses the HA-RBD, designated HA63-286-RBD, was expressed in Escherichia coli as inclusion bodies, refolded and purified, and used as a vaccine to produce immunity in ferrets (2). In this report, we show that this construct behaves as a stable, structured protein in solution, can be readily crystallized, and indeed adopts a structure that is virtually indistinguishable from that in the H1N1 HA protein ectodomain (41).
MATERIALS AND METHODS
Cloning and protein production.
The details of HA63-286-RBD protein production have been described elsewhere (2). Briefly, a codon-optimized synthetic gene corresponding to A/H1N1/2009 influenza virus hemagglutinin residues 63 to 286 (GenBank accession number ACQ99608) (residues 55 to 271 in H3 numbering) was cloned into the pJexpress404 vector. HA63-286-RBD was expressed in Escherichia coli as insoluble protein in inclusion bodies. The protein was purified under denaturing conditions, refolded on a metal affinity purification column, and eluted with standard elution buffer. A baculovirus-expressed and purified construct of the full-length A/CA/04/2009 H1 HA protein (HA-full) was obtained from BEI Resources (catalog no. NR-15258). A baculovirus-expressed and purified construct of the ectodomain of A/CA/04/2009 H1 HA protein (HA-ecto) was prepared as previously described (41).
HA63-286-RBD protein size estimation.
HA63-286-RBD protein was further purified by size exclusion chromatography using a Superdex 200 10/300 column (Pharmacia) and 10 mM Tris-Cl (pH 8.0)-100 mM NaCl as running buffer. Gel filtration standards (Bio-Rad) were used to estimate the molecular weight of HA63-286-RBD. Protein eluting as a monomer was concentrated to ∼4 mg/ml. The purification tag was not removed for these studies.
Crystal structure determination.
HA63-286-RBD protein crystals were grown by the hanging-drop vapor diffusion method at 18°C by combining 1 μl protein solution with 1 μl well solution (20% PEG 2000MME and 0.1 M Tris-HCl, pH 8.5). Crystals were transferred to a reservoir solution containing 25% glycerol for 1 to 2 min before being frozen in liquid nitrogen. Diffraction data were collected to a 2.10-Å resolution at cryogenic temperature at the Advanced Light Source (ALS) Synchrotron beamline 8.2.1 at an X-ray wavelength at 1.00 Å. Data processing and reduction were carried out with HKL2000 (27). Data collection statistics are presented in Table 1.
TABLE 1.
Data collection and refinement statisticsa
| Parameter | Value |
|---|---|
| Data collection | |
| Space group | P21 |
| a, b, c (Å) | 39.86, 74.07, 75.18 |
| α, β, γ (°) | 90.00, 94.73, 90.00 |
| Resolution (Å) | 40.0-2.1 (2.18-2.10) |
| Rmerge | 0.127 (0.373) |
| I/σI | 12.7 (3.8) |
| Completeness (%) | 100 (100) |
| Redundancy | 5.0 (3.9) |
| Refinement | |
| Resolution (Å) | 40.0-2.1 |
| No. of reflections | 24,258 |
| Rwork/Rfreeb | 0.177/0.206 |
| No. of atoms | |
| Protein | 3382 |
| Water | 193 |
| Glycerol | 36 |
| Average B-factors (Å2) | |
| Protein | 16.1 |
| Water | 23.3 |
| Glycerol | 26.8 |
| Ramachandran (%) | |
| Favored | 95.9 |
| Allowed | 4.1 |
| Outliers | 0 |
| Rms deviations | |
| Bond lengths (Å) | 0.008 |
| Bond angles (Å) | 1.074 |
Data were collected from a single crystal. Values for the highest-resolution shell are shown in parentheses.
Rfree was calculated using 5% of the reflections.
The HA63-286-RBD structure was determined by molecular replacement (MR) using Phaser (25). An initial solution was obtained using residues 50 to 273 from the crystal structure of the 1930 H1 swine hemagglutinin protein (Protein Data Bank [PDB] entry 1RUY) (17). Inspection of the initial electron density maps revealed a number of loops with little to no density, so the MR model was trimmed. MR was repeated and showed improved results (Z score, 37.2; log-likelihood gain, 1,808). After we “mutated” the model to the correct amino acid sequence, model building was performed with Coot (16), followed by iterative rounds of simulated annealing with Phenix (1) and restrained refinement CCP4-REFMAC5 (26). Refinement was monitored by following Rfree calculated for a random subset (5%) of reflections omitted from refinement. The final model was validated using MolProbity (15). Data refinement statistics are listed in Table 1.
ELISA.
Specific binding to antibodies from serum samples of influenza virus A/H1N1/2009 convalescent patients was determined by a specific enzyme-linked immunosorbent assay (ELISA) protocol using protein HA63-286-RBD as an antigen as described previously (2). All patients provided written informed consent for the collection of samples and subsequent analysis at the moment that the blood sample was taken. This study was conducted according to the principles expressed in the Declaration of Helsinki. The study was approved by the Institutional Review Board of the School of Biotechnology and Health at Tecnológico de Monterrey at Monterrey, México. Exposure to A/H1N1/2009 was confirmed in patients by reverse transcription (RT)-PCR. Unexposed samples were collected from patients in 2008.
Sequence accession number.
The coordinates for the HA63-286-RBD structure have been deposited at the Protein Data Bank as PDB entry 3MLH.
RESULTS
Analysis of the HA receptor-binding domain in solution.
The choice of construct and the production of the hemagglutinin HA-RBD from a swine origin pandemic A/H1N1/2009 influenza virus are described in a recently published report (2). Briefly, HA residues 63 to 286 (residues 55 to 271 in H3 numbering) were selected because they represent the N- and C-terminal boundaries of the HA-RBD lectin fold and were hypothesized to adopt the correct three-dimensional structure in isolation. To be consistent with the previous report, this construct will be referred to as HA63-286-RBD. HA63-286-RBD was expressed in E. coli as an ∼25-kDa His-tagged insoluble protein in inclusion bodies, purified under denaturing conditions on a Ni2+-chelation column, refolded on the column, and finally eluted. While the HA protein ectodomain forms a trimer in solution, few of the trimeric interactions involve the receptor-binding domains. Therefore, we anticipated that HA63-286-RBD would be monomeric in solution, in keeping with results from other published studies (3). To determine the oligomeric state, the refolded HA63-286-RBD was subjected to size exclusion chromatography and found to elute as a single sharp peak at ∼25 kDa (Fig. 1), consistent with the monomeric species. This procedure not only served to further purify the sample but also suggested that HA63-286-RBD is well folded into a homogenous population suitable for structural analyses.
FIG. 1.

Size exclusion chromatography of recombinant HA63-286-RBD. The isolated HA receptor-binding domain elutes as a well-folded monomer (labeled black peak) with an apparent molecular size of ∼25 kDa compared to molecular size standards (labeled gray peaks) on a Superdex 200 size exclusion chromatography column. The purified peak was collected and yielded well-diffracting crystals. Abs, absorbance.
Crystal structure analysis of the HA receptor-binding domain.
HA63-286-RBD readily formed clusters of plate-like crystals in a number of conditions, and the best crystals grew in PEG 2000MME at pH 8.5 and diffracted well despite being very thin (∼200 by 10 by 2 μm3). The crystals are in space group P21, and the structure was solved to 2.1-Å resolution by molecular replacement using the receptor-binding domain of 1930 swine H1 HA protein (PDB entry 1RUY) as a search model (17). This search model was used because the structure of the A/H1N1/2009 HA ectodomain (41, 42) had not been published at the time of these studies. The structure was refined to high accuracy, and the crystallographic and refinement statistics are shown in Table 1. Our structure (Fig. 2 A) reveals that isolated and refolded HA63-286-RBD adopts the same three-dimensional fold that occurs in the A/H1N1/2009 HA ectodomain structure that was generated by baculovirus expression in insect cells (Fig. 2B) (41). The two receptor-binding domain structures overlay very well, with an overall root mean square deviation (RMSD) of 0.32 Å (Fig. 2C). Notably, both disulfide bonds are formed correctly between Cys59 and Cys71 and Cys94 and Cys139 (H3 numbering) (Fig. 2C), and these were subsequently confirmed by a simulated annealing omit map in which the cysteines were replaced by alanines.
FIG. 2.

Structural comparison of the HA receptor-binding domain in the HA63-286-RBD (A) and the A/H1N1/2009 HA ectodomain (B). In these two panels, the domain is colored blue to red, N to C terminus, to facilitate the chain trace. Glycosylation at Asn94 and Asn278 in the HA ectodomain is shown as a ball-and-stick model. (C) Overlay of HA63-286-RBD (green) and equivalent residues in HA ectodomain (blue). The locations of the receptor-binding pocket and two disulfide bonds are highlighted. (D and E) Detail of boxed regions in panels A and B, respectively, corresponding to the interface region between the receptor-binding domain and the helical stalk region that undergoes pH-dependent conformational changes. Note that the isoleucine-rich third β-strand of the interfacial β-sheet in panel E is disordered in panel D. The coordinates for HA63-286-RBD have been deposited at the Protein Data Bank as PDB entry 3MLH. The coordinates for the A/H1N1/2009 HA ectodomain are from PDB entry 3LZG. All residues are labeled using H3 numbering.
HA63-286-RBD forms a dimer in the crystallographic asymmetric unit, but we believe that this is a crystal artifact because the dimer interface is mediated by hydrophilic interactions and a number of ordered water molecules (Fig. 3). Compared to the trimer subunits in the HA ectodomain structure, the dimer subunits in HA63-286-RBD are rotated by ∼180°, and the dimer interface has no relationship to the native trimeric interface. As a result, the receptor-binding pockets are facing opposite directions in HA63-286-RBD, in contrast to the structurally related dimeric bovine coronavirus hemagglutinin-esterase protein in which the receptor-binding pockets are facing the same direction (44). Our size exclusion chromatography data support HA63-286-RBD as being monomeric in solution (Fig. 1), and previous analytical ultracentrifugation analyses found that HA63-286-RBD exists mainly as a monomer in solution (2).
FIG. 3.

HA63-286-RBD forms a crystallographic dimer with a hydrophilic interface. (A) The asymmetric unit of HA63-286-RBD is shown as surface-filled and ribbon models for chain A (purple) and chain B (green). Waters (red) at the interface are shown as spheres. Note the interactions of Glu216 with Lys211 and Tyr233. (B) Electrostatic surface of HA63-286-RBD. The chains are turned 90° to the right or left to reveal the electrostatic surface at the interface of the two chains. The acidic patch at Glu216 interacts with the basic patch at Lys211 in the opposite chain. All residues are labeled using H3 numbering.
Unsurprisingly, one difference between the isolated HA63-286-RBD structure and the complete ectodomain version occurs close to the N and C termini where the insertion of the RBD into the stalk of the HA protein occurs. A close-up view of this region reveals that the C-terminal 5 residues in HA63-286-RBD are disordered compared to the same residues in the HA ectodomain (compare Fig. 2D and E). In the HA ectodomain, these residues form a β-strand that contains three sequential isoleucine residues and packs against the interhelical loop of the stalk (Fig. 2E). This region of the molecule is relatively distant from the receptor-binding pocket and the antigenic sites, and this structural difference in the HA63-286-RBD construct is unlikely to affect its binding specificity or immunological properties. However, the structural difference may be important in terms of potentially optimizing the domain for expression in E. coli.
Receptor-binding pocket.
To verify that the structure of HA63-286-RBD retains an intact receptor-binding pocket, the conformations of the amino acids lining this region of the HA63-286-RBD were analyzed. The sialic acid-binding pocket in HA63-286-RBD is structurally very similar to that in the A/H1N1/2009 HA ectodomain (Fig. 4 A) (41). A sialic acid molecule was successfully modeled into this site based on the structure of the 1934 H1 HA protein bound to a human sialic acid receptor analog (PDB entry 1RVZ) (Fig. 4B) (17). Glycerol was used as a cryoprotectant for the HA63-286-RBD crystals prior to freezing, and we observed clear electron density in the receptor-binding pocket of HA63-286-RBD that was successfully interpreted and refined as a bound glycerol molecule (Fig. 4C). Interactions between the glycerol molecule and residues Tyr98, His183, Asp190, and Gln226 (in H3 numbering) suggest that the glycerol molecule may be mimicking the 8-OH and 9-OH arm of the sialic acid molecule, as seen by comparing Fig. 4B and C.
FIG. 4.

Analysis of the receptor-binding pocket of HA63-286-RBD. (A) Overlay of the receptor-binding pockets of HA63-286-RBD (green) and the A/H1N1/2009 HA ectodomain (blue). The coordinates for the A/H1N1/2009 HA ectodomain are from PDB entry 3LZG. (B) Structural model of a sialic acid molecule within the receptor-binding pocket of HA63-286-RBD. The model is based on the structure of the 1934 H1 HA ectodomain bound to a human sialic acid receptor analog (PDB entry 1RVZ). (C) Observed binding of a glycerol molecule in the receptor-binding pocket of HA63-286-RBD. Glycerol occupies the site that recognizes the 8-OH and 9-OH moiety of sialic acid. All residues are labeled using H3 numbering.
Antigenic epitopes.
For a protein to be an effective antigen in a protein-based vaccine, it is important for antigenic epitopes to have the same three-dimensional structures as found on the surface of the virus. There are four distinct antigenic sites on the H1 HA protein, called Sa, Sb, Ca, and Cb (5, 8). The three antigenic sites Sa, Sb, and Cb are contained within a single protomer of the trimer. The antigenic site Ca spans a large cleft between two adjacent RBDs in the HA trimer and is composed of two subsites, Ca1 and Ca2, in each protomer. Figure 5 shows the locations of the antigenic sites in HA63-286-RBD. Significantly, all of the antigenic sites are structurally conserved compared to those in the HA ectodomain. Amino acid backbones overlay with RMSDs of 0.32 Å (Sa site), 0.33 Å (Sb site), 0.41 Å (Ca1 subsite), 0.09 Å (Ca2 subsite), and 2.22 Å (Cb site). The relatively large RMSD for the Cb site is due to its location in a flexible loop that is known to adopt variable backbone conformations. These differences in the Cb site are observed even between different chains within the same crystal structure, such as in the two HA63-286-RBD monomers and in the six RBDs in the HA ectodomain (two trimers) (Fig. 5F). Thus, we contend that all of the antigenic sites in HA63-286-RBD, including the Cb site, are structurally conserved between the E. coli-expressed RBD construct and the insect cell-expressed trimeric ectodomain construct.
FIG. 5.

Analysis of the antigenic sites on HA63-286-RBD. (A) Conformational antigenic sites Sa, Sb, Ca, and Cb are mapped onto the HA63-286-RBD crystal structure (dark gray secondary structure elements within a semitransparent surface representation). The Ca site is formed from two subsites, Ca1 and Ca2, and they form a single contiguous antigenic site only when two protomers are adjacent in the HA trimer. (B to F) Zoomed-in views show an overlay of HA63-286-RBD (green) and the A/H1N1/2009 HA ectodomain (blue) at each antigenic site. The Cb site adopts variable backbone conformations, as seen in the two molecules of HA63-286-RBD (green), and the six molecules of A/H1N1/2009 HA ectodomain (blue) in their respective crystal structures. Similarly, some surface-exposed hydrophilic amino acid side chains (e.g., Q192 in the Sb site and R208 in the Ca1 site) can adopt variable conformations. The coordinates for A/H1N1/2009 HA ectodomain are from PDB entry 3LZG. All residues are labeled using H3 numbering as done in reference 5.
The Sa antigenic site forms a three-dimensional antigenic site composed of two loops. The Sa sites of the 1918 and 2009 pandemic influenza H1 virus HA proteins are highly conserved in sequence and in structure and do not have glycosylation sites nearby (41). An Sa site-specific antibody (2D1) isolated from a survivor of the 1918 pandemic has high affinity and cross-reactivity to both the 1918 and 2009 pandemic influenza viruses (24, 41, 43). Seasonal human H1 isolates gradually obtained up to three glycosylation sites in the Sa region from 1930 to 2007 that mask the Sa antigenic site from antibodies that bind this site. These data suggest that the Sa antigenic site may be at least part of the reason for age-related immunity to 2009 H1N1 virus. An overlay of amino acid backbone and side chains in the Sa antigenic site between the HA63-286-RBD and HA ectodomain shows the high degree of structural similarity in the two constructs (Fig. 5B). Thus, an intact and exposed Sa site in the E. coli-expressed construct might allow for the generation of neutralizing antibodies to this immunologically important epitope.
HA63-286-RBD-specific recognition of antibodies from H1N1-infected subjects.
The high degree of structural similarity between the antigenic sites of HA63-286-RBD and HA ectodomain suggests that vaccination with HA63-286-RBD will elicit antibodies to these antigenic sites and will provide protection against viral infection. The ability of HA63-286-RBD to elicit immunity in vivo is consistent with recently published data which show that HA63-286-RBD is immunogenic and has protective capacity in ferrets (2).
To confirm that antigenic epitopes are intact in HA63-286-RBD protein, an ELISA was used to measure binding of antibodies in human serum samples to HA63-286-RBD protein. Sera from patients positive for infection by the pandemic A/H1N1/2009 influenza virus were found to bind HA63-286-RBD protein at significantly higher levels (two-tailed P value, <0.0001 by an unpaired t test) than sera collected in 2008 before the emergence of the pandemic virus (Fig. 6). Thus, the data demonstrate that the E. coli-expressed protein is recognized with specificity by serum antibodies from infected humans.
FIG. 6.
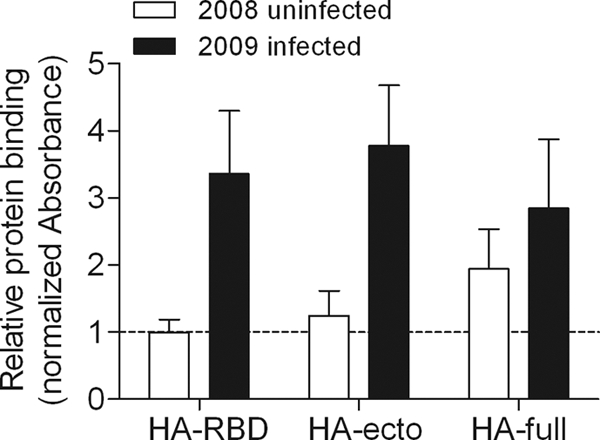
Specific recognition of HA protein constructs by sera from patients infected with influenza virus A/H1N1/2009. ELISAs were performed using either sera collected between March and May of 2008 from 10 nonexposed subjects or sera collected in 2009 from 16 PCR-confirmed A/H1N1/2009-infected subjects. The protein constructs used were E. coli-expressed HA63-286-RBD (HA-RBD), baculovirus-expressed HA ectodomain (HA-ecto), and baculovirus-expressed full-length HA protein (HA-full). All of the absorbance levels from ELISAs were normalized to a value of 1 for the binding of sera from 2008 uninfected subjects to the HA63-286-RBD protein. The reactivity of HA63-286-RBD to sera from 2009 infected subjects was statistically similar to the reactivities of HA ectodomain and full-length HA protein (two-tailed P values of 0.2069 and 0.1519, respectively, from unpaired t tests). For the HA63-286-RBD protein, the difference in binding between the two groups of sera from either uninfected or infected subjects was found to be statistically significant (two-tailed P value, <0.0001 by an unpaired t test).
Polyclonal sera can react with antigen in degraded or unfolded forms, though often with lower affinity than structurally native protein. Moreover, it is possible that a lack of glycosylation in the E. coli-expressed HA63-286-RBD protein could decrease antigenic reactivity. Therefore, we compared the antigenic reactivity of the E. coli-expressed and refolded HA63-286-RBD protein to baculovirus-expressed and glycosylated constructs of the full-length HA protein (HA-full) and the HA protein ectodomain (HA-ecto). The HA-ecto protein construct had slightly higher reactivity to sera from patients positive for infection by the pandemic A/H1N1/2009 influenza virus than the HA63-286-RBD protein (Fig. 6). Although the difference was not statistically significant (two-tailed P value, 0.2069 by an unpaired t test), the slightly higher reactivity of the HA-ecto protein could be due to the presence of additional epitopes located within the stalk region. In addition, the average antigenic reactivities of the HA63-286-RBD protein and HA-full protein constructs were also statistically similar (two-tailed P value, 0.1519 by an unpaired t test).
DISCUSSION
We have shown for the first time that an isolated HA-RBD of the influenza virus expressed in E. coli, and refolded from inclusion bodies, adopts the same three-dimensional structure that is found on the surface of infectious virions. We have specifically studied the domain that encompasses residues 63 to 286 (HA63-286-RBD) of the A/H1N1/2009 strain that emerged from Mexico in April of 2009. However, the highly conserved structure of the domain suggests that the HA-RBD of any influenza virus strain can be studied in a similar manner. Significantly, the structure of the isolated HA-RBD, including the receptor-binding pocket, antigenic epitopes, and disulfide bridges, of the E. coli-expressed HA-RBD match exactly those of the domain in the HA protein ectodomain structure that has recently been characterized (41). This is consistent with recently published data which show that the isolated domain retains immunogenicity, despite lacking glycosylation sites and forming monomers in solution (2).
Pandemic influenza viruses of the last century have contained few glycosylation sites in their HA-RBDs. A key finding reported here is that glycosylation is not required for the proper folding of this domain, and this posttranslational modification may be for other purposes, such as immune evasion or receptor-binding affinity (36). While currently circulating human H3N2 influenza viruses contain seven glycosylation sites in the HA-RBD, the 1968 Hong Kong H3N2 pandemic virus contained only two before acquiring five additional glycosylation sites while circulating in humans over the past 40 years. Similarly, seasonal human H1N1 influenza viruses have up to four or five glycosylation sites in their HA-RBDs, yet the 1918 H1N1 Spanish flu pandemic virus contained only one glycosylation site in its HA-RBD. The 2009 swine origin H1N1 pandemic virus also contains only one RBD glycosylation at N94 (Fig. 2B), distant from its antigenic sites and receptor-binding pocket. The results described here show that this single glycosylation is not required for the proper folding of the RBD from the 2009 pandemic virus.
Considering how the HA protein functions during membrane fusion and the manner in which the HA-RBD is inserted into the stalk of the full-length HA protein, it is perhaps not surprising that the HA-RBD can fold independently. This is in stark contrast to the membrane fusion-mediating stalk domain that spontaneously refolds into its postfusion structure if expressed alone in E. coli (10, 11). The main role of the HA-RBD is to recognize and bind sialic acid receptors on the surface of the target cell and to initiate endocytosis. The actual membrane fusion process involves a major conformation change of the HA trimer stalk during which the three HA-RBDs are essentially discarded yet remain structurally intact (3, 38). In terms of structure, the HA-RBD is a lectin superfamily member (4) and can also be considered a β-sandwich “fold family 1” carbohydrate-binding module (CBM) (4). Typical of CBMs, the sialic acid-binding site of the HA-RBD is populated with aromatic residues (Tyr98, Trp153, and His183, in H3 numbering) that act as platforms for the sugar ring (4). Evidently, during the evolution of influenza viruses, a recombination event occurred in which the gene for a primordial lectin molecule was incorporated into a membrane fusion domain to provide sugar recognition abilities without compromising the fusion mechanism. Thus, as is typical for an independently folded domain, the N- and C-termini remain spatially close, and the HA-RBD/HA-RBD interfaces within the trimeric structure are very loose and mainly hydrophilic in nature. The results presented here are consistent with the previous observation that distinct hemagglutinin and esterase domains are inserted into the membrane fusion domain of the influenza C virus HEF protein (29). Our data suggest that other viral fusion proteins may also contain inserted domains that fold independently and could be used for structural studies.
Significant to the prevention and control of influenza, these results provide a structural basis supporting the use of isolated HA-RBD constructs generated in E. coli as candidates for protein-based vaccines. Most significantly, the refolded HA-RBD has intact antigenic sites: three complete antigenic sites (Sa, Sb, Cb) and a fourth Ca site that spans adjacent RBDs in the context of the trimer and is split into two subsites (Ca1 and Ca2) in the monomer. We support these structural findings with data that show that human serum antibodies from A/H1N1/2009 influenza virus-exposed patients are able to recognize the isolated HA-RBD. In practical terms, the isolated HA-RBD offers four therapeutic advantages. (i) It provides an avenue to quickly characterize the HA-RBD from any new virus strain at the structural level. Our structural analysis was completed in 3 weeks (from size exclusion chromatography to completed structure), whereas the technically more challenging H1N1 HA protein ectodomain structural analysis has taken considerably longer to complete. (ii) The lack of glycosylation sites on our sample might serve to increase immunogenicity by increasing the exposure of antigenic sites to the immune system, as long as vaccine efficacy is not reduced by the immune response also targeting epitopes masked by glycosylation on the native protein (37). (iii) The isolated domain will facilitate the search for small-molecule inhibitors that can block the binding of sialic acid. (iv) Most importantly, the HA-RBD offers the potential to develop influenza vaccines rapidly from bacterially expressed protein. E. coli-expressed constructs of the HA1 receptor-binding domains from H3, H5, and H7 subtype viruses have also been found to bind monoclonal antibodies and retain immunogenicity in several other recent studies (12, 20, 22, 23, 31, 34). The metastable folding of the prefusion form of the HA protein (7, 32) means that virus-free generation of the entire HA protein for vaccine production and biological characterization remains a challenge. Also, soluble fragments of the HA2 membrane fusion domain expressed in E. coli spontaneously fold into their postfusion conformation, making them less suitable for vaccine development (7, 10, 11).
Finally, it should be emphasized that neither the HA63-286-RBD construct described here nor equivalent constructs from other strains are likely to be the most optimal for expression in E. coli. Therefore, we intend to investigate other constructs by protein engineering and crystallography. For example, the unfolded C-terminal β-strand that contains three sequential isoleucine residues packs against the interhelical loop of the stalk region in the HA protein ectodomain (Fig. 2E) and represents the one hydrophobic interface involving the HA-RBD. Further truncating the C terminus to remove this β-strand or identifying a more hydrophilic sequence that stabilizes this β-strand and the associated β-sheet are potential avenues for improving yield and/or overall protein stability. In addition, it may be possible to remove the disulfide bridges to improve yield and solubility in E. coli-based expression systems. The fact that the HA-RBD can be refolded into its native structure and subsequently forms correct disulfide bonds suggests that these are created late during folding. We hypothesize that mutation of the four cysteines to alanines or serines will not seriously affect the final structure but may facilitate expression of soluble protein in E. coli. If this is correct, the refolding step of our current production procedure may no longer be necessary.
Acknowledgments
Funding for this research was provided by the National Institutes of Health, National Institute of Allergy and Infectious Diseases, under contract no. HHSN266200700005C, Cancer Center core grant CA21765, the American Lebanese Syrian Associated Charities (ALSAC), and the Children's Infection Defense Center (CIDC) at St. Jude Children's Research Hospital. We thankfully acknowledge the financial support and unconditional trust of the Zambrano-Hellion family, FEMSA, and Tecnológico de Monterrey (seed fund CAT-122). Portions of this research were conducted at the Advanced Light Source, a national user facility operated by Lawrence Berkeley National Laboratory on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The Berkeley Center for Structural Biology is supported in part by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences.
We thank Richard Heath and Muralidhar Reddivari for assistance in the production of HA-ecto protein. We also thank Brenda Schulman and Eric Enemark for generosity and technical assistance and Ian Wilson and colleagues for providing the coordinates of the full-length A/H1N1/2009 HA ectodomain structure prior to publication.
Footnotes
Published ahead of print on 10 November 2010.
REFERENCES
- 1.Adams, P. D., P. V. Afonine, G. Bunkoczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L. W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, J. S. Richardson, T. C. Terwilliger, and P. H. Zwart. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66:213-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aguilar-Yáñez, J. M., R. Portillo-Lara, G. I. Mendoza-Ochoa, S. A. García-Echauri, F. López-Pacheco, D. Bulnes-Abundis, J. Salgado-Gallegos, I. M. Lara-Mayorga, J. Webb-Vargas, F. O. León Angel, R. E. Rivero-Aranda, M. Y. Oropeza-Almazán, G. M. Ruiz-Palacios, M. I. Zertuche-Guerra, R. M. DuBois, S. W. White, S. Schultz-Cherry, C. J. Russell, and M. M. Alvarez. 2010. An influenza A/H1N1/2009 hemagglutinin vaccine produced in Escherichia coli. PLoS One 5:e11694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bizebard, T., B. Gigant, P. Rigolet, B. Rasmussen, O. Diat, P. Bosecke, S. A. Wharton, J. J. Skehel, and M. Knossow. 1995. Structure of influenza virus haemagglutinin complexed with a neutralizing antibody. Nature 376:92-94. [DOI] [PubMed] [Google Scholar]
- 4.Boraston, A. B., D. N. Bolam, H. J. Gilbert, and G. J. Davies. 2004. Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem. J. 382:769-781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brownlee, G. G., and E. Fodor. 2001. The predicted antigenicity of the haemagglutinin of the 1918 Spanish influenza pandemic suggests an avian origin. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356:1871-1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bullough, P. A., F. M. Hughson, J. J. Skehel, and D. C. Wiley. 1994. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 371:37-43. [DOI] [PubMed] [Google Scholar]
- 7.Carr, C. M., C. Chaudhry, and P. S. Kim. 1997. Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc. Natl. Acad. Sci. U. S. A. 94:14306-14313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caton, A. J., G. G. Brownlee, J. W. Yewdell, and W. Gerhard. 1982. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell 31:417-427. [DOI] [PubMed] [Google Scholar]
- 9.Chen, J., K. H. Lee, D. A. Steinhauer, D. J. Stevens, J. J. Skehel, and D. C. Wiley. 1998. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 95:409-417. [DOI] [PubMed] [Google Scholar]
- 10.Chen, J., J. J. Skehel, and D. C. Wiley. 1999. N- and C-terminal residues combine in the fusion-pH influenza hemagglutinin HA(2) subunit to form an N cap that terminates the triple-stranded coiled coil. Proc. Natl. Acad. Sci. U. S. A. 96:8967-8972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen, J., S. A. Wharton, W. Weissenhorn, L. J. Calder, F. M. Hughson, J. J. Skehel, and D. C. Wiley. 1995. A soluble domain of the membrane-anchoring chain of influenza virus hemagglutinin (HA2) folds in Escherichia coli into the low-pH-induced conformation. Proc. Natl. Acad. Sci. U. S. A. 92:12205-12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiu, F. F., N. Venkatesan, C. R. Wu, A. H. Chou, H. W. Chen, S. P. Lian, S. J. Liu, C. C. Huang, W. C. Lian, P. Chong, and C. H. Leng. 2009. Immunological study of HA1 domain of hemagglutinin of influenza H5N1 virus. Biochem. Biophys. Res. Commun. 383:27-31. [DOI] [PubMed] [Google Scholar]
- 13.Connor, R. J., Y. Kawaoka, R. G. Webster, and J. C. Paulson. 1994. Receptor specificity in human, avian, and equine H2 and H3 influenza virus isolates. Virology 205:17-23. [DOI] [PubMed] [Google Scholar]
- 14.Daniels, R. S., J. C. Downie, A. J. Hay, M. Knossow, J. J. Skehel, M. L. Wang, and D. C. Wiley. 1985. Fusion mutants of the influenza virus hemagglutinin glycoprotein. Cell 40:431-439. [DOI] [PubMed] [Google Scholar]
- 15.Davis, I. W., A. Leaver-Fay, V. B. Chen, J. N. Block, G. J. Kapral, X. Wang, L. W. Murray, W. B. Arendall III, J. Snoeyink, J. S. Richardson, and D. C. Richardson. 2007. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35:W375-W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Emsley, P., and K. Cowtan. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126-2132. [DOI] [PubMed] [Google Scholar]
- 17.Gamblin, S. J., L. F. Haire, R. J. Russell, D. J. Stevens, B. Xiao, Y. Ha, N. Vasisht, D. A. Steinhauer, R. S. Daniels, A. Elliot, D. C. Wiley, and J. J. Skehel. 2004. The structure and receptor binding properties of the 1918 influenza hemagglutinin. Science 303:1838-1842. [DOI] [PubMed] [Google Scholar]
- 18.Garten, W., F. X. Bosch, D. Linder, R. Rott, and H. D. Klenk. 1981. Proteolytic activation of the influenza virus hemagglutinin: the structure of the cleavage site and the enzymes involved in cleavage. Virology 115:361-374. [DOI] [PubMed] [Google Scholar]
- 19.Godley, L., J. Pfeifer, D. Steinhauer, B. Ely, G. Shaw, R. Kaufmann, E. Suchanek, C. Pabo, J. J. Skehel, D. C. Wiley, et al. 1992. Introduction of intersubunit disulfide bonds in the membrane-distal region of the influenza hemagglutinin abolishes membrane fusion activity. Cell 68:635-645. [DOI] [PubMed] [Google Scholar]
- 20.Ho, H. T., H. L. Qian, F. He, T. Meng, M. Szyporta, N. Prabhu, M. Prabakaran, K. P. Chan, and J. Kwang. 2009. Rapid detection of H5N1 subtype influenza viruses by antigen capture enzyme-linked immunosorbent assay using H5- and N1-specific monoclonal antibodies. Clin. Vaccine Immunol. 16:726-732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang, Q., R. Opitz, E. W. Knapp, and A. Herrmann. 2002. Protonation and stability of the globular domain of influenza virus hemagglutinin. Biophys. J. 82:1050-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jeon, S. H., and R. Arnon. 2002. Immunization with influenza virus hemagglutinin globular region containing the receptor-binding pocket. Viral Immunol. 15:165-176. [DOI] [PubMed] [Google Scholar]
- 23.Khurana, S., A. L. Suguitan, Jr., Y. Rivera, C. P. Simmons, A. Lanzavecchia, F. Sallusto, J. Manischewitz, L. R. King, K. Subbarao, and H. Golding. 2009. Antigenic fingerprinting of H5N1 avian influenza using convalescent sera and monoclonal antibodies reveals potential vaccine and diagnostic targets. PLoS Med. 6:e1000049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krause, J. C., T. M. Tumpey, C. J. Huffman, P. A. McGraw, M. B. Pearce, T. Tsibane, R. Hai, C. F. Basler, and J. E. Crowe, Jr. 2010. Naturally occurring human monoclonal antibodies neutralize both 1918 and 2009 pandemic influenza A (H1N1) viruses. J. Virol. 84:3127-3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCoy, A. J., R. W. Grosse-Kunstleve, L. C. Storoni, and R. J. Read. 2005. Likelihood-enhanced fast translation functions. Acta Crystallogr. D Biol. Crystallogr. 61:458-464. [DOI] [PubMed] [Google Scholar]
- 26.Murshudov, G. N., A. A. Vagin, and E. J. Dodson. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53:240-255. [DOI] [PubMed] [Google Scholar]
- 27.Otwinowski, Z., D. Borek, W. Majewski, and W. Minor. 2003. Multiparametric scaling of diffraction intensities. Acta Crystallogr. A 59:228-234. [DOI] [PubMed] [Google Scholar]
- 28.Rogers, G. N., J. C. Paulson, R. S. Daniels, J. J. Skehel, I. A. Wilson, and D. C. Wiley. 1983. Single amino acid substitutions in influenza haemagglutinin change receptor binding specificity. Nature 304:76-78. [DOI] [PubMed] [Google Scholar]
- 29.Rosenthal, P. B., X. Zhang, F. Formanowski, W. Fitz, C. H. Wong, H. Meier-Ewert, J. J. Skehel, and D. C. Wiley. 1998. Structure of the haemagglutinin-esterase-fusion glycoprotein of influenza C virus. Nature 396:92-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Russell, C. J., and R. G. Webster. 2005. The genesis of a pandemic influenza virus. Cell 123:368-371. [DOI] [PubMed] [Google Scholar]
- 31.Shien, J. H., L. F. Fu, J. R. Wu, M. C. Cheng, H. K. Shieh, and P. C. Chang. 2008. Development of blocking ELISA for detection of antibodies against avian influenza virus of the H7 subtype. J. Microbiol. Immunol. Infect. 41:369-376. [PubMed] [Google Scholar]
- 32.Skehel, J. J., P. M. Bayley, E. B. Brown, S. R. Martin, M. D. Waterfield, J. M. White, I. A. Wilson, and D. C. Wiley. 1982. Changes in the conformation of influenza virus hemagglutinin at the pH optimum of virus-mediated membrane fusion. Proc. Natl. Acad. Sci. U. S. A. 79:968-972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skehel, J. J., and D. C. Wiley. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69:531-569. [DOI] [PubMed] [Google Scholar]
- 34.Song, L., Y. Zhang, N. E. Yun, A. L. Poussard, J. N. Smith, J. K. Smith, V. Borisevich, J. J. Linde, M. A. Zacks, H. Li, U. Kavita, L. Reiserova, X. Liu, K. Dumuren, B. Balasubramanian, B. Weaver, J. Parent, S. Umlauf, G. Liu, J. Huleatt, L. Tussey, and S. Paessler. 2009. Superior efficacy of a recombinant flagellin:H5N1 HA globular head vaccine is determined by the placement of the globular head within flagellin. Vaccine 27:5875-5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steinhauer, D. A., and J. J. Skehel. 2002. Genetics of influenza viruses. Annu. Rev. Genet. 36:305-332. [DOI] [PubMed] [Google Scholar]
- 36.Vigerust, D. J., and V. L. Shepherd. 2007. Virus glycosylation: role in virulence and immune interactions. Trends Microbiol. 15:211-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei, C. J., J. C. Boyington, K. Dai, K. V. Houser, M. B. Pearce, W. P. Kong, Z. Y. Yang, T. M. Tumpey, and G. J. Nabel. 2010. Cross-neutralization of 1918 and 2009 influenza viruses: role of glycans in viral evolution and vaccine design. Sci. Transl Med. 2:24ra21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weissenhorn, W., A. Dessen, L. J. Calder, S. C. Harrison, J. J. Skehel, and D. C. Wiley. 1999. Structural basis for membrane fusion by enveloped viruses. Mol. Membr. Biol. 16:3-9. [DOI] [PubMed] [Google Scholar]
- 39.WHO. 2009. Pandemic (H1N1) 2009—update 68. Accessed 13 April 2010. www.who.int/csr/don/2009_10_02/en.
- 40.Wilson, I. A., J. J. Skehel, and D. C. Wiley. 1981. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature 289:366-373. [DOI] [PubMed] [Google Scholar]
- 41.Xu, R., D. C. Ekiert, J. C. Krause, R. Hai, J. E. Crowe, Jr., and I. A. Wilson. 2010. Structural basis of preexisting immunity to the 2009 H1N1 pandemic influenza virus. Science 328:357-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang, H., P. Carney, and J. Stevens. 2010. Structure and receptor binding properties of a pandemic H1N1 virus hemagglutinin. PLoS Curr. 2:RRN1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu, X., T. Tsibane, P. A. McGraw, F. S. House, C. J. Keefer, M. D. Hicar, T. M. Tumpey, C. Pappas, L. A. Perrone, O. Martinez, J. Stevens, I. A. Wilson, P. V. Aguilar, E. L. Altschuler, C. F. Basler, and J. E. Crowe, Jr. 2008. Neutralizing antibodies derived from the B cells of 1918 influenza pandemic survivors. Nature 455:532-536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeng, Q., M. A. Langereis, A. L. van Vliet, E. G. Huizinga, and R. J. de Groot. 2008. Structure of coronavirus hemagglutinin-esterase offers insight into corona and influenza virus evolution. Proc. Natl. Acad. Sci. U. S. A. 105:9065-9069. [DOI] [PMC free article] [PubMed] [Google Scholar]