

Abstract
The IR3 transcript of equine herpesvirus-1 (EHV-1) harbors 117 nts antisense to the immediate-early (IE) mRNA, suggesting it plays a regulatory role. Here, we show that the IR3 transcript downregulates IE gene expression and that the absence of IR3 expression altered EHV-1 biological properties and virulence in mice. Reporter assays revealed that the IR3/IE overlapping sequences [IR3(+226/+342)] and an additional IR3(+343/+433) region are necessary for the IR3 RNA to downregulate IE expression. Experiments with the ΔIR3 EHV-1 showed that the IR3 gene is dispensable for EHV-1 replication. Protein expression of the IE and representative EHV-1 genes was increased in cells infected with ΔIR3 EHV-1 as compared to that of cells infected with wt EHV-1. The ΔIR3 EHV-1 exhibited increased virulence in mice as compared to the parent virus. The finding that the IR3 transcript affects IE gene expression extends the role of RNA as a regulatory molecule in alphaherpesvirus infection.
Keywords: equine herpesvirus-1, IR3 RNA, regulatory RNA, herpesvirus
Introduction
EHV-1 is a member of genus Varicellovirus, subfamily Alphaherpesvirinae, and its genome of 155 kbp is comprised of a unique long (Ul) region and a short (S) region that is comprised of a unique segment (Us) bracketed by identical diploid internal and terminal repeat (IR and TR) sequences (Henry et al., 1981; Telford et al., 1992). As is the case with other alphaherpesviruses, the regulated expression of EHV-1 genes occurs in a program of immediate-early, early, and late stages (Caughman et al., 1985; Gray et al., 1987). During productive viral replication, the sole IE gene is trans-activated by the multi-functional virion-associated ETIF protein (ETIFP) (Elliott, 1994; Elliott and O’Hare, 1995; von Einem et al., 2006) to generate the IE protein (IEP) that trans-activates early and some late viral genes (Smith et al., 1992, 1994; Kim et al., 1997; Garko-Buczynski et al., 1998) and represses its own expression (Kim et al., 1995). The IEP interacts with early viral regulatory proteins such as the IR4 protein (IR4P), EICP0 protein (EICP0P) and UL5 protein (UL5P), and these protein complexes synergistically trans-activate EHV-1 genes (Smith et al., 1993; Kim et al., 1997; Derbigny et al., 2000, 2002; Albrecht et al., 2004, 2005). Recent studies showed that the early IR2 protein (IR2P) encoded within the IE gene functions as a negative regulatory protein (Kim et al., 2006). In addition, the EHV-1 unique IR3 gene was predicted to be a potential seventh regulatory gene. Previous studies showed that the IR3 gene generates a 1 kb transcript antisense to the 5’ untranslated region (UTR) of the IE mRNA, such that 117 nts of the IR3 transcript overlap the 5’UTR of the IE mRNA (Holden et al., 1992). These data also tentatively identified a TATA box, a 5’UTR, an open reading frame (ORF), and a poly (A) signal. Recently, we revealed that the TATA box is essential for IR3 promoter activation, and that the IEP trans-activates the IR3 promoter by direct binding to the IR3 promoter region and acts synergistically with the EICP0 and IR4 proteins to increase IR3 promoter activity (Ahn et al., 2007). Additionally, an IR3 protein (IR3P) was only detected as an in vitro transcription/translation product, but not in EHV-1 infected or IR3 expression vector transfected cells, raising the possibility that the IR3 transcript is a non-coding RNA. The observation that the IR3 transcript harbors 117 nts antisense to the 5’UTR (untranslated region) of the IE mRNA suggests that the IR3 transcript may affect IE gene expression in a RNA-RNA fashion. Here, we show that the non-coding IR3 transcript downregulates IE gene expression as a regulatory RNA molecule, and that ΔIR3 EHV-1 possesses altered biological properties in cell culture and in the EHV-1 mouse model.
Results
IR3(+226/+433) of the IR3 transcript is essential to downregulate IE gene expression
The non-coding transcript of the IR3 gene that lies antisense to the IE gene harbors 117 nts overlapping the 5’UTR of the IE mRNA (Fig. 1A; Holden et al., 1992; Ahn et al., 2007). This observation led us to hypothesize that the IR3 transcript may affect IE gene expression in a RNA-RNA fashion. To determine whether the IR3 transcript downregulates IE gene expression, we applied a luciferase reporter system. As shown in Fig.1, the IE promoter region harboring the IE 5’UTR was cloned into the pGL3-Basic plasmid (Promega, Madison, WI) as a reporter vector [pIE(-993/+627-luc)], such that the luciferase gene was under the control of the IE promoter and IE 5’UTR (Fig. 1A), and different regions of the IR3 transcript were cloned into the pSVSPORT1 plasmid (Gibco, BRL, Gaithersburg, MD) under the control of the SV40 promoter as the effecter vectors (Fig. 1B). Cotransfection assays showed that effector vectors [pIR3(+1/+443) and pIR3(+1/+728)] harboring the IR3/IE overlapping sequences downregulated expression of the luciferase gene from the reporter vector in a dose dependent manner, but effector vector pIR3(+444/+728) lacking the IR3/IE overlapping sequences did not affect the expression of the luciferase gene (Fig. 2A). This observation suggested that the IR3(+1/+443) region harboring 117 nts of IR3 transcript [IR3(+226/+342)] antisense to 5’UTR of IE mRNA is responsible for the downregulation of the IE gene. To examine whether IR3(+226/+342) complementary to the IE 5’UTR are the only sequences responsible for luciferase gene downregulation, we dissected the IR3(+1/+443) region, cloned these sequences into the pSVSPORT1 vector (Fig.1B), and then performed cotransfection assays using the pIE(-993/+627-luc) reporter vector. Effector vectors pIR3(+1/+226) and pIR3(+343/+443) that lack the IR3/IE overlapping sequences did not downregulate luciferase gene expression, possibly due to the lack of sequences antisense to IE mRNA. Next, we tested pIR3(+1/+284) that contains 59 nts of the 5’ IR3 transcript within the 117 nts IR3/IE overlapping sequences, pIR3(+226/+343) that contains the entire IR3/IE overlapping sequences, and pIR3(+285/+443) that contains 58 nts of the 3’ IR3 transcript within the IR3/IE overlapping sequences as effector vectors. None of these three effector vectors affected luciferase gene expression (Fig. 2B). However, expression of pIR3(+226/+443) that contains both the entire IR3/IE overlapping sequences [IR3(+226/+343)] and additional IR3 transcript sequences [IR3(+344/+443)] lacking any of the IR3/IE overlapping sequence led to the downregulation of luciferase gene expression (Fig. 2B). To examine which sequences within [IR3(+344/+443)] are essential for the regulatory function of the IR3 transcript, pSVIR3(+226/+403) and pSVIR3(+226/+433) were assayed in the luciferase reporter assay. Expression of IR3(+226/+433) and the positive control IR3(+1/+443) downregulated luciferase gene expression from the reporter vector, but expression of the reporter luciferase gene was not affected by IR3(+226/+403) or the negative control IR3(+444/+728) transcript (Fig. 2C).
Fig. 1.
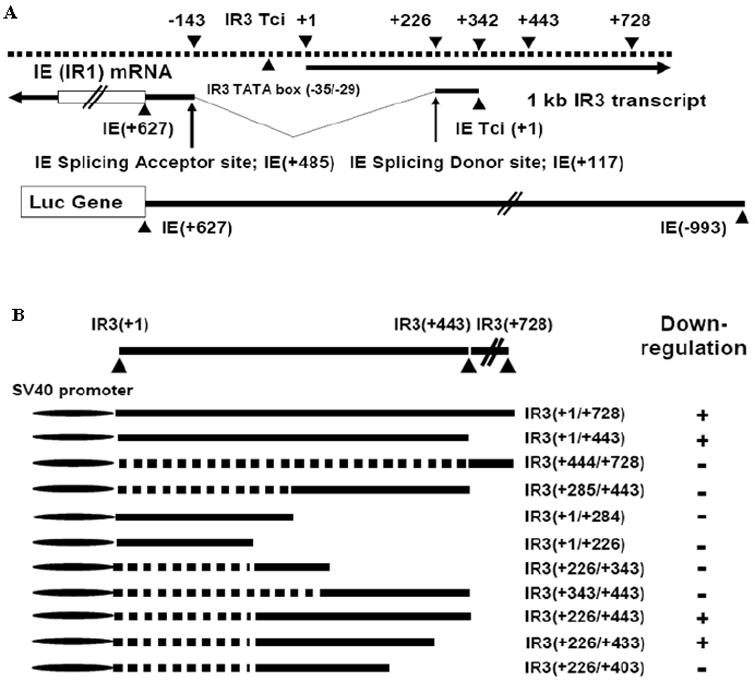
Reporter assays to test IR3 transcript sequences for the ability to downregulate luciferase gene expression controlled by IE gene promoter sequences.
(A). Location of IR3 and IE transcripts, and the IE reporter vector. The IE promoter region harboring the IE 5’UTR was cloned into pGL3-Basic (Promega). Tci, transcription initiation site. “+” and “v” line indicate nt distance from the transcript initiation site and IE intron region, respectively. (B) The different regions of the IR3 transcript cloned into pSPORT1 as effector vector; + indicates downregulation of the luciferase IE reporter vector by IR3 effector vectors. The different regions of IR3 transcript were cloned into pSPORT1 (Gibco, BRL), such that expression of IR3 transcript was under the control of SV40 promoter. “+” within parenthesis indicates nucleotide distances from IR3 transcript initiation site.
Fig. 2.
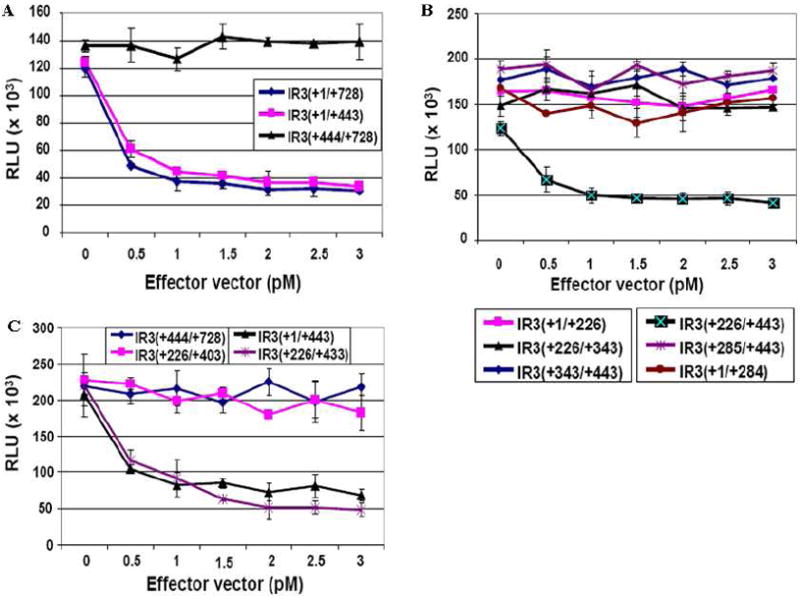
Reporter assays to identify IR3 transcript sequences that downregulate the expression of the luciferase gene controlled by IE promoter sequences. Cotransfection assays were performed by using 1 pM of the pIE(-993/+627)-Luc reporter vector and different concentrations of effector vectors expressing regions of the IR3 transcript as described in Materials and Methods. Error bars represent standard deviations. (A) Luciferase reporter assays using effector vectors [pSVIR3(+1/+728), pSVIR3(+1/+443) and pSVIR3(+444/+728)]. (B) Luciferase reporter assays using effector vectors [pSVIR3(+1/+226), pSVIR3(+226/+443), pSVIR3(+226/+343), pSVIR3(+343/+443), pSVIR3(+1/+284), and pSVIR3(+284/+443) and]. (C) Luciferase reporter assays using effector vectors [pSVIR3(+226/+403) and pSVIR3(+226/+433)]. pSVIR3(+1/+443) and pSVIR3(+444/+728) were used as positive and negative control effector vectors, respectively.
The IR3 gene is dispensable for viral replication
Downregulation of lE gene expression by the IR3 RNA molecule suggests that the absence of expression of the IR3 transcript may affect the biological properties of EHV-1. To examine this hypothesis, ΔIR3 EHV-1 was constructed by using BAC technology (Rudolph et al., 2002) as described in Materials and Methods. The EHV-1 IR3 gene lies antisense to the sole IE gene and overlaps sequences within the IE promoter region (Fig. 1A; Holden et al., 1992). Therefore, deletion of the entire IR3 gene would disrupt the IE promoter and would be lethal because the IE gene is essential for EHV-1 replication (Garko-Buczynski et al., 1998). As an alternative approach, the entire 371 nt IE intron, in which the IR3 TATA box sequence and IEP binding sites essential for IR3 promoter activation are located (Ahn et al., 2007), was replaced with a kanR or zeoR marker without removing the authentic IE splice donor and acceptor site sequences. This approach suggests that the presence of a marker within the IE intron sequence would not affect IE gene expression because the transcript of the marker would be spliced out as an intron from the primary IE mRNA.
Insertion of the kanR or zeoR markers was confirmed by PCR amplification of marker flanking regions (Fig. 3A) and DNA sequence analysis (data not shown). Primer sets specific for the marker flanking regions amplified the expected size of PCR product from pΔIR3 EHV-1 BAC (Fig. 3A, lanes 1, 3, 5 and 7), but not from the parent pRacL11 EHV-1 BAC (Fig. 3A, lanes 2, 4, 6 and 8). BamHI digestion patterns were used to examine the insertion of markers into pRacL11 EHV-1 BAC and showed that two different fragments were observed from pΔIR3 EHV-1 BAC (Fig. 3B, lane 2 indicated as arrows). Additionally, the presence of the two markers in pΔIR3 EHV-1 BAC was demonstrated by a southern blot assay using BamHI digested pΔIR3 EHV-1 BAC and a probe specific for the zeoR (Fig. 3C) or kanR (Fig. 3D) marker. Each radiolabeled probe bound to one fragment (Fig. 3C, lane 2, and Fig. 3D, lane 2), but did not bind to any BamHI digested DNA fragments of pRacL11 EHV-1 BAC (Fig. 3C, lane 1, and Fig. 3D, lane 1). Once insertion of markers into pRacL11 EHV-1 BAC was confirmed, ΔIR3 EHV-1 was generated from pΔIR3 EHV-1 BAC by cotransfection of pΔIR3 EHV-1 BAC and the DNA fragment harboring the Us4 gene into RK13 cells as described in Materials and Methods. To confirm that the ΔIR3 EHV-1 does not express the IR3 transcript, northern blot analysis was performed using total RNA of ΔIR3 EHV-1 infected RK13 cells and a probe specific for the IR3 transcript. The IR3 transcript was detected in RK13 cells infected with RacL11 EHV-1 (Fig. 3E, lane 3), but not in RK13 cells infected with ΔIR3 EHV-1 (Fig. 3E, lane 2). As a positive control, the IR5 transcript was detected by the IR5 gene-specific probe in RK13 cells infected with ΔIR3 EHV-1 and RacL11 EHV-1. These results indicate that ΔIR3 EHV-1 does not express the IR3 transcript and that the IR3 gene is dispensable for EHV-1 replication. To construct the IR3 rescued EHV-1, galK BAC technology was performed as described in Materials and Methods.
Fig. 3.
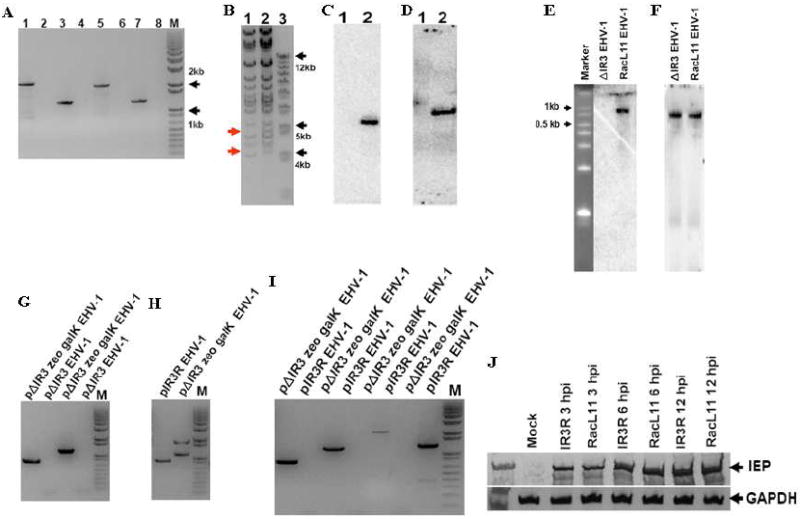
Insertion of zeoR and kanR markers into the EHV-1 genome, and confirmation that ΔIR3 EHV-1 lacks expression of the IR3 transcript. PCR, BamHI digestion, northern blot, and southern blot analyses were performed as described in Materials and Methods. (A) PCR amplification of marker flanking sequences of pΔIR3 EHV-1. pΔIR3 EHV-1 template (lanes 1, 3, 5, and 7). pRacL11 EHV-1 template (lanes 2, 4, 6, and 8). KanR left flanking region amplification primer set (lanes 1 and 2). KanR right flanking region amplification primer set (lanes 3 and 4). ZeoR left flanking region amplification primer set (lanes 5 and 6). ZeoR right flanking region amplification primer set (lanes 7 and 8). “M” is DNA size marker. (B) BamHI digestion pattern of pΔIR3 EHV-1 BAC DNA. Lanes 1 and 2 represent pRacL11 EHV-1 BAC and pΔIR3 EHV-1 BAC, respectively. (C and D) Southern blot analyses of BamHI digested pΔIR3 EHV-1 BAC DNA by using probes specific for zeoR (C) and kanR (D) genes. Lanes 1 and 2 indicate pRacL11 EHV-1 and pΔIR3 EHV-1 BAC DNA, respectively. (E and F) Northern blot analysis to confirm the absence of the IR3 transcript in ΔIR3 EHV-1 infected cells by using a probe specific for the IR3 transcript (E), and a probe specific to IR5 transcript as a positive control (F). (G, H and I) The PCR amplifications of marker flanking sequences to confirm 1). the insertion of galK marker by using galK marker left and right flanking sequence specific primer sets (G), 2). the removal of galK and zeoR markers by using IE intron flanking sequence specific primer set (H) and 3). the presence of galK and zeoR markers by using primer sets specific to galK and zeoR marker flanking sequences (I). (J) Comparison of the IE protein expression from RK13 cells infected with RacL11 EHV-1 and IR3R EHV-1 (at m.o.i of 10) during immediate-early, early, and late times of EHV-1 replication.
First, replacement of the kanR marker with galK marker was confirmed by the PCR amplification of galK marker flanking regions. Left and right primer sets specific to the galK marker flanking sequences detected the expected sizes of PCR amplicons from pΔIR3 zeo galK EHV-1 (Fig. 3G, lanes 1 and 3), but not from pΔIR3 EHV-1 (Fig. 3G, lanes 2 and 4). Next, galK and zeoR markers were replaced with the IE intron containing EHV-1 deletion sequences by using galK counter selection. The replacement was selected by the PCR amplification of IE intron spanning sequences. The primer set specific to the IE intron spanning sequences detected a PCR amplicon of the expected size from pIR3R EHV-1 (Fig. 3H, lane 1), and two PCR amplicons of the expected sizes from pΔIR3 zeo galK EHV-1 (Fig. 3H, lane 2) followed by the sequence analysis of the PCR amplicons. Complete replacement of the two markers with native IE intron sequences was confirmed by the PCR amplification using the galK and zeoR marker flanking sequence specific primer sets. The galK marker left and right flanking sequence specific primer sets detected PCR amplicons of the expected sizes from pΔIR3 zeo galK EHV-1, respectively (Fig. 3I, lanes 1 and 3), but not from pIR3R EHV-1 (Fig. 3I, lanes 2 and 4). In addition, the zeoR marker left and right flanking sequence specific primer sets detected PCR amplicons of the expected sizes from pΔIR3 zeo galK EHV-1, respectively (Fig. 3I, lanes 5 and 8), but not from pIR3R EHV-1 (Fig. 3I, lanes 6 and 8), indicating that the IE intron deletion sequences were completely restored. Once IR3R EHV-1 was generated from pIR3R EHV-1 as described above, the protein expression of RacL11 EHV-1 and IR3R EHV-1 was directly compared to exclude the possibility of a secondary mutation(s) of ΔIR3 EHV-1. IE protein expression levels of the RacL11 EHV-1 and IR3R EHV-1 were identical during immediate-early, early, and late time of EHV-1 replication (Fig. 3J), indicating that there was no secondary mutation in the ΔIR3 EHV-1 mutant.
Serial passage of ΔIR3 EHV-1 in permissive cells leads to the sequence recombination between IR and TR sequences
The IR and TR sequences of the EHV-1 short genomic region harbor identical sequences (Henry et al., 1981; Telford et al., 1992). Previously, we observed that deletion of one gene within the IR or TR region is recovered by identical sequences from the other inverted repeat during reproductive replication in permissive cells, such that both IR and TR sequences become identical (data not shown). In addition, homologous genomic recombination between two different viral species has been described for alphaherpesviruses (Muerens et al., 2004, Umene, 1985). These observations led us to examine whether the IR and TR segments harboring each of the two different marker genes, respectively, become identical diploid sequences during the serial passages of the ΔIR3 mutant virus in permissive cells. First, the presence of marker genes in the ΔIR3 EHV-1 was examined by PCR analysis using DNA from ΔIR3 EHV-1 infected RK13 cells, and primer sets to detect the kanR and zeoR markers as described in Materials and Methods. Interestingly, the expected size PCR product was amplified from primer sets to detect the zeoR marker (Fig. 4A, lanes 3 and 4), but not from primer sets to detect the kanR marker (Fig. 4A, lanes 1 and 2). Further, the presence of the zeoR marker and the absence of the kanR marker were confirmed by dot blot hybridization analysis with a probe specific for the kanR or zeoR marker as described in Materials and Methods. The DNA probe specific for the kanR marker (Fig. 4B) bound to only pΔIR3 EHV-1 BAC DNA (no. 3), but not to the empty spot (no. 1), pRacL11 EHV-1 BAC DNA (no. 2), RK13 cell DNA (no. 4), RacL11 EHV-1 infected RK13 DNA (no. 5) or ΔIR3 EHV-1 infected RK13 cell DNA (no. 6). The probe specific to zeoR marker (Fig. 4C) to determine if the zeoR marker was retained in the ΔIR3 EHV-1 genome bound to both pΔIR3 EHV-1 BAC DNA (no. 3) and also to DNA of ΔIR3 EHV-1 infected RK13 cells (no. 6). However, the zeoR probe (Fig. 4B) did not bind to control empty DNA (no. 1), pRacL11 EHV-1 BAC DNA (no. 2), RK13 cell DNA (no. 4), or DNA of RacL11 EHV-1 infected RK13 cells (no. 5).
Fig. 4.
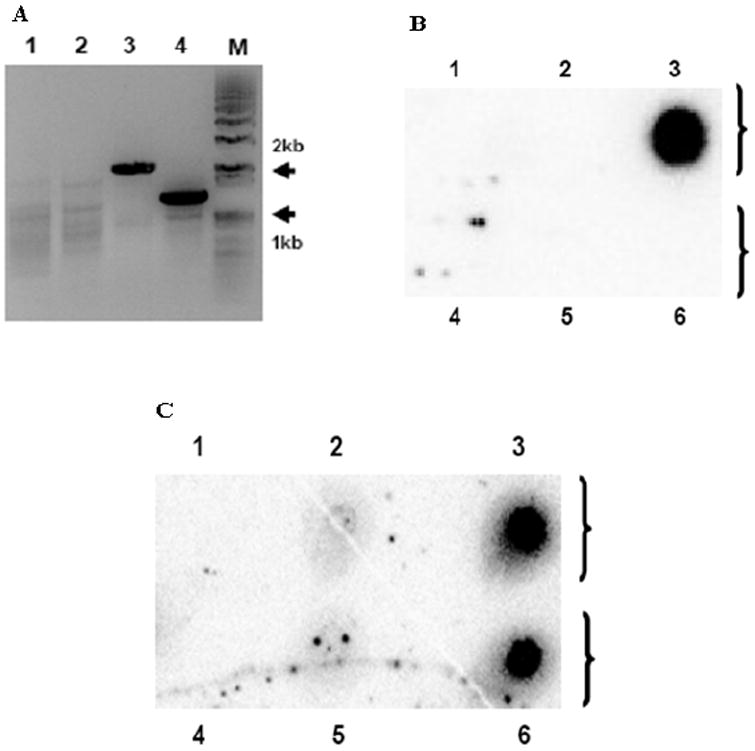
The kanR marker was replaced with the zeoR marker during serial passage of ΔIR3 EHV-1. PCR and dot blot hybridization analyses were performed as described in Materials and Methods. (A) PCR amplification of flanking regions of markers inserted into ΔIR3 EHV-1 genome. ΔIR3 EHV-1 infected RK13 DNA was used as template. KanR left flanking region amplification primer set (lane 1). KanR right flanking region amplification primer set (lane 2). ZeoR left flanking region amplification primer set (lane 3). ZeoR right flanking region amplification primer set (lane 4). “M” is DNA size marker. (B and C) Dot blot hybridization analyses with probes specific for the kanR and zeoR markers, respectively. Lanes 1 to 6 represent mock DNA, pRacL11 EHV-1 DNA, pΔIR3 EHV-1 DNA, RK13 DNA, RacL11 EHV-1 infected RK13 DNA, and ΔIR3 EHV-1 infected RK13 DNA, respectively.
ΔIR3 EHV-1 has a larger plaque size, but exhibits host cell tropism and growth kinetics in RK13 cells similar to those of the parent virus
Next, experiments were carried out to determine whether the absence of expression of the IR3 transcript affected the biological properties of ΔIR3 EHV-1 in cell culture. Initial assays revealed that the plaque size of ΔIR3 EHV-1 was increased by more than 50% compared to those of RacL11 EHV-1 and IR3R EHV-1 (Fig. 5A and B) and that the change in plaque size was statistically significant (p=0.0006). As a next step, we examined the cell tropism and growth kinetics of ΔIR3 EHV-1 as compared to those of the parent RacL11 EHV-1 (Fig.6A). The ΔIR3 EHV-1 like the parent virus was capable of replication in cells of mouse, rabbit, equine, monkey, and human origin (Fig. 6A), and IR3R EHV-1 also exhibited the cell tropism identical to that of RacL11 and ΔIR3 EHV-1 (data not shown). Both RacL11 and ΔIR3 EHV-1 replicated to much greater titers in the equine NBL-6, rabbit RK13, and mouse LM cells as compared to titers achieved in human HeLa and monkey Vero cells. In addition, RacL11 EHV-1 and ΔIR3 EHV-1 exhibited virtually identical growth kinetics in RK13 cells infected at a multiplicity of infection of both 5 (Fig. 6B) and 0.1 (Fig. 6C) pfu/cell.
Fig. 5.
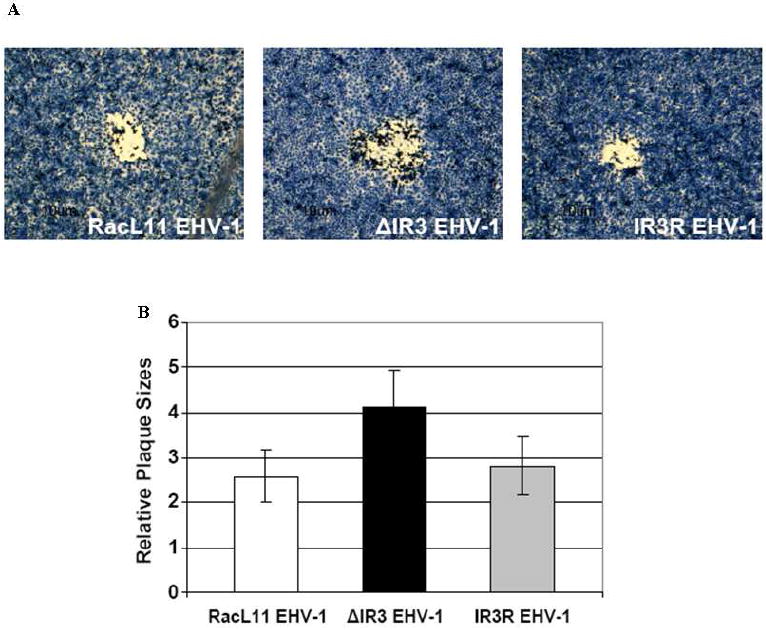
Comparison of plaque size of RacL11 EHV-1, ΔIR3 EHV-1 and IR3R EHV-1 in RK13 cells. (A) Morphology of plaques in RK13 cells infected with RacL11 EHV-1, ΔIR3 EHV-1 and IR3R EHV-1. (B) Comparison of plaque size in RK13 cells infected with RacL11 EHV-1, ΔIR3 EHV-1 and IR3R EHV-1. Relative plaque sizes were measured by using the ImageJ software program; the two-tailed Student’s t test was used for data analysis. Error bars indicate standard deviation.
Fig. 6.
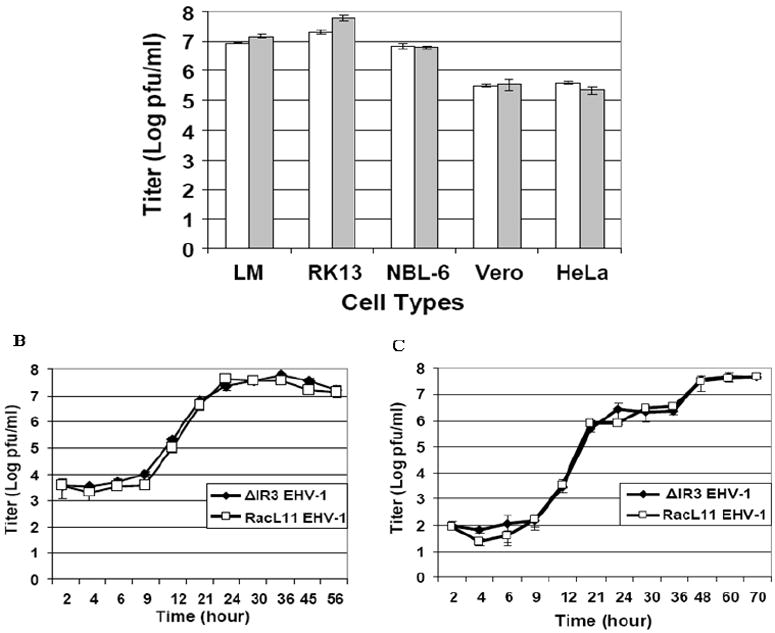
Cell tropism and growth kinetics. (A) Cell tropism and viral titers of ΔIR3 EHV-1 and RacL11 EHV-1 in mouse LM, rabbit RK13, equine NBL-6, simian Vero, and human HeLa cells. Cell monolayers were infected at a moi of 5 moi with ΔIR3 EHV-1 or RacL11 EHV-1, harvested at 72hpi, and titered as described in Materials and Methods. White and gray bars indicate RacL11 EHV-1 and ΔIR3 EHV-1 titers, respectively. (B) Viral growth kinetics of RacL11 EHV-1 and ΔIR3 EHV-1 in RK13 cells infected at a moi of 5. (C) Growth kinetics of RacL11 EHV-1 and ΔIR3 EHV-1 in RK13 cells infected at a moi of 0.1. Cell monolayers were infected with RacL11 EHV-1 or ΔIR3 EHV-1, and aliquots were harvested at designated time points. Virus was titered as described in Materials and Methods. Error bars indicate standard deviations.
ΔIR3 EHV-1 showed increased protein expression of EHV-1 representative genes in RK13 cells
The observation that the IR3 transcript downregulated expression of the luciferase gene under the control of the IE 5’UTR and promoter suggests that the absence of IR3 transcript may affect expression of the IE gene in infected cells. To examine whether any consequences of the lack of IR3 gene expression occur in EHV-1 gene programming, expression of the sole IE gene and representative early and late genes was monitored at different time points after infection of RK13 cells. The IEP was detected at 3 hours post infection (hpi) in both RacL11 EHV-1 and ΔIR3 EHV-1 infected RK13 cells (Fig. 7A, lanes 3 and 4), and IEP levels in ΔIR3 EHV-1 infected cells were slightly higher during early and late stages of replication than those of RacL11 EHV-1 infected cells (Fig. 7A, lanes 5-8). Protein expression from the IR4 early gene was barely detected in RK13 cells infected with RacL11 or ΔIR3 EHV-1 at 3 hpi (Fig. 7B, lanes 3 and 4), and protein expression levels of ΔIR3 EHV-1 infected cells were higher than those of RacL11 EHV-1 infected cells during early and late time points of replication (Fig. 7B, lanes 5-8). As another early gene product, the UL5P was observed at 3 hpi, and UL5P expression levels in RK13 cells infected with ΔIR3 EHV-1 were higher than those of RacL11 EHV-1 infected cells through all stages of replication (Fig. 7C, lanes 3-8). Protein expression of EICP0, a potent early regulatory gene, was barely detected at 3 hpi (Fig. 7D, lanes 3 and 4), and protein expression levels were higher in RK13 cells infected with ΔIR3 EHV-1 than in RK13 cells infected with RacL11 EHV-1 during early and late stages of replication (Fig. 7D, lanes 5-8). As expected, the late gene product glycoprotein D (gDP) was first detected at late times (12 hpi; Fig. 7E, lanes 7 and 8), not at earlier times (Fig. 7E, lanes 3-6), and the amount of gDP in ΔIR3 EHV-1 infected RK13 cells was greater than that in RK13 cells infected with RacL11 EHV-1 (Fig. 7E, lanes 7-8). The multiple repeat experiments also showed identical results in the protein expression. In addition, the difference of the protein expression between parent virus and mutant virus infected RK13 cells was further determined by using ImageJ software program. Comparison of protein expression of EHV-1 representative genes showed that EHV-1 protein expression levels from ΔIR3 EHV-1 infected RK13 cells were higher than those from RacL11 EHV-1 infected RK13 cells (Fig. 7F).
Fig. 7.
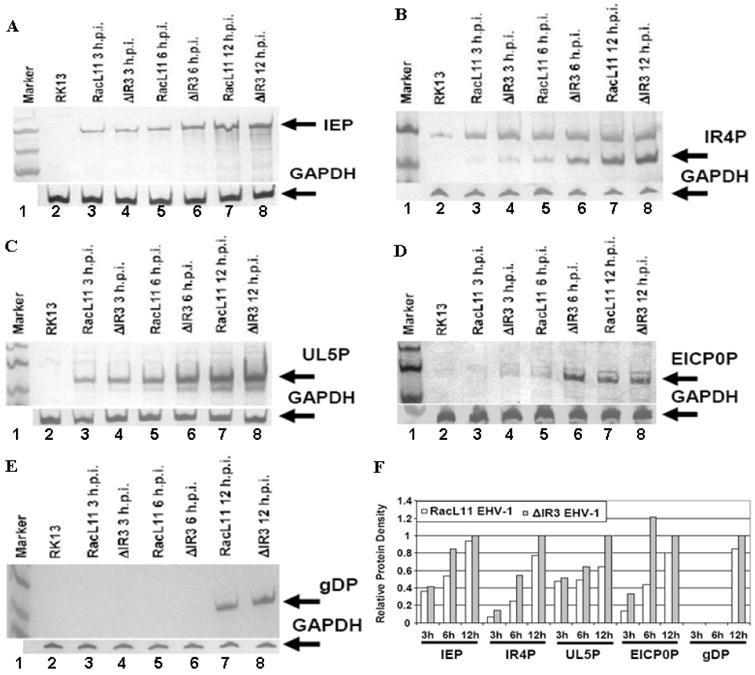
Expression levels of EHV-1 IE, early, and late proteins. RK13 cells infected with ΔIR3 EHV-1 or RacL11 EHV-1 at a moi of 10 were harvested at 3, 6, and 12 hpi, and protein expression was examined by western blot analysis as described in Materials and Methods. Detection of the (A) immediate early protein, (B) early IR4 protein, (C) early UL5 protein, (D) early EICP0 protein, and (E) late glycoprotein. (F) Relative protein density of EHV-1 representative genes in RacL11 and ΔIR3 EHV-1 infected RK13 cells. Protein density was compared by using the ImageJ software program (http://rebweb.nih.gov/ij/). White and gray bars indicate proteins of RK13 cells infected with RacL11 EHV-1 and ΔIR3 EHV-1, respectively. Relative protein density was the protein density of each protein relative to the amount of that protein in RK13 cells infected with ΔIR3 EHV-1 at 12hpi.
ΔIR3 EHV-1 retained full virulence in the CBA mouse model
To examine the possibility that the absence of expression of the IR3 transcript affected EHV-1 virulence, animal experiments in the well-characterized CBA mouse model were performed as described in Materials and Methods. As shown in Fig. 8A, body weight of mice infected with ΔIR3 EHV-1, IR3R EHV-1, or RacL11 EHV-1 were significantly decreased compared to those of control group mice (all p values < 0.001). In addition, the correlation of the body weight loss among mouse groups infected with ΔIR3 EHV-1, IR3R EHV-1, or RacL11 EHV-1 was significant (p values < 0.01), indicating there was no difference of body weight loss among mouse groups infected with ΔIR3 EHV-1, IR3R EHV-1, or RacL11 EHV-1. However, a plot of the percent survival in Fig. 8B showed that there was a significant difference (p=0.003) between the RacL11 EHV-1 and ΔIR3 EHV-1, and there was no significant difference between the RacL11 EHV-1 and IR3R EHV-1 (p=0.14), indicating an increase in the virulence of the mutant virus as compared to that of the parent RacL11 EHV-1.
Fig. 8.
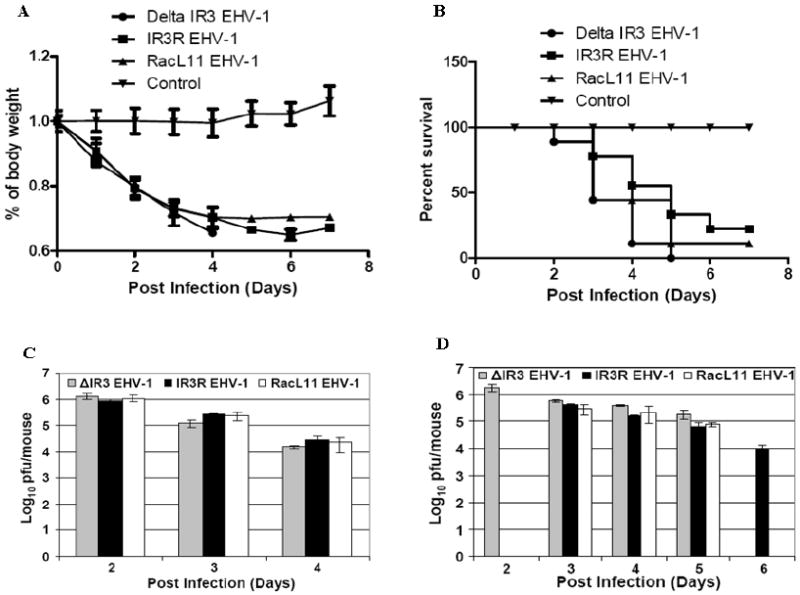
Percentage change in body weight of CBA mice infected with mock inoculum, RacL11 EHV-1, and ΔIR3 EHV-1, and EHV-1 titers of mouse lungs. Mice were intranasally inoculated with sterile medium as control or with 2×106 PFU/mouse of either wt RacL11 EHV-1 or ΔIR3 EHV-1, and total virus was isolated from mouse lungs and titered as described in Materials and Methods. Body weight was measured daily, and the two-tailed Student’s t test was used for data analysis to compare body weight between groups. Error bars indicate standard deviation. (A) Percentage change in body weight of CBA mice infected with ΔIR3 EHV-1 (n=9), IR3R (n=9) or RacL11 EHV-1 (n=9). (B) Percent survival of the mice infected with RacL11 EHV-1 (n=9), IR3R EHV-1 (n=9), ΔIR3 EHV-1 (n=9), or mock (n=9). (C) Viral titers of lungs of live mice infected with RacL11 EHV-1 (n=3, white bars), IR3R EHV-1 (n=3, black bars), or ΔIR3 EHV-1 (n=3, gray bars) at 2, 3, and 4 dpi. (D) Viral titers of lungs from dead mice infected with RacL11 EHV-1 (white bars), IR3R EHV-1 (black bars) or ΔIR3 EHV-1 (gray bars) at 2, 3, 4, 5, and 6 dpi. Each bar indicates the average viral titer of mice succumbed at the same day. The sample number(s) of each bar from at 2 dpi to 6 dpi are n=1 (bar 1), n=4 (bar 2), n=2 (bar 3), n=2 (bar 4), n=3 (bar 5), n=2 (bar 6), n=3 (bar 7), n=1 (bar 8), n=2 (bar 9), n=3 (bar 10), and n=1 (bar 11), respectively.
To determine whether the early death of mice infected with ΔIR3 EHV-1 was related to viral load, we examined the viral titers of lungs from EHV-1 infected live (Fig. 8C) and dead (Fig. 8D) mice. Viral titers in the lungs of live mice were the highest at early dpi and decreased at later dpi (Fig. 8C). Decrease of the viral load in mice infected with RacL11, IR3R, or ΔIR3 EHV-1s was similar at each dpi. Assay of the viral titers in lungs of dead mice (Fig. 8D) revealed that viral lung titers also decreased as time progressed. However, the lung viral titers of all ΔIR3 EHV-1 infected mice that succumbed early to infection were at the peak titer at these early times, suggesting that the IR3 mutant virus is capable of rapid and efficient replication in the murine respiratory tract.
Discussion
Here, we presented findings that the non-coding IR3 transcript has a regulatory function in EHV-1 replication and its absence affects EHV-1 biological properties and virulence in a mouse model. Our observation that part of the IR3 transcript is antisense to IE mRNA led us to examine the possibility that the IR3 transcript may regulate IE gene expression as a RNA molecule. Cotransfection assays showed that the IR3 transcript [(IR3(+1/+443)] harboring IR3/IE overlapping sequences downregulated IE gene expression in a dose dependent manner. Additional experiments revealed that in addition to the IR3/IE overlapping sequences at [IR3(+226/+342), IR3 sequences +344/+433 outside the antisense region are essential for IR3 RNA to downregulate IE gene expression. Also, the expression of the IR3 transcript from all IR3 effector vectors in transfected RK13 cells at 12 hpt was confirmed by RT-PCR analyses (data not shown). Thus, the lack of the IE gene downregulation by some of the IR3 effector vectors was not due to the lack of its IR3 transcript expression. Overall, these results indicate that EHV-1 gene expression is influenced not only by the six known viral regulatory proteins, but also by EHV-1 encoded RNA as a regulatory molecule.
Next, we employed a genetic approach to examine the biological functions of the IR3 transcript and generated a virus mutant that lacked the ability to express the IR3 transcript. Since deletion of the entire IR3 gene sequence would remove the promoter region of the IE gene (see Fig. 1) that is essential for EHV-1 replication (Garko-Buczynski et al., 1998), an alternative approach to overcome this obstacle was required. To this end, 371 nts of the IE intron (Harty et al. 1989; Grundy et al., 1989, Fig.1A) that harbors the IR3 TATA box essential for IR3 promoter activity (Ahn et al., 2007) were replaced with a kanR or zeoR maker by using BAC technology. The marker within the IE intron sequence would not affect IE gene expression because the transcript of the marker would be spliced out as an intron of the primary IE mRNA. As expected, the resulting ΔIR3 EHV-1 generated from the pΔIR3 EHV-1 BAC was replication competent, indicating that the IR3 gene as a negative regulator is dispensable for EHV-1 replication.
In alphaherpesviruses, homologous genomic recombination between distinguishable strains has been described for herpes simplex viruses 1 and 2 (Umene 1985), pseudorabies virus (Dangler et al., 1993), feline herpesvirus 1 (Fujita et al., 1998), varicella-zoster virus (Dohner et al., 1988), and bovine herpesviruses 1 and 5 (Meurens et al., 2004). The presence of identical IR and TR sequences in the EHV-1 genome led us to examine whether one marker located within either the IR or TR of the ΔIR3 EHV-1 genome could be replaced with the other marker sequence by homologous recombination during serial passage in permissive cells. Interestingly, this was the case as the kanR marker was replaced by the zeoR marker sequences, which was confirmed by PCR amplification, sequence analysis, and dot blot hybridization analyses (Fig. 3). Considering that the flanking sequences of the two markers within the ΔIR3 EHV-1 genome are identical and the frequency of homologous genomic recombination in alphaherpesviruses, it was not surprising that one marker was replaced with the other marker in permissive RK13 cells. We hypothesize that this replacement that favored the selection of zeoR over kanR was due to the size difference between the two foreign DNA sequences, about 0.5kb and 1.5kb for zeoR and kanR , respectively.
The effect of preventing the expression of the IR3 transcript on plaque size, cell tropism, growth kinetics, and protein expression levels of representative genes was examined in cells infected with the ΔIR3 EHV-1. The increased plaque size of the ΔIR3 EHV-1 was not an unexpected result because the absence of a negative regulatory molecule could accelerate the trans-activation of EHV-1 genes, leading to more rapid virus spread. Also, that the cell tropism of ΔIR3 EHV-1 was identical to that of the RacL11 EHV-1 was not surprising since the absence of the IR3 transcript was not predicted to prevent the expression of viral envelope glycoprotein(s) responsible for attachment and penetration and thereby influence host range. Interestingly, protein expression levels of representative EHV-1 genes were increased in ΔIR3 EHV-1 infected cells, and these observations could be explained by the following facts. The IR3 transcript as a negative regulator downregulates expression of the IE gene that directly or indirectly upregulates expression of the EICP0, IR4, UL5 and gD genes. As a consequence of the lack of IR3 transcript, expression of the IE gene would be upregulated, and then protein expression of early and late genes that are trans-activated to a large extent by the IEP would be increased. The upregulation of the IEP was not only shown in the RK13 cells as presented in our results, but also observed in the equine origin NBL-6 cells (data not shown), supporting the role of the IR3 transcript in the EHV-1 gene expression. In addition, that deletion of an EHV-1 regulatory gene affects expression levels of EHV-1 genes has been described in EHV-1 mutants such as ΔEICP0 EHV-1 (Yao et al., 2003), and ΔIR4 EHV-1 (Breitenbach et al., 2009). These observations are consistent with the suggestion that increased protein expression in ΔIR3 EHV-1 may be due to the lack of the IR3 transcript that reduces IEP production, even though we cannot exclude the possibility that the IE intron plays a role. . When growth kinetics were assayed in RK cells infected at a moi of 5 or 0.1, similar growth patterns were observed for both RacL11 and ΔIR3 EHV-1. These findings showed that the increase in the levels of the IEP and representative early and late proteins in cells infected with the IR3 mutant virus did not result in enhanced virus growth in cell culture. However, in the CBA mouse model, intranasal infection with the ΔIR3 EHV-1 resulted in an increase in virulence as this group of mice succumbed more rapidly to infection and virus titers in the lung peaked much earlier than was the case for mice infected with RacL11 EHV-1.
EHV-1 encodes only one immediate-early gene, and its 1,487 amino acid protein product is the major viral regulatory protein that by itself and in concert with other viral regulatory protein orchestrates the gene program (O’Callaghan and Osterrieder, 2008). Because of the importance of the IEP in EHV-1 replication, at least three mechanisms operate to govern the IE gene: 1. the IEP itself is autoregulatory and is capable of downregulating the IE gene by binding to a target binding site that overlaps the transcription start site (Kim et al., 1995); 2. the early IR2 protein that is generated from an early gene that maps with the IE gene (Harty and O’Callaghan. 1991) is a major negative regulatory protein and functions to downregulate IE gene expression (Kim et al. 2006); and 3. the IR3 transcript harbors sequences antisense to the IE gene (Holden et al., 1992; Ahn et al., 2007) and as shown in this study has the ability to affect IE gene expression. The findings that expression of the EHV-1 unique IR3 gene is not essential for viral replication in cell culture and that overall the regulatory IR3 RNA appears to have a modest role in virus growth in cell culture raise the question of the importance of this gene in EHV-1 infection of its natural hosts. One possibility that merits consideration is that the IR3 regulatory RNA might contribute to suppression of IE gene expression in EHV-1 latently infected cells and thus serve a function as a latency-associated transcript. This possibility and questions about the specific mechanism by which IR3 RNA mediates its regulatory function are topics for ongoing investigations.
Materials and methods
Cell culture
All cell cultures were maintained with Eagle’s minimal essential medium (EMEM) supplemented with 100 units of penicillin/ml, 100 μg of streptomycin/ml, nonessential amino acids, and 5% (or 10%) fetal bovine serum.
Construction of reporter and effector plasmids for luciferase assays
The PCR products were amplified using Accuprime pfx polymerase (Invitrogen, Carlsbad, CA), template plasmid containing IR region flanking sequences, and primers containing proper restriction enzyme sites at the 5’ ends. The PCR products were digested with appropriate restriction enzymes and cloned into proper vectors using standard cloning methods (Sambrook et al., 1989). All clones were confirmed by DNA sequence analysis. For the luciferase reporter assay, reporter and effector vectors were constructed. To generate the reporter vector, the IE promoter region [IE(+627/-993)] including the IE 5’UTR was amplified (primers, 5’ aag gtg ggt acc ggg cat ctc c 3’, 5’ aat aga tct ggc gtg cta gct ccg gc 3’), and the PCR product that was digested with KpnI and BglII was cloned into pGL3-Basic (Promega) digested with KpnI and BglII (named pIE(+627/-993)-Luc). Different regions of the IR3 transcript were PCR-amplified and cloned into pSVSPORT1 (Gibco, BRL). The PCR product of IR3(+1/+226) (primers, 5’att ggt acc tcc gcc gag gca gaa gcc g 3’, 5’ taa tct aga caa cgg cca atc aca atc gat agt gtg 3’), IR3(+1/+284) (primers, 5’att ggt acc tcc gcc gag gca gaa gcc g 3’, 5’ tta tct aga act gat cgc atc ccg gaa ag 3’), IR3(+1/+443) (primers, 5’att ggt acc tcc gcc gag gca gaa gcc g 3’, 5’ taa tct aga caa cgg cca atc aca atc gat agt gtg 3’), IR3(+1/+728) (primers, 5’att ggt acc tcc gcc gag gca gaa gcc g 3’, 5’ agc tct aga tca cga acc ccg ttg gcg cga cgc 3’), IR3(+226/+343) (primers, 5’ ttt ggt acc ctt acc tcc gca agc cg 3’, 5’ttt tct aga gta gca tcg cgg act aga cac 3’), IR3(+226/+443) (primers, 5’ ttt ggt acc ctt acc tcc gca agc cg 3’, 5’ taa tct aga caa cgg cca atc aca atc gat agt gtg 3’), IR3(+285/+443) PCR product (primers, 5’ tta ggt acc agt cct cga agg ctg gct gg 3’, 5’ taa tct aga caa cgg cca atc aca atc gat agt gtg 3’), IR3(+444/+728) (primers, 5’ tag ggt acc gcg acc atg ggc ggt ggg cgt gta gcg 3’, 5’ agc tct aga tca cga acc ccg ttg gcg cga cgc 3’), IR3(+343/+443) (primers, 5’ tat ggt acc gat ggg taa gca aca ggt g 3’, 5’ taa tct aga caa cgg cca atc aca atc gat agt gtg 3’), IR3(+226/+403) (primers, 5’ ttt ggt acc ctt acc tcc gca agc cg 3’ 5’ ttt tct aga cac tag ggg gaa ggc 3’), or IR3(+226/+433) (primers, 5’ ttt ggt acc ctt acc tcc gca agc cg 3’ 5’ ttt tct aga tca caa tcg ata gtg tgg g 3’) was digested with KpnI and XbaI, and cloned into pSVSPORT1 vector (Gibco, BBL) digested with KpnI and XbaI. The above cloned plasmids were named pSVIR3(+1/+226), pSVIR3(+1/+284), pSVIR3(+1/+443), pSVIR3(+1/+728), pSVIR3(+226/+342), pSVIR3(+226/+443), pSVIR3(+285/+443), pSVIR3(+444/+728), pSVIR3(+343/+443), pSVIR3(+226/+403), and pSVIR3(+226/+433), respectively, and were used as effector vectors in the luciferase reporter assays.
Luciferase reporter assays
The luciferase reporter assay was performed as previously described (Ahn et al., 2007). Briefly, 80% confluent RK13 cells were prepared in 24 well plates, and 1 pM of reporter vector [pIE(+627/-993)-Luc] and different concentration (0 to 3 pM) of effector vectors [pSVIR3(+1/+226), pSVIR3(+1/+284), pSVIR3(+1/+443), pSVIR3(+1/+728), pSVIR3(+226/+343), pSVIR3(+226/+443), pSVIR3(+343/+443), pSVIR3(+285/+443), pSVIR3(+226/+403), pSVIR3(+226/+433), and pSVIR3(+444/+728)] were used. Six microliters of lipofectin (Invitrogen) was mixed with 100 μl of Opti-MEM medium (Gibco, BRL), and incubated for 45 min at room temperature. The effector and reporter plasmids were mixed with 100 μl of Opti-MEM medium (Gibco, BRL), and total DNA was adjusted to the same amount with pSVSPORT1 DNA (Gibco, BRL). The mixtures were combined and incubated at room temperature for 15 min, and 460 μl of Opti-MEM medium (Gibco, BRL) was added. One-third of the total mixture was transferred into each of three wells of 24 well plates of RK13 cell monolayers after washing with serum-free and antibiotics-free E-MEM. At 8 hours post transfection (hpt), normal growth medium was added. Luciferase activity was measured at 48 hpt using a luciferase assay kit (Promega) and POLARstar OPTIMA plate reader (BMG LABTECH Inc., Burham, NC) according to the manufacturer’s directions.
RT-PCR assay to verify IR3 RNA synthesis
Each of the effector vectors expressing the IR3 transcript or portions of this mRNA was transfected into the 80% confluent RK13 cells by using lipofectin (Invitrogen, Carlsbad, CA) according to the manufacturer’s directions. At 12 hour post transfection, total RNAs were isolated by using RNA Bee RNA isolation kit (Tel-Test, Inc., Friendwood, TX). The first strand cDNA synthesis was performed by using total RNA template, 10 pM/μ reverse primer, and Superscript III reverse transcriptase kit (Invitrogen) according to the manufacturer’s directions. The second round DNA amplification of the specific IR3 transcript region was performed by using the Acuprime pfx supermix (Invitrogen), primers specific to each IR3 transcript region, and 2 μl of the first strand cDNA. Amplification of the IR3 transcript region was visualized on 1% agarose gels containing ethidium bromide.
Generation of ΔIR3 EHV-1 and IR3R EHV-1
To delete the IR3 TATA box sequences, the PCR product of the IE intron left flanking sequences (primers, 5’ agt tct gca gcc agg aca tgg ctt cgc gg 3’, 5’ tta tct aga aaa aga tct ata act tcg tat aat gta tgc tat acg aag tta tgc ttt tcc agg gta gag aag cgg 3’) was digested with PstI and XbaI, and cloned into pGEM-3Z(+) (Promega) digested with PstI and XbaI (named pIR3L). The PCR fragment of the IE intron right flanking sequences (primers, 5’ tta tct aga ata act tcg tat agc ata cat tat acg aag tta tgg gag ctc tta cct ccg caa gat gct acg atg ggt aag caa cag g 3’, 5’ tgg acg tgg gtg gaa gga ggg tac cgg 3’) was digested with XbaI and KpnI, and cloned into pIR3L digested with XbaI and KpnI (named pIR3L-R) . The PCR product of zeoR (primers, 5’ tta tct aga atc gaa atc tcg tag cac gtg 3’, 5’ gtt aga tct tta gga gct tgt ata tcc att ttc gg 3’) from plasmid pSVZeo2 (Invitrogen)] was digested with BglII and XbaI, and cloned into pIR3L-R digested with BglII and XbaI (named pIR3LR-zeoR). The PCR product of kanR (primers, 5’ att aga tct ttt cgg gga aat gtg cg 3’, 5’ tta tct aga aat ctc gtg atg gca ggt tgg 3’) from plasmid pEGF-N1 (Clontech, Mountain View, CA) was digested with BglII and XbaI, and cloned into pIR3L-R digested with BglII and XbaI (named pIR3LR-kanR). The flanking sequences of both kanR and zeoR markers that were cloned into the pIR3LR vector included authentic IE splicing donor and acceptor sites. Deletion of the IE intron to construct the ΔIR3 EHV-1 was performed by using BAC technology (Rudolph et al., 2002). First, the DNA fragment with the kanR gene with flanking sequences of the IE intron was obtained from pIR3LR-kanR by KpnI and PstI digestion, and was electroporated into E. coli DY380 containing pRacL11 EHV-1 BAC (parent EHV-1 BAC) previously grown at 42° C for 20 min to induce RED recombinase. Electroporated cultures were grown at 30° C on agar plates containing kanamycin (30 μg/ml) and chloramphenicol (30 μg/ml), and selected colonies were confirmed by PCR amplification of junctions between the EHV-1 genomic sequences and the antibiotic resistant marker (primers, 5’ ggc atc aga gca gcc gat tgt ctg ttg tg 3’/5’ tga act tct ggc aga act gcg cca ggt tct gg 3’, 5’ gct atc agg aca tag cgt tgg cta ccc gtg ata 3’/5’ 5’ ttt act cgg tac aac gat tgt gcg tgt gtg cgg 3’) and sequence analysis (named pΔIR3 kanREHV-1 BAC). The second copy of the IE intron was deleted in the same manner. The DNA fragment harboring the zeoR gene with flanking sequence of the IE intron was obtained from pIR3LR-zeoR by KpnI, ScaI, and PstI digestion, and was electrophorated into E. coli DY380 containing pΔIR3 kanREHV-1 BAC. Then, positive colonies were selected on agar plates containing kanamycin (30 μg/ml), chloramphenicol (30 μg/ml), and zeomycin (30 μg/ml). Complete replacement of the IE intron with antibiotic marker genes was confirmed by BamHI restriction digestion, northern blot analysis using probes specific for the kanR or zeoR marker, and PCR amplification of junctions between the EHV-1 genomic sequences and antibiotic resistant markers (primers, 5’ tcc gac cac tcg gcg tac agc tcg tcc agg 3’/5’ tga act tct ggc aga act gcg cca ggt tct gg 3’, 5’ tgg acg agc tgt acg ccg agt ggt cgg 3’/5’ ttt act cgg tac aac gat tgt gcg tgt gtg cgg 3’) followed by DNA sequence analysis (named pΔIR3 EHV-1 BAC).
To construct IR3 gene rescued (IR3R) EHV-1, galK BAC technology was used according to the previously described method (Worming et al., 2005). First, pΔIR3 EHV-1 was transformed into SW106 E. coli, and galK gene PCR product flanked by kan gene flanking sequences (primers, 5’ ttt cgg gga aat gtg cgc gga acc cct att tgt tta ttt ttc taa cct gtt gac aat taa 3’/5’ aat ctc gtg atg gca ggt tgg gcg tcg ctt ggt cgg tca ttt cga act gtc ctg ctc ctt 3’) was transformed into SW106 E. coli containing pΔIR3 EHV-1. First, replacement of the kan gene with galK gene was screened on galK positive agar plate containing chloramphenicol and zeocine, and then confirmed by PCR amplification of galK left (primers, 5’ atg atc ccg cag tta cag cct aca aac tgg 3’/5’ tag ctc cgg ctt cgg ggt cga g 3’) and right (primers, 5’ tga aac atc tgc aac tgc gta aca aca gct tcg g 3’/5’ ata tga gag gcg cta ttc ggc atc ccg 3’) flanking sequences (named pΔIR3 zeo galK EHV-1). The PCR product harboring the entire IE intron and its flanking sequences (primers, 5’ tag ctc cgg ctt cgg ggt cga g 3’/5’ ata tga gag gcg cta ttc ggc atc ccg 3’) was transformed into SW106 E. coli harboring pΔIR3 zeo galK EHV-1. Replacement of the galK and zeoR markers with the IE intron sequences in pΔIR3 zeo galK EHV-1 was screened by galK counter selection and then confirmed by the PCR amplification (primers, 5’ tag ctc cgg ctt cgg ggt cga g 3’/5’ ata tga gag gcg cta ttc ggc atc ccg 3’) of the IE intron sequences (named pIR3R EHV-1). In addition, recovery of the deleted IE intron sequences was confirmed by PCR amplification using galK left (primers, 5’ atg atc ccg cag tta cag cct aca aac tgg 3’/5’ tag ctc cgg ctt cgg ggt cga g 3’) and right (primers, 5’ tga aac atc tgc aac tgc gta aca aca gct tcg g 3’/5’ ata tga gag gcg cta ttc ggc atc ccg 3’) flanking sequences, and zeoR left (primers, 5’ tga act tct ggc aga act gcg cca ggt tct gg 3’/5’ tgg acg agc tgt acg ccg agt ggt cgg 3’) and right (primers, 5’ ttt act cgg tac aac gat tgt gcg tgt gtg cgg 3’/5’ aag ttc gtg gac acg acc tcc gac cac tcg 3’) flanking sequences followed by the sequence analysis of the PCR amplicons.
To generate the ΔIR3 EHV-1 and IR3R EHV-1, purified pΔIR3 EHV-1 BAC (or pIR3R EHV-1) and Us4 gene DNA in which the BAC DNA was inserted were cotransfected into 80% confluent RK13 cells by using the BD CalPhos Mammalian transfection kit (Clontech) according to the manufacturer’s directions. The supernatant was harvested at 3 days post transfection and was transferred to plates of 80% confluent RK13 cells followed by low melting agar overlay. Plaques lacking green fluorescence were selected and purified by three rounds of plaque assays (named ΔIR3 EHV-1 and IR3R EHV-1, respectively).
Southern blot analyses
To confirm the insertion of the zeoR and kanR markers into pRacL11 EHV-1 BAC, BamHI digested pΔIR3 EHV-1 BAC was separated on 0.3% agarose gel and transferred onto a positively charged nylon membrane (Ambion, Austin, TX) by using a semi-dry electroblotter (Bio-Rad Laboratories, Hercules, CA). DNA transferred to the membrane was placed on a piece of blot paper saturated with 0.5M NaOH for 15 min, briefly washed with 2x SSC, and baked at 80°C for 1 hour. The PCR fragment of the kanR [primers, 5’ taa aac atg aat gaa caa gat gga ttg cac g 3’, 5’ tta tct aga aat ctc gtg atg gca ggt tgg 3’, template, pEGFP-N1 (Clontech)] or zeoR gene [(primers, 5’ tta tct aga atc gaa atc tcg tag cac gtg 3’, 5’ gtt aga tct tta gga gct tgt ata tcc att ttc gg 3’; template, pSVZeo2 (Invitrogen)] was end-labeled with [γ-32P]ATP (New England Nuclear Corporation, Boston, MA) by T4 polynucleotide kinase (Promega) according to the manufacturer’s directions. Radiolabeled probe was denatured by adding 10% 3M NaOH for 10 min at room temperature and then neutralized by adding an equal volume of 1 M Tris-HCl (pH 7.0). Prehybridization, hybridization and washing were performed using the NorthernMax Kit (Ambion) followed by autoradiography using a phosphorimage screen and the molecular imager FX system (Bio-Rad Laboratories).
Dot blot hybridization
To confirm the presence of the kanR or zeoR marker in pΔIR3 EHV-1 BAC and ΔIR3 EHV-1 genome, dot blot hybridization was performed. pΔIR3 EHV-1 BAC DNA and total DNA that was extracted from ΔIR3 EHV-1 infected RK13 cells by using DNAzol reagent (Molecular Research Center, Inc., Cincinnati, OH) were placed on a positively charge nylon membrane (Ambion). DNA denaturation, hybridization using radiolabeled kanR marker PCR product, washing, and autoradiography were performed under the same conditions previously described.
Northern blot analysis
For the detection of the IR3 transcripts, the DNA probe [IR3(+443/+728) PCR product] was radiolabeled as before. RNA samples from EHV-1 infected RK13 cells were prepared by using RNA Bee (Tel-Test, Inc., Friendwood, TX), separated by electrophoresis in 5% denaturing acrylamide gel (8M urea), and transferred to a nylon membrane (Ambion) by using a semi-dry electroblotter (Bio-Rad Laboratories). Prehybridization, hybridization, washing, and autoradiography were performed as previously described.
Western blot analysis
For protein detection, RK13 cells were infected with RacL11 EHV-1, IR3R EHV-1, or ΔIR3 EHV-1 at a moi of 10, and cells were harvested at 3, 6, and 12 hpi. The whole cell lysates of virus infected cells at the immediate-early, early and late stage of replication were separated by SDS-PAGE, and then transferred to a nitrocellulose membrane (Ambion) by using a semi-dry electroblotter (Bio-Rad Laboratories). The immediate-early (IE), early (IR4, EICP0, UL5), and late (gD) proteins were detected by using anti-EHV-1 protein rabbit polyclonal antibodies (IEP, Caughman et al., 1995; IR4P, Holden et al., 1994; EICP0P, Bowles et al., 1997; UL5P, Zhao et al., 1995; and gDP, Flowers and O’Callaghan, 1992) as primary antibodies and anti-rabbit IgG[Fc]-alkaline phosphatase conjugated (Promega) as secondary antibody. Proteins were visualized by incubating the membrane containing blotted protein in AP conjugate substrate (AP conjugate substrate kit, Bio-Rad Laboratories) according to manufacturer’s directions. Protein density was measured by using ImageJ software program (http://rsbweb.nih.gov/ij/), and the relative protein density was compared.
Determination of plaque morphology, growth kinetics, and cell tropism
For the plaque assay, confluent RK13 cells in 6 well plates were infected with serial dilutions of RacL11 EHV-1, IR3R EHV-1, or ΔIR3 EHV-1. After 1 hour attachment at 4° C, virus infected cells were covered with medium containing 1.5% methylcellulose and incubated in 5% CO2 at 37°C. After 4 dpi, plaques were fixed with 10% formalin, stained with 0.5% crystal violet (or methylene blue), and counted (Perdue et al., 1974). For the comparison of plaque size, plaque sizes of RacL11 EHV-1, IR3R EHV-1, or ΔIR3 EHV-1 in RK13 cells were measured by using the ImageJ software program ((http://rebweb.nih.gov/ij/). For single step growth kinetics, confluent RK13 cells in 25 mm flask were infected with RacL11 EHV-1 or ΔIR3 EHV-1 at a moi of 0.1 or 5. After one hour viral attachment at 4° C, virus infected cells were washed with PBS, and then 4 ml of growth medium was added. Cell samples were harvested at designated time points followed by a freeze and thaw cycle, and virus was titered as described above. To determine cell tropism of ΔIR3 EHV-1, different cell types (LM, RK13, NBL-6, Vero and HeLa cells) were infected with RacL11 EHV-1 or ΔIR3 EHV-1 at a moi of 1. After one hour virus attachment at 4° C, virus infected cells were washed with cold PBS followed by adding normal growth medium, and total viral titers were determined at 3 dpi.
Animal experiments and statistical analysis
Animal experiments were conducted as described previously (Osterrieder et al., 1996; von Einem et al., 2004). Groups (n=12 for mock infection, n=12 for ΔIR3 EHV-1, n=12 for IR3R EHV-1, and n=12 for RacL11 EHV-1) of four-week-old CBA mice were intranasally inoculated with 2×106 pfu of ΔIR3 EHV-1 or RacL11 EHV-1; sterile medium was inoculated as mock infection. Mice were observed daily and weighed from prior to inoculation until 7 dpi. Virus isolation from the lungs of dead or live mouse was performed by using silica beads and BeadBeater (BioSpec Products, Inc., Bartlesville, OK) according to the manufacturer’s directions. Virus from lungs of mice infected with RacL11 EHV-1 (n=3), IR3R EHV-1 (n=3) or ΔIR3 EHV-1 (n=3) at 2, 3 and 4 dpi for live mice, and at the time points of death for each dead mouse was titered as described above. For the statistical analysis to compare body weight of mice and plaque sizes between two groups, two-tailed Student’s-t test was performed by using the Excel software program. The percent survival of mice infected with wild type or the mutant virus was plotted, and statistical differences were analyzed by the log-rank (Mantel-Cox) test.
Acknowledgments
We thank Mrs. Suzanne Zavecz for excellent technical assistance. These investigations were supported by research grant AI-22001 from the National Institute of Allergy and Infectious Diseases and research grant P20-RR018724 from the National Center for Research Resources of the National Institutes of Heath.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn BC, Breitenbach JE, Kim SK, O’Callaghan DJ. The equine herpesvirus-1 IR3 gene that lies antisense to the sole immediate-early (IE) gene is trans-activated by the IE protein, and is poorly expressed to a protein. Virology. 2007;316:15–25. doi: 10.1016/j.virol.2007.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht RA, Kim SK, O’Callaghan DJ. The EICP27 protein of equine herpesvirus 1 is recruited to viral promoters by its interaction with the immediate-early protein. Virology. 2005;333:74–87. doi: 10.1016/j.virol.2004.12.014. [DOI] [PubMed] [Google Scholar]
- Albrecht RA, Kim SK, Zhang Y, Zhao Y, O’Callaghan DJ. The equine herpesvirus 1 EICP27 protein enhances gene expression via an interaction with TATA box-binding protein. Virology. 2004;324:311–326. doi: 10.1016/j.virol.2004.03.040. [DOI] [PubMed] [Google Scholar]
- Bowles DE, Holden VR, Zhao Y, O’Callaghan DJ. The ICP0 protein of equine herpesvirus type 1 is an early protein that independently transactivates expression of all classes of viral promoters. J Virol. 1997;71:4904–4914. doi: 10.1128/jvi.71.7.4904-4914.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitenbach JE, Ebner PD, O’Callaghan DJ. The IR4 auxiliary regulatory protein expands the in vitro host range of equine herpesvirus 1 and is essential for pathogenesis in the murine model. Virology. 2009;383:188–194. doi: 10.1016/j.virol.2008.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughman GB, Lewis JB, Smith RH, Harty RN, O’Callaghan DJ. Detection and intracellular localization of equine herpesvirus 1 IR1 and IR2 gene products by using monoclonal antibodies. J Virol. 1995;69:3024–3032. doi: 10.1128/jvi.69.5.3024-3032.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughman GB, Staczek J, O’Callaghan DJ. Equine herpesvirus type 1 infected cell polypeptides: evidence for immediate early/early/late regulation of viral gene expression. Virology. 1985;145:49–61. doi: 10.1016/0042-6822(85)90200-4. [DOI] [PubMed] [Google Scholar]
- Dangler CA, Henderson LM, Bowman LA, Deaver RE. Direct isolation and identification of recombinant pseudorabies virus strains from tissues of experimentally coinfected swine. Am J Vet Res. 1993;54:540–545. [PubMed] [Google Scholar]
- Derbigny WA, Kim SK, Caughman GB, O’Callaghan DJ. The EICP22 protein of equine herpes virus 1 physically interacts with the immediate-early protein and with itself to form dimers or higher-ordered complexes. J Virol. 2000;74:1425–1435. doi: 10.1128/jvi.74.3.1425-1435.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbigny WA, Kim SK, Jang HK, O’Callaghan DJ. EHV-1 EICP22 protein sequences that mediate its physical interaction with the immediate-early protein are not sufficient to enhance the trans-activation activity of the IE protein. Virus Res. 2002;84:1–15. doi: 10.1016/s0168-1702(01)00377-x. [DOI] [PubMed] [Google Scholar]
- Dohner DE, Adams SG, Gelb LD. Recombination in tissue culture between varicella-zoster virus strains. J Med Virol. 1988;24:329–341. doi: 10.1002/jmv.1890240310. [DOI] [PubMed] [Google Scholar]
- Elliott GD. The extreme carboxyl terminus of the equine herpesvirus 1 homolog of herpes simplex virus VP16 is essential for immediate-early gene activation. J Virol. 1994;68:4890–4897. doi: 10.1128/jvi.68.8.4890-4897.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott GD, O’Hare P. Equine herpesvirus 1 gene 12, the functional homologue of herpes simplex virus VP16, transactivates via octamer sequences in the equine herpesvirus IE gene promoter. Virology. 1995;213:258–262. doi: 10.1006/viro.1995.1568. [DOI] [PubMed] [Google Scholar]
- Flower CC, O’Callaghan DJ. equine herpesvirus 1 glycoprotein D: mapping of the transcript and a neutralization epitope. J Virol. 1992;66:6451–6460. doi: 10.1128/jvi.66.11.6451-6460.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita K, Maeda K, Yokoyama N, Miyazawa T, Kai C, Mikami T. In vitro recombination of feline herpesvirus type 1. Arch Virol. 1998;143:25–34. doi: 10.1007/s007050050265. [DOI] [PubMed] [Google Scholar]
- Garko-Buczynski KA, Smith RH, Kim SK, O’Callaghan DJ. Complementation of a replication-defective mutant of equine herpesvirus type 1 by a cell line expressing the immediate-early protein. Virology. 1998;248:83–94. doi: 10.1006/viro.1998.9247. [DOI] [PubMed] [Google Scholar]
- Gray WL, Baumann RP, Robertson AT, Caughman GB, O’Callaghan DJ, Staczek J. Regulation of equine herpesvirus type l gene expression: characterization of immediate early, early, and late transcription. Virology. l987;158:79–87. doi: 10.1016/0042-6822(87)90240-6. [DOI] [PubMed] [Google Scholar]
- Grundy FJ, Baumann RP, O’Callaghan DJ. DNA sequence and comparative analyses of the equine herpesvirus type 1 immediate early gene. Virology. 1989;172:223–236. doi: 10.1016/0042-6822(89)90124-4. [DOI] [PubMed] [Google Scholar]
- Harty RN, Colle CF, Grundy FJ, O’Callaghan DJ. Mapping the termini and introns of the spliced immediate-early transcript of equine herpesvirus 1. J Virol. 1989;63:5101–5110. doi: 10.1128/jvi.63.12.5101-5110.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harty RN, O’Callaghan DJ. An early gene maps within and is 3’ co-terminal with the immediate-early gene of equine herpesvirus 1. J Virol. 1991;65:3829–3838. doi: 10.1128/jvi.65.7.3829-3838.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry BE, Robinson RA, Dauenhauer SA, Atherton SS, Hayward GS, O’Callaghan DJ. Structure of the genome of equine herpesvirus type 1. Virology. 1981;115:97–114. doi: 10.1016/0042-6822(81)90092-1. [DOI] [PubMed] [Google Scholar]
- Holden VR, Caughman GB, Zhao Y, Harty RN, O’Callaghan DJ. Identification and characterization of the ICP22 protein of equine herpesvirus 1. J Virol. 1994;68:4329–4340. doi: 10.1128/jvi.68.7.4329-4340.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden VR, Harty RN, Yalamanchili RR, O’Callaghan DJ. The IR3 gene of equine herpesvirus type 1: a unique gene regulated by sequences within the intron of the immediate-early gene. DNA Seq. 1992;3:143–152. doi: 10.3109/10425179209034010. [DOI] [PubMed] [Google Scholar]
- Kim SK, Ahn BC, Albrecht RA, O’Callaghan DJ. The unique IR2 protein of equine herpesvirus 1 negatively regulates viral gene expression. J Virol. 2006;80:5041–5049. doi: 10.1128/JVI.80.10.5041-5049.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Holden VR, O’Callaghan DJ. The ICP22 protein of equine herpesvirus 1 cooperates with the IE protein to regulate viral gene expression. J Virol. 1997;71:1004–1012. doi: 10.1128/jvi.71.2.1004-1012.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Smith RH, O’Callaghan DJ. Characterization of DNA binding properties of the immediate-early gene product of equine herpesvirus type1. Virology. 1995;213:46–56. doi: 10.1006/viro.1995.1545. [DOI] [PubMed] [Google Scholar]
- Meurens F, Keil GM, Muylkens B, Gogev S, Schynts F, Negro S, Wiggers L, Thiry E. Interspecific recombination between two ruminant alphaherpesviruses, bovine herpesviruses 1 and 5. J Virol. 2004;78(18):9828–36. doi: 10.1128/JVI.78.18.9828-9836.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Callaghan DJ, Osterrieder N. Herpesviruses of horses. In: Mahy B, Van Regenmortel M, editors. Encyclopedia of Virology. 3. Elsevier Ltd.; Oxford, UK: 2008. pp. 411–420. [Google Scholar]
- Osterrieder N, Neubauer A, Brandmüller C, Kaaden OR, O’Callaghan DJ. The equine herpesvirus 1 IR6 protein influences virus growth at elevated temperature and is a major determinant of virulence. Virology. 1996;226(2):243–51. doi: 10.1006/viro.1996.0652. [DOI] [PubMed] [Google Scholar]
- Perdue ML, Kemp MC, Randall CC, O’Callaghan DJ. Studies of the molecular anatomy of the L-M cell strain of equine herpes virus type 1: proteins of the nucleocapsid and intact virion. Virology. 1974;59(1):201–16. doi: 10.1016/0042-6822(74)90216-5. [DOI] [PubMed] [Google Scholar]
- Rudolph J, O’Callaghan DJ, Osterrieder N. Cloning of the genomes of equine herpesvirus type 1 (EHV-1) strains KyA and RacL11 as bacterial artificial chromosomes (BACs) J Vet Med B Infect Dis Vet Public Health. 2002;49(1):31–36. doi: 10.1046/j.1439-0450.2002.00534.x. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- Smith RH, Caughman GB, O’Callaghan DJ. Characterization of the regulatory functions of the equine herpesvirus 1 immediate-early gene product. J Virol. 1992;66:936–945. doi: 10.1128/jvi.66.2.936-945.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RH, Zhao Y, O’Callaghan DJ. The equine herpesvirus 1 (EHV-1) UL3 gene, an ICP27 homolog, is necessary for full activation of gene expression directed by an EHV-1 late promoter. J Virol. 1993;67:1105–1109. doi: 10.1128/jvi.67.2.1105-1109.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RH, Zhao Y, O’Callaghan DJ. The equine herpesvirus type 1 immediate-early gene product contains an acidic transcriptional activation domain. Virology. 1994;202:760–770. doi: 10.1006/viro.1994.1398. [DOI] [PubMed] [Google Scholar]
- Telford EA, Watson MS, McBride K, Davison AJ. The DNA sequence of equine herpesvirus-1. Virology. 1992;189:304–316. doi: 10.1016/0042-6822(92)90706-u. [DOI] [PubMed] [Google Scholar]
- Umene K. Intermolecular recombination of the herpes simplex virus type 1 genome analysed using two strains differing in restriction enzyme cleavage sites. J Gen Virol. 1985;66:2659–2670. doi: 10.1099/0022-1317-66-12-2659. [DOI] [PubMed] [Google Scholar]
- von Einem J, Schumacher D, O’Callaghan DJ, Osterrieder N. The α-TIF (VP16) homologue (ETIF) of equine herpesvirus 1 (EHV-1) is essential for secondary envelopment and virus egress. J Virol. 2006;80:2609–2620. doi: 10.1128/JVI.80.6.2609-2620.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Einem J, Wellington J, Whalley JM, Osterrieder K, O’Callaghan DJ, Osterrieder N. The truncated form of glycoprotein gp2 of equine herpesvirus 1 (EHV-1) vaccine strain KyA is not functionally equivalent to full-length gp2 encoded by EHV-1 wild-type strain RacL11. J Virol. 2004;78:3003–3013. doi: 10.1128/JVI.78.6.3003-3013.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 2005;33(4):e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, Osterrieder N, O’Callaghan DJ. Generation and characterization of an EICP0 null mutant of equine herpesvirus 1. Virus Res. 2003;98:163–172. doi: 10.1016/j.virusres.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Holden VR, Smith RH, O’Callaghan DJ. Regulatory function of the equine herpesvirus 1 ICP27 gene product. J Virol. 1995;69:2786–2793. doi: 10.1128/jvi.69.5.2786-2793.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]