

Summary
To determine whether insulin action on endothelial cells promotes or protects against atherosclerosis, we generated apolipoprotein E null mice in which the insulin receptor gene was intact or conditionally deleted in vascular endothelial cells. Insulin sensitivity, glucose tolerance, plasma lipids, and blood pressure were not different between the two groups, but atherosclerotic lesion size was more than 2-fold higher in mice lacking endothelial insulin signaling. Endothelium-dependent vasodilation was impaired and endothelial cell VCAM-1 expression was increased in these animals. Adhesion of mononuclear cells to endothelium in vivo was increased 4-fold compared with controls, but reduced to below control values by a VCAM-1 blocking antibody. These results provide definitive evidence that loss of insulin signaling in endothelium, in the absence of competing systemic risk factors, accelerates atherosclerosis. Therefore, improving insulin sensitivity in the endothelium of patients with insulin resistance or type 2 diabetes may prevent cardiovascular complications.
Introduction
Insulin stimulates signal transduction in endothelial cells (Zeng and Quon, 1996) and augments endothelial vasodilator function (Steinberg et al., 1994). Multiple studies have shown that systemic insulin resistance is associated with impaired vascular insulin signaling (Jiang et al., 1999) and blunted vascular effects of insulin (Steinberg et al., 1996). Insulin resistance (Howard et al., 1996) and the resultant hyperinsulinemia (Despres et al., 1996) are independent risk factors for vascular disease. These associations could be due to excessive insulin action on endothelial cells during hyperinsulinemia or decreased endothelial insulin action due to vascular insulin resistance, but the relative roles of such effects have been controversial (Rask-Madsen and King, 2007).
Both anti-atherosclerotic and pro-atherosclerotic mechanisms activated by insulin have been described in vascular endothelial cells. Anti-atherogenic actions of insulin include increased production of nitric oxide (NO) (Kuboki et al., 2000; Steinberg et al., 1994; Zeng and Quon, 1996), decreased endothelial cell apoptosis (Hermann et al., 2000), decreased production of reactive oxygen species (Du et al., 2006), and increased expression of genes with anti-oxidant effects, like heme oxygenase-1 (HO-1) (Geraldes et al., 2008). Conversely, increased expression of endothelin-1 (Cardillo et al., 1999; Oliver et al., 1991) and plasminogen activator inhibitor-1 (PAI-1) (Grenett et al., 1998; Nordt et al., 1998) are among pro-atherogenic actions of insulin on vascular endothelial cells. There have been reports of both increased (Madonna et al., 2004; Okouchi et al., 2002) and decreased (Aljada et al., 2000; Booth et al., 2001) expression of adhesion molecules after insulin stimulation.
Despite mechanistic information about presumed anti- or pro-atherosclerotic effects of insulin signaling in endothelial cells, the net effect of endothelial insulin action on the development of atherosclerosis is unknown. In order to improve managenement of the cardiovascular risk profile particular to patients with insulin resistance or diabetes (Reusch and Draznin, 2007), it is critical to understand the effects of insulin directly on endothelial cells and the consequences of insulin resistance in endothelium. Thus, if hyperinsulinemia is detrimental, intensive insulin treatment could potentially have adverse effects in the vascular endothelium, thereby blunting the effects of improved control of glucose and lipid metabolism. Conversely, if the direct action of insulin on the endothelium has an anti-atherosclerotic effect, therapies that prevent or improve insulin resistance in endothelial cells could decrease the risk of atherosclerosis. These local effects may be different from the results of targeting insulin resistance at the whole-body level. For example, we have previously shown that treatment with an inhibitor specific for the β isoform of protein kinase C normalized the severely compromised insulin-stimulated NO production in the aorta of insulin resistant rats, but had no effect on whole-body insulin sensitivity (Naruse et al., 2006).
To determine whether the loss of insulin action on endothelial cells, similar to what is observed in patients with insulin resistance (Rask-Madsen et al., 2001; Steinberg et al., 1996), promotes or retards atherosclerosis, we generated endothelial insulin receptor and apolipoprotein E knockout (EIRAKO) mice, using the Cre/loxP system to achieve a conditional knockout of the insulin receptor gene targeted to endothelial cells. These animals were compared with littermate apolipoprotein E (apoE) knockout mice with intact insulin receptor genes (“controls”). Our results show that atherosclerotic disease progression is more severe in EIRAKO mice compared with controls, despite there being no differences in whole-body insulin sensitivity, glucose tolerance, plasma lipids, or blood pressure between the two groups. These results provide the first definitive evidence that loss of insulin signaling in the endothelium, in the absence of competing systemic risk factors, accelerates atherosclerosis.
Results
Insulin receptor expression and insulin signaling
EIRAKO mice were bred as described in Experimental Procedures and maintained on regular chow with 22% of calories provided by fat. A PCR product representing Cre-mediated recombination was amplifed from tail biopsies from EIRAKO mice, but not from hair, which contains no endothelial cells (Supplemental Figure S1A, see also Supplemental Experimental Procedures). In most organs, endothelial cells constitute only a small fraction of the total cell number. Accordingly, insulin receptor-β (IRβ) protein expression and insulin-stimulated Akt Ser473 phosphorylation did not differ between control and EIRAKO mice in skeletal muscle (Figure 1A), liver (Figure 1B), or adipose tissue (Figure 1C). In lung tissue, IRβ protein was decreased by 50% (Figure 1D), likely because of the high content of endothelial cells in this tissue. Lung endothelial cells were isolated using magnetic microbeads complexed to ICAM-2 antibody during two sequential passages, which resulted in >99% CD31+ cells (Supplemental Figure S1B). IRβ protein was clearly expressed in cultures from control mice but was not detectable in cultures from EIRAKO mice (Supplemental Figure S1C, limit of detection described in Supplemental Methods). Insulin at a concentration of 10 nM increased Akt Ser473 phosphorylation more than 6-fold in lung endothelial cells from control animals, but had no effect in cells from EIRAKO mice (Supplemental Figure S1C and D).
Figure 1. Insulin receptor expression and insulin signaling.
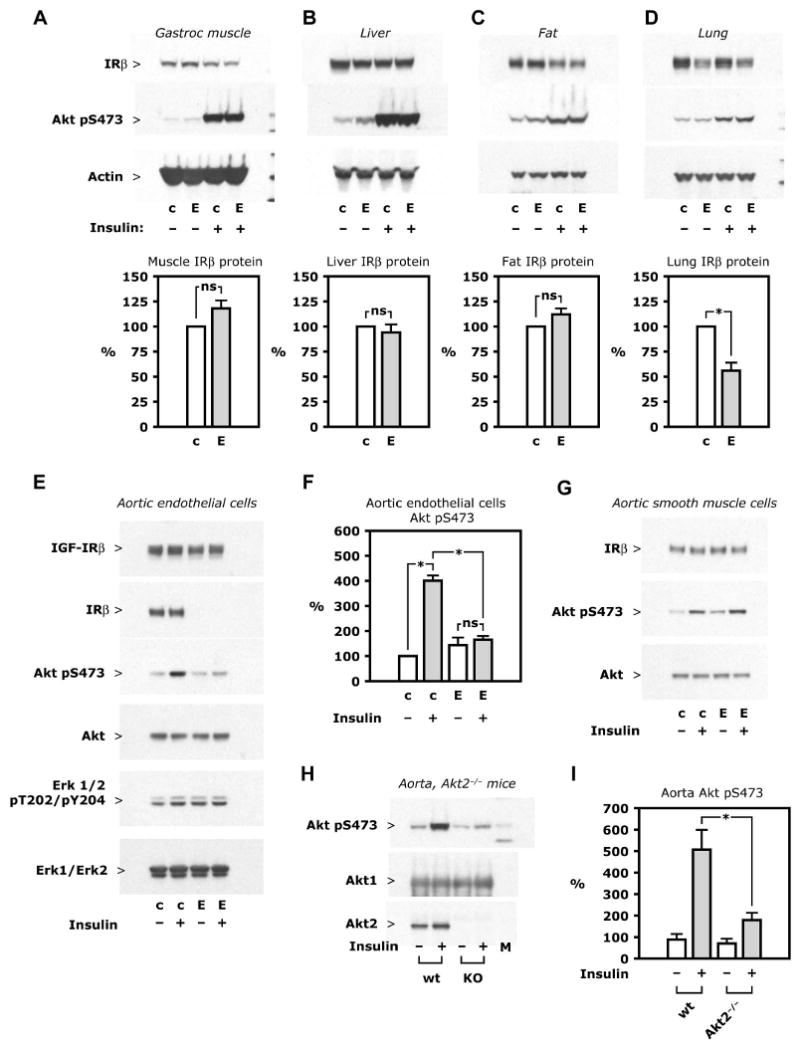
A-D. Insulin 5 mU/g i. v. was given to mice fasted overnight and tissues collected after 5 minutes. Representative Western blots (top) show insulin receptor-β (IRβ) protein expression and insulin-stimulated Akt Ser473 phosphorylation in lysate of the tissues indicated; also shown are mean values of IRβ protein after densitometry of Western blots from 3 independent experiments (bottom). E-F. Aorta was digested by collagenase and endothelial cells were isolated by immunoselection with magnetic microbeads complexed with ICAM2 antibody, with aortic smooth muscle cells grown from the ICAM2-negative fraction. Cultures were treated with insulin (10 nM, 5 minutes) after 24 hours of serum starvation. E. Representative Western blots from whole cell lysate of primary culture of aortic endothelial cells are shown. F. Mean values of Akt Ser473 phosphorylation was based on densitometry of Western blots from 3 independent experiments. G. Western blots from whole cell lysate of aortic smooth muscle cells representative of 3 independent experiments. H-I. Insulin 10 mU/g was given i. v. in wild-type and Akt2−/− mice. After 5 minutes, the aorta was isolated and snap frozen. H. Representative Western blots of aorta lysate. I. Mean values of Akt Ser473 phosphorylation based on data from 10 wild-type and 13 Akt2−/− mice. See also Figure S1. Abbreviations: c, control mice; E, EIRAKO mice; wt, wild-type mice.
Since lung endothelial cells are mostly of microvascular origin, we also examined macrovascular endothelial cells, isolated with the same approach of immuno-magnetic sorting as described above. As with microvascular endothelial cells, no IRβ protein was detectable in cultured aortic endothelial cells from EIRAKO mice (Figure 1E). Insulin had no effect on Akt Ser473 phosphorylation in these cells compared with a 4-fold increase in Akt phosphorylation in cells from control animals after insulin treatment (Figure 1E and F, p<0.001). Similarly, insulin treatment caused a 50±18% increase in Erk1/2 phosphorylation in aortic endothelial cells from control mice, but had no effect on cells from EIRAKO mice (Figure 1E). Of note, unstimulated Erk phosphorylation was increased by 71±7% in cells from EIRAKO mice compared with control cells (Figure 1E). A similar increase in unstimulated Erk phosphorylation was found in lung endothelial cells, where Erk phosphorylation in cells from EIRAKO was 55±20% higher than in cultures from control animals (p=0.03, data not shown). Insulin-like growth factor-I receptor β (IGF-IRβ) protein was not different in lung or aortic endothelial cells from EIRAKO compared to control mice (Figure 1E). In cultures of aortic smooth muscle cells, IRβ protein and insulin-stimulated Akt phosphorylation were not different between the two groups of mice (Figure 1G).
A recent publication showed that in Akt1−/− mice, insulin-stimulated Akt phosphorylation was intact in whole lysate and in Akt2 immunoprecipitates from vascular tissue (Symons et al., 2009). We injected insulin intravenously in Akt2−/− and control mice and found that in the aorta, Akt phosphorylation at Ser473 increased by 5.7±1.0 fold in wild-type mice, but only by 2.0±0.4 fold in Akt2−/− mice (Figure H and I, p=0.003 for the stimulated value in wild-type compared to Akt2−/− mice). Therefore, a major part of vascular insulin signaling is mediated by Akt2.
Metabolic parameters and blood pressure
Body mass was not different between EIRAKO mice and their littermate controls (37.3±0.8 versus 37.3±1.6 g at 24 weeks of age, 43.9±3.7 versus 42.7±4.8 g at 52 weeks of age). Furthermore, no difference was seen in random fed blood glucose (136±8 and 134±9 mg/dl at 12 weeks and 152±24 and 149±12 mg/dl at 52 weeks in EIRAKO and control mice, respectively, p>0.8) or fasting plasma glucose (data not shown). Intraperitoneal glucose tolerance tests showed no difference in blood glucose (Figure 2A) or plasma insulin (Figure 2B) between EIRAKO and control mice, and intraperitoneal insulin tolerance tests did not differ with respect to glucose levels between the two groups (Figure 2C).
Figure 2. Whole-body glucose tolerance and insulin sensitivity, plasma lipids, blood pressure.
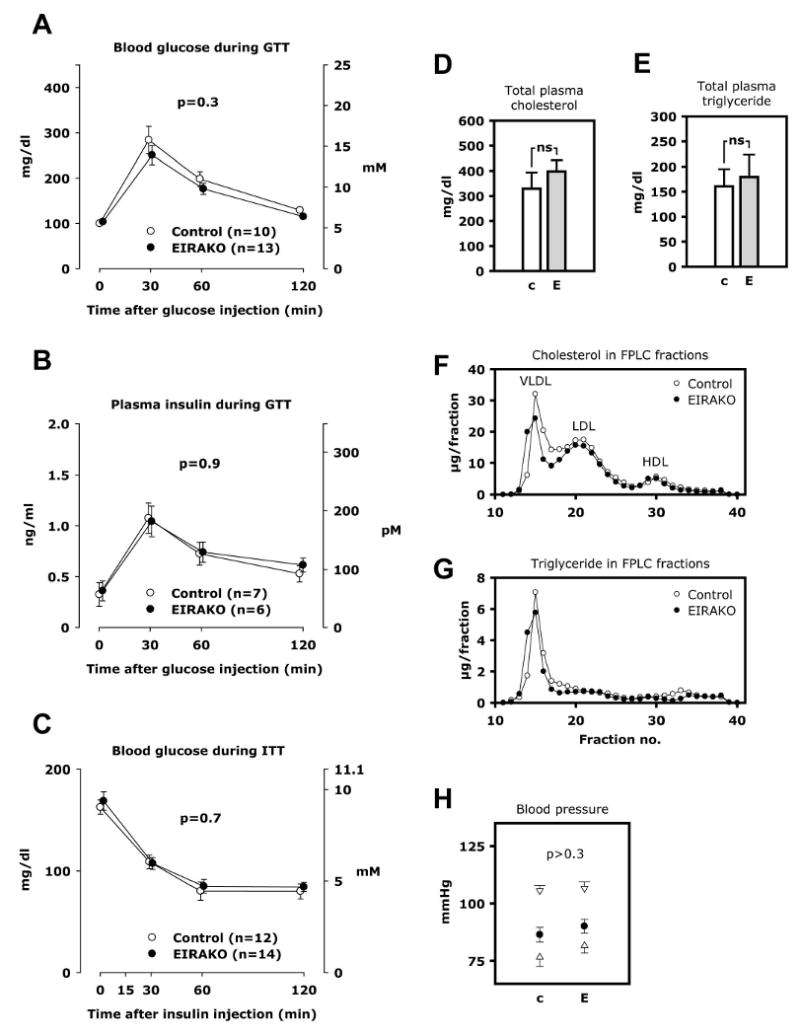
A. Blood glucose during glucose tolerance test. B. Plasma insulin during glucose tolerance test. C. Plasma glucose during insulin tolerance test. D-G. Plasma lipids were measured in 12 EIRAKO mice and 11 littermate controls after a 4-6 hours fast. Plasma fractions were obtained by fast protein liquid chromatography (FPLC) in a subset of 3 EIRAKO mice and 3 littermate controls. D. Total cholesterol in plasma. E. Total triglycerides in plasma. F. Cholesterol concentration in FPLC fractions of plasma. G. Triglyceride concentration in FPLC fractions of plasma. H. Blood pressure was measured by tail vein plethysmography in 14 EIRAKO mice and 12 littermate controls. Mean values for systolic and diastolic blood pressure (open triangles) as well as mean blood pressure (closed circles) are shown.
Total cholesterol and triglyceride concentrations in plasma from animals fasted 4-6 hours were not different between EIRAKO and control mice at 24 weeks of age (Figure 2D and E). Similarly, total plasma cholesterol and triglyceride showed no significant differences in mice fasted overnight or mice fasted 4-6 hours or overnight at 12 weeks of age (data not shown). Cholesterol and triglyceride concentrations were also measured in lipoprotein fractions separated by fast protein liquid chromatography (FPLC), and were similar between the two groups of animals (Figure 2F and G).
Blood pressure and pulse were measured by tail vein plethysmography. There were no significant differences in systolic blood pressure, diastolic blood pressure, or mean blood pressure between the two groups (Figure 2H). Thus, the loss of insulin receptors in the endothelium did not change whole-body glucose tolerance, systemic insulin sensitivity, plasma lipids, or blood pressure.
Atherosclerosis in the aorta and brachiocephalic artery
Atherosclerotic lesions were evaluated in several different ways. The entire aorta was stained with Sudan IV to visualize atherosclerotic plaques in flat preparations (en face preparations) (Figure 3A and B). Atherosclerotic plaques were not observed in vascular endothelial insulin receptor knockout (VENIRKO) mice (Vicent et al., 2003), which have no apoE gene mutation (data not shown). In EIRAKO mice, the mean atherosclerotic lesion area, expressed relative to total aortic luminal area, was 2.1-fold greater at 24 weeks than in littermate controls (3.5±1.1 and 1.6±0.4%, respectively, p=0.01, Figure 3C) and 2.9-fold greater at 52 weeks (34.1±4.1 and 11.8±2.0%, respectively, p=0.00007, Figure 3D). Lesion area normalized to values for littermate controls was also analyzed. By this approach, when lesion area in each EIRAKO mice belonging to a given litter was expressed as a fraction of the average lesion area in control animals belonging to that litter, results were similar (lesion area in EIRAKO mice 2.7±0.5 times higher than controls at 24 weeks, p=0.02; 4.4±1.6 times higher than controls at 52 weeks, p=0.02).
Figure 3. Atherosclerotic lesion size in the aorta.
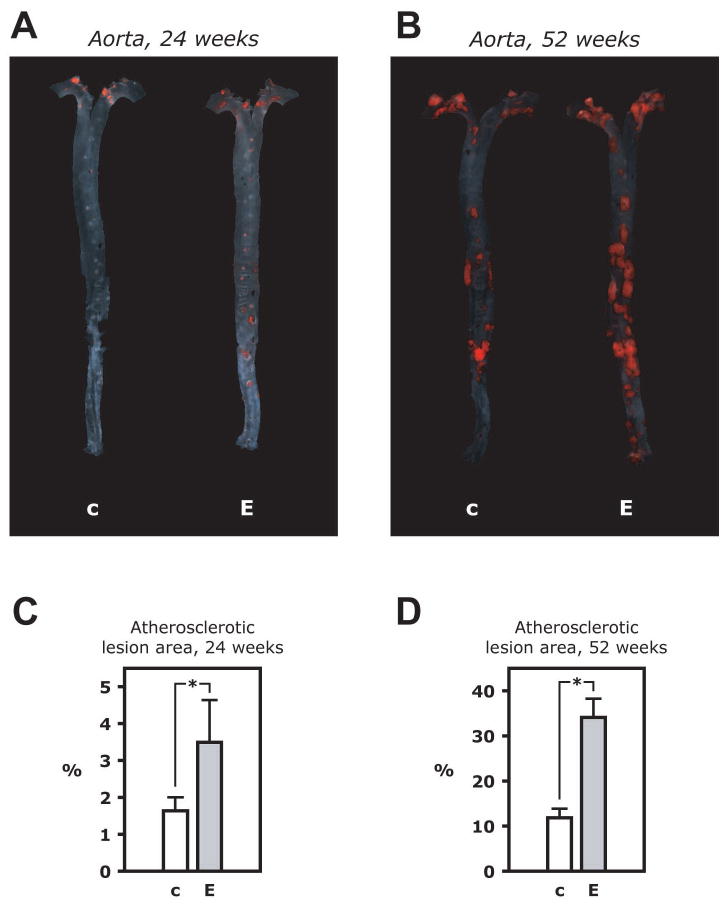
A. Microphotographs of aortas from an EIRAKO mouse and its littermate control at 24 weeks of age in the en face flat preparation after staining with Sudan IV. B. Summary data from quantitation of atherosclerotic lesion area relative to total area of the aorta in 10 EIRAKO mice and 8 littermate controls at 24 weeks of age. C. Stained aortas at 52 weeks of age. D. Summary data from 9 EIRAKO animals and 11 littermate controls at 52 weeks of age
Atherosclerotic lesions were also analyzed in the brachiocephalic artery in mice 36 weeks of age by quantitatively extracting lipids from the brachiochephalic artery with chloroform/methanol followed by measurement of cholesteryl ester content by mass spectroscopy. As cholesteryl ester is a major component of the lipid accumulated in atherosclerotic plaques, this provided a measure of plaque volume. Cholesteryl ester content in the brachiocephalic artery was more than 2-fold higher in EIRAKO mice compared to controls (9.6±3.1 and 4.5±0.7 nmoles, respectively, p=0.03, Figure 4A) and more than 3-fold higher at 52 weeks (25.7±7.4 and 8.1±1.4 nmoles, respectively, p=0.048, Figure 4B). After lipid extraction, the brachiocephalic arteries were analyzed histologically. Planimetry of the area covered by plaques in histological cross-sections showed that plaque area was larger in EIRAKO mice, although this did not reach statistical significance (38,578±11,245 and 17,091±9,506 μm2 in EIRAKO mice and their controls, respectively, p=0.4, Figure 4C). Collagen, stained blue by Masson trichrome, was distributed throughout plaques in both groups (Figure 4C). There were significantly more vascular smooth muscle cells per plaque cross-section in EIRAKO mice compared to controls (17.3±8.3 and 2.9±2.3 α-actin-positive cells per section, respectively, p=0.04, Figure 4D), indicating that EIRAKO mice had more complex atherosclerotic lesions. The area occupied by macrophages tended to be higher in EIRAKO mice (12,664±6,473 μm2 and 5,724±1,444, respectively, p=0.16, Figure 4E).
Figure 4. Brachiocephalic artery lesion size and atherosclerotic plaques histology.
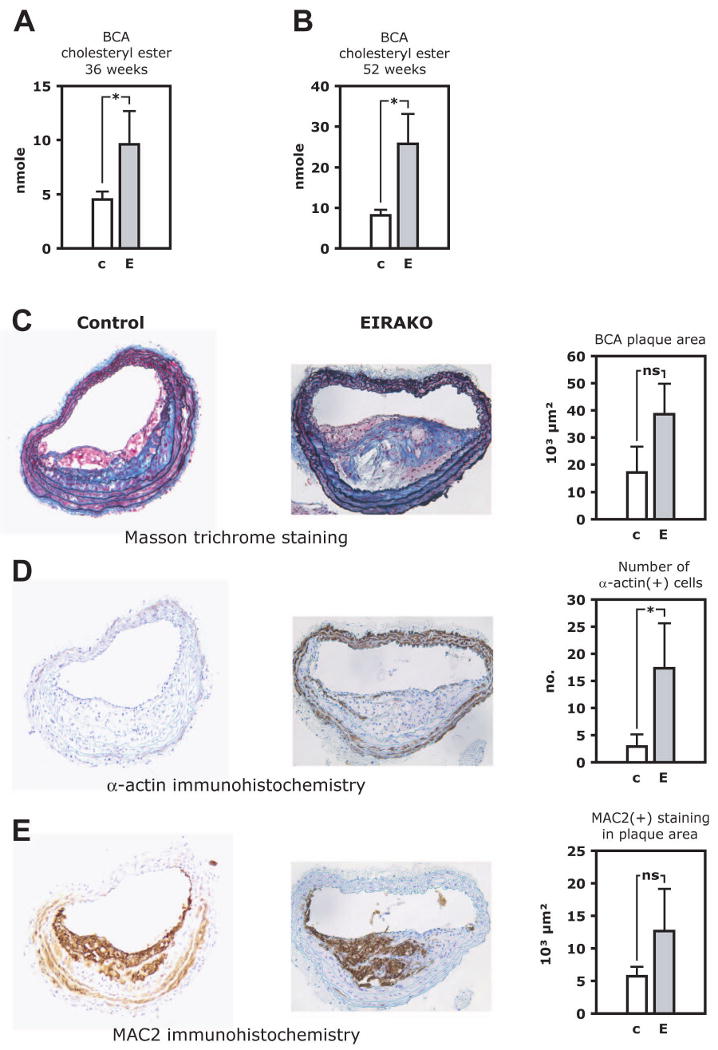
Lipids were quantitatively extracted from the brachiocephalic artery, which was then paraffin embedded and cross-sectioned. Cholesteryl ester was measured in lipid extracts by mass spectrometry. A. Cholesteryl ester content in the brachiocephalic artery at 36 weeks. B. Colesteryl ester content in the brachiocephalic artery at 52 weeks. C-E. Histological staining and immunohistochemistry of brachiocephalic artery cross-sections. Representative images are shown (left), as are mean values of quantitative analysis (right). C. Masson trichrome stain, with collagen staining blue. D. MAC-2 immunohistochemistry to visualize plaque area occupied by macrophages. E. α-smooth muscle cell actin immunohistochemistry to identify vascular smooth muscle cells. See also Figure S2.
Atherosclerotic lesions in cross-sections of the aortic sinus were analyzed at 52 weeks of age. The mean cross-sectional area was not different in EIRAKO and control mice (389,841±77,547 and 359,791±58,034 μm2, respectively, Figure S2A, B, & I, p=0.8). Similarly, there was no difference in plaque area positive for macrophage immunostaining ((Figure S2C, D & J) or number of smooth muscle cells identified by immunostaining (Figure S2E & F) in the two groups. Most of the difference in atherosclerotic lesion area in the aorta appeared to be in the descending aorta, which may explain why no difference was seen in the aortic sinus. Subocclusive plaques in coronary arteries were observed in animals from both groups (Figure S2G & H).
eNOS regulation
In control mice, intravenous injection of insulin resulted in a 69±13% increase in eNOS Ser1177 phosphorylation in the aorta compared to mice injected with vehicle (p=0.007, Figure 5A and B). By contrast, insulin injection did not not change eNOS phosphorylation in the aorta of EIRAKO mice compared to vehicle injection (Figure 5A and B). eNOS phosphorylation was not different in aortas from EIRAKO and control mice injected with vehicle (Figure 5A and B). In lung an aortic endothelial cells isolated from control mice, eNOS phosphorylation increased 2-fold after treatment with insulin (5C and D, p=0.01 for aortic endothelial cells). Insulin treatment did not increase eNOS phosphorylation in cells from EIRAKO mice, although phosphorylation state tended to be higher in the unstimulated condition (68±34% higher in unstimulated aortic endothelial cells from EIRAKO mice than in unstimulated cells from control mice, p=0.11, Figure 5C and D). Therefore, insulin-stimulated eNOS phosphorylation is lost in EIRAKO mice, both in isolated endothelial cells and in the aorta in vivo.
Figure 5. eNOS regulation.
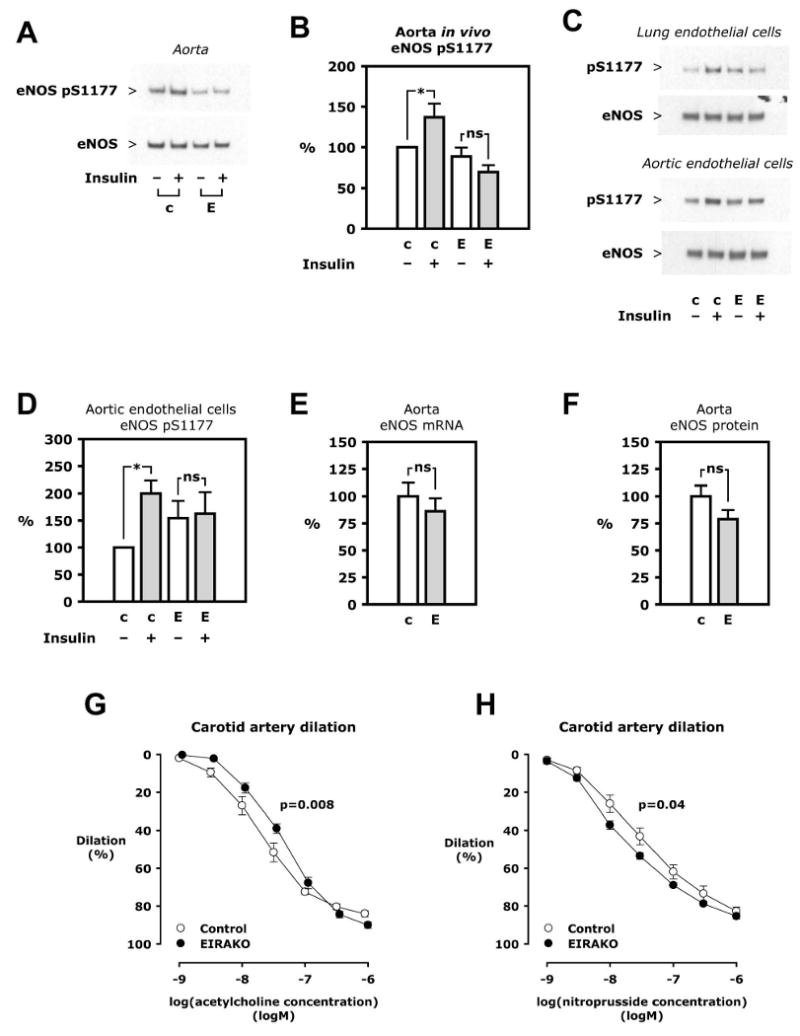
A. Insulin 5 mU/g was injected i. v. and the aorta removed after 5 minutes. Western blotting was performed on aorta lysate. B. Mean values of eNOS Ser1177 phosphorylation relative to eNOS based on densitometry of Western blots of aorta from 6 sets of 4 animals. C. Primary cultures of lung and aortic endothelial cells isolated from an EIRAKO mouse and its littermate control were treated with insulin (100 nM in lung endothelial cells, 10 nM in aortic endothelial cells, 5 minutes). Representative Western blots are shown. D. Mean values of eNOS Ser1177 phosphorylation relative to eNOS expression in aortic endothelial cells from 3 independent experiments. E. eNOS mRNA expression in aorta measured by real-time PCR, mean results from 11 EIRAKO mice and 9 controls. F. eNOS protein in aorta based on densitometry of Western blots and normalized to actin, mean results from 11 EIRAKO mice and 8 controls. G-H. The carotid artery was isolated, mounted, and pressurized in a myograph. Graphs show mean vasodilation using arteries from 6 control and 6 EIRAKO mice. G. Concentration-response study using acetylcholine. H. Concentration-response study using sodium nitroprusside.
eNOS expression is decreased in aorta and endothelial cells from vascular endothelial insulin receptor knockout (VENIRKO) mice (Vicent et al., 2003). In EIRAKO mice, however, no changes in expression of eNOS mRNA or protein in the aorta were observed when compared with their controls (Figure 5E and F). In a pro-atherosclerotic environment, eNOS gene expression is likely upregulated by factors other than insulin, such as oxidative stress (Drummond et al., 2000). Indeed, mRNA expression of the anti-oxidant enzyme HO-1 (Hmox1) was significantly increased in EIRAKO mice (to 165±25% of the level in controls, p=0.04), likely reflecting that oxidative stress in a more pro-atherosclerotic milieu is quantitatively more important for its regulation than insulin signaling. The expression of other anti- and pro-oxidant genes, including Sod1, Cybb (Nox2/gp91phox), Ncf1 (p47phox), and Nox4, were not changed (data not shown). Also, no change was observed in aortic endothelin-1 (Edn1) expression (expression in EIRAKO mice 91±8 of and 109±20% of controls at 12 and 20 weeks, respectively, 20 vs. 20 and 9 vs. 12 animals, respectively, p>0.4).
To study the functional consequences of the changes in eNOS regulation in EIRAKO mice, we measured vasodilation stimulated by acetylcholine, most of which is dependent on endothelium-derived NO. Isolated carotid arteries from EIRAKO mice had decreased sensitivity to acetylcholine, as the EC50 in EIRAKO mice was almost twice as high as in controls (44±5.6 and 23±3.8 nM, respectively, Figure 5G, p=0.008). Vasodilator responses to exogenous NO, measured during concentration-response studies with sodium nitroprusside, showed that arteries from EIRAKO mice were more NO-sensitive (with an EC50 of 20±2.5 and 39±8.5 nM in EIRAKO and control mice, respectively, Figure 5H, p=0.04). These experiments establish that NO-mediated vasodilation is impaired in EIRAKO mice compared to controls, but indicate that vascular smooth muscle cell sensitivity to NO is increased, perhaps as a compensatory mechanism.
Leukocyte-endothelial cell interaction in vivo
Decreased eNOS activity plays a major role in the development of atherosclerosis, in part by decreasing the interaction of monocytes and other leukocytes with endothelium. We therefore evaluated leukocyte interaction with endothelium in vivo, using intravital microscopy of the mesenteric circulation after labeling of leukocytes with a fluorescent dye. In 12-week old EIRAKO animals, the number of rolling leukocytes increased by 1.8-fold compared to controls (26.8±2.9 and 48.7±2.3 rolling leukocytes per 100 μm in EIRAKO and controls, respectively, p=0.004, Figure 6A). The number of firmly adhering leukocytes increased by 4.1-fold (1.5±0.3 and 6.2±0.9 adhering leukocytes per 100 μm, respectively, p<0.001, Figure 6B).
Figure 6. Leukocyte-endothelial cell interaction in vivo.
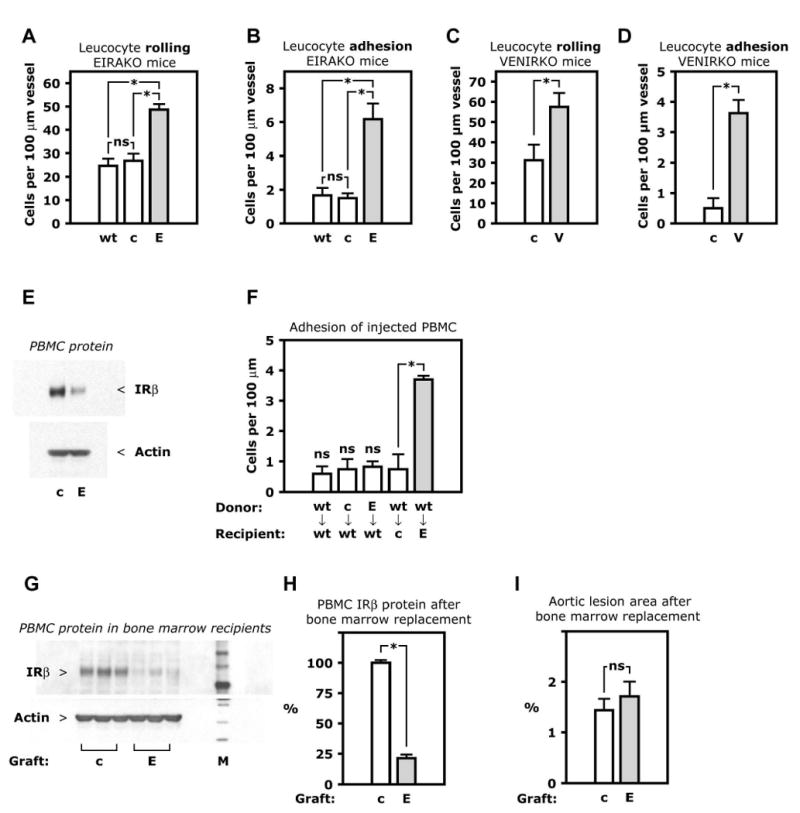
A-D. Circulating leukocytes were fluorescently labeled by i. v. injection of rhodamine and visualized in peri-intestinal, post-capillary venules by intravital microscopy. Mean numbers of rolling or firmly adhering leukocytes in 3-4 animals are shown. A. Leukocyte rolling in EIRAKO mice and controls. B. Leukocyte adhesion in EIRAKO mice. C. Leukocyte rolling in VENIRKO mice and controls, both wild-type for apoE. D. Leukocyte adhesion in VENIRKO mice and controls. E. Western blotting of lysate of peripheral blood mononuclear cells (PBMC). F. Adhesion in vivo of transferred mononuclear cells. PBMC were freshly isolated from one donor animal, labeled with rhodamine ex vivo, and injected i. v. in a single recipient animal. Mean numbers of firmly adhering PBMC, with genotypes of donor and recipient animals indicated, are shown (n=3-5). G-I. Bone marrow transplantation in apoE knockout mice, which were lethally irradiated and recieved a graft of bone marrow cells from EIRAKO or control mice. An atherogenic “Western” diet was started the day after transplantation. G. Western blotting of PBMC lysate 8 weeks after transplantation. H. Mean values for insulin receptor-β protein based on densitometry of Western blots of PBMC from 3 mice in each group of apoE knockout mice with EIRAKO or control bone marrow replacement. See also Figure S3. I. Mean values for atherosclerotic lesion area in the aorta in 11 recipients of EIRAKO mice bone marrow and 11 recipients of control bone marrow 8 weeks after bone marrow transplantation. Abbreviations: E, endothelial insulin receptor and apoE knockout (EIRAKO) mice; V, vascular endothelial receptor knockout (VENIRKO) mice; PBMC, peripheral blood mononuclear cells.
We have previously shown an increase in leukocyte rolling and adherence in apoE null compared to wild-type mice while fed a regular chow diet, using the same intravital preparation as in the current paper (Scalia et al., 2001). However, rolling and adhesion were not different between EIRAKO controls and wild-type mice (Figure 6A and B), likely because mice used in the previous study were up to twice as old (Scalia et al., 2001). The apoE null phenotype, however, could modify the effects of endothelial insulin receptor knockout on leukocyte-endothelium interaction in EIRAKO mice. Therefore, we performed experiments in VENIRKO animals and their controls, both of which are wild-type for apoE. Leukocyte rolling was increased by 1.8-fold in VENIRKO mice compared with their littermate controls (57.5±6.9 and 31.3±7.5 rolling leukocytes per 100 μm vessel length, respectively (Figure 6C, p=0.03). As in EIRAKO mice, leukocyte adhesion was increased dramatically in VENIRKO mice, by 7.3-fold compared with their controls (3.6±0.4 and 0.5±0.3 adhering leukocytes per 100 μm, respectively, Figure 6D, p=0.001). Thus, increased leukocyte-endothelial cell interaction resulting from a loss of insulin signaling occurs in the abscence of hypercholesterolemia.
Mononuclear cell transfer and EIRAKO bone marrow transplantation
When it was first characterized, the Tie2-Cre transgene used in the creation of EIRAKO mice did not result in Cre-mediated recombination in embryonic hematopoietic cells (Kisanuki et al., 2001). However, a recent study showed that Tie2 was expressed in 20% of human blood monocytes (Murdoch et al., 2007). We therefore measured insulin receptor expression in peripheral blood mononuclear cells (PBMC) from EIRAKO mice and found that IRβ protein was reduced by 68% compared with controls (Figure 6E). IRβ was also reduced in thioglycollate-induced peritoneal macrophages (Figure S3) and in bone marrow (data not shown). To determine whether the large increase in firmly adhering leukocytes in EIRAKO mice was due to decreased insulin receptor expression in mononuclear cells, PBMC were isolated from wild-type mice, from EIRAKO mice, or from their littermate controls and subsequently injected intravenously into wild-type recipients after being labeled ex vivo with a fluorescent dye. The adherence of PBMC to wild-type endothelium was not different whether the donor PBMC were from wild-type mice, EIRAKO mice, or their littermate controls (0.60±0.25, 0.83±0.17, or 0.75±0.32 adhering leukocytes per 100 μm, respectively, Figure 6F). Each of these rates of adherence was not different from the adherence of PBMC from wild-type mice when injected into control mice (0.75±0.48 adhering leukocytes per 100 μm, Figure 6F). These data demonstrate that PBMC from both EIRAKO mice and their controls were functionally normal with respect to their adherence to endothelial cells. In contrast, when PBMC were isolated from wild-type mice and injected into EIRAKO recipients, the number of adhering donor cells was more than 4-fold higher than in any of the other transfer experiments (3.7±0.1 adhering PBMC per 100 μm, Figure 6F, p<0.001). Therefore, the abnormally high leukocyte-endothelial cell adhesion in EIRAKO mice is caused by a functional abnormality in the endothelium rather than in leukocytes.
Once resident in the vascular wall, leukocytes with reduced insulin receptor expression could contribute to plaque development differently than leukocytes with normal expression of insulin receptor protein. We therefore studied whether bone marrow-derived cells from EIRAKO mice would cause different atherosclerosis progression than control bone marrow cells. apoE null mice were lethally irradiated and received bone marrow grafts from either EIRAKO mice or littermate controls. Chimerism after bone marrow replacement was evident by a reduction of IRβ protein by 79±3% in PBMC isolated from apoE null mice transplanted with EIRAKO bone marrow grafts compared to recipients of control bone marrow (Figure 6G and H). Despite a reduction of PBMC insulin receptor expression comparable to the level seen in EIRAKO mice, aortic atherosclerotic lesion area 8 weeks after transplantation was no different in recipients of EIRAKO bone marrow than in recipients of control bone marrow (Figure 6I).
Regulation of VCAM-1
Endothelial cells affect leukocyte adhesion, and to some extent leukocyte rolling, by expression levels of ICAM-1 and VCAM-1. In lung endothelial cells from EIRAKO mice, VCAM-1 protein expression was 199±29% of the level seen in cells isolated from control mice (Figure 7A and B, p=0.01), whereas ICAM-1 protein expression was no different (Figure 7A). This suggested that insulin might negatively regulate VCAM-1. When MS1 mouse endothelial cells were treated with 10 nM insulin, VCAM-1 protein expression was reduced by 22±8.3% after 24 hours (p=0.06), 46±7% after 48 hours (p=0.01) and by 57±2% after 72 hours (p=0.01) compared with cultures grown under identical conditions for the same duration without insulin (Figure 7C and D). No changes were seen either 4 or 8 hours after insulin treatment (data not shown).
Figure 7. VCAM-1 regulation.
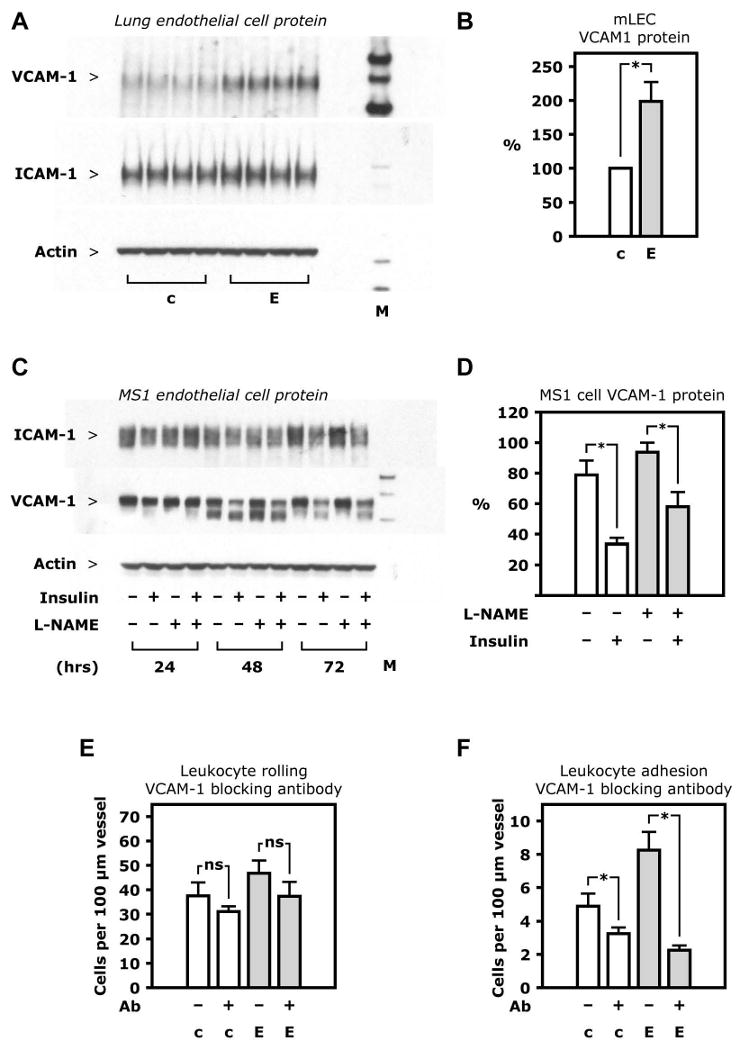
A. Western blotting of whole cell lysate from lung endothelial cells isolated from one EIRAKO mouse and one control, with each sample loaded as 4 replicates. B. Mean values for VCAM-1 protein based on densitometry of Western blots of lung endothelial cell lysate from 4 pairs of EIRAKO mice and controls. C. Western blotting of whole cell lysate from MS1 endothelial cells. D. Mean values for ICAM-1 and VCAM-1 protein based on densitometry of Western blots from 3 independent experiments in MS1 endothelial cells. E-F. Leukocyte rolling on and firm adhesion to endothelium of peri-intestinal, post-capillary venules visualized by intravital microscopy after fluorescent labeling by i. v. injection of rhodamine. Measurements were repeated 60 minutes after i. v. injection of a VCAM-1 blocking antibody. Mean values from 4 pairs of animals 20-23 weeks of age are shown. E. Leukocyte rolling before and after VCAM-1 injection F. Leukocyte adhesion before and after VCAM-1 injection.
To demonstrate that increased expression of VCAM-1 was the cause of increased leukocyte-endothelial cell interaction in vivo, we used a VCAM-1 antibody with documented ability to block leukocyte adhesion to endothelial cells. Leukocyte rolling and adhesion was monitored by intravital microscopy before and 60 minutes after intravenous injection of the antibody. This treatment had no significant effect on leukocyte rolling in EIRAKO or control mice (Figure 7E, p>0.2). However, the VCAM-1 antibody decreased leukocyte adhesion in EIRAKO mice by 72.6±1.0%, to below control levels (Figure 7F, p=0.01). These results indicate that VCAM-1 upregulation in EIRAKO mice contributes significantly to the increased leukocyte-endothelial adhesion observed in these animals.
Discussion
The current study provides definitive evidence that a loss of insulin signaling in the vascular endothelium without changes in systemic metabolism promotes early events in atherogenesis and accelerates the progression of advanced atherosclerotic disease. These outcomes support the hypothesis previously examined by us (Jiang et al., 1999) and others (Steinberg et al., 1996), proposing that in patients with obesity or type 2 diabetes, insulin resistance in endothelial cells may contribute to increased risk for development of cardiovascular disease.
Our results show that systemic insulin action in EIRAKO mice is no different than in control mice. Thus, endothelial insulin resistance did not cause changes in whole-body glucose tolerance, circulating insulin concentrations, or insulin sensitivity in EIRAKO mice compared to controls. This is consistent with what has previously been reported for VENIRKO mice (Vicent et al., 2003) and with findings in transgenic mice with a mutant insulin receptor targeted to endothelium (Duncan et al., 2008). Furthermore, the similarity in cholesterol and triglyceride content in lipoproteins between EIRAKO mice and their controls shows that insulin signaling in endothelial cells does not significantly alter regulation of lipoprotein metabolism in the liver or adipose tissue. This lack of differences in systemic insulin sensitivity or lipoprotein profile between control and EIRAKO mice is essential in order for us to conclude that the accelerated atherosclerosis in EIRAKO mice is a direct result of the loss of insulin action on the endothelium.
Leukocyte rolling on and adhesion to endothelial cells in vivo were increased in both EIRAKO mice, which are apoE null, and VENIRKO mice, which are apoE wild-type. These findings correspond with results showing upregulation of VCAM-1 expression in endothelial cells isolated from EIRAKO mice, and decreased VCAM-1 protein expression in endothelial cells treated with insulin. The functional significance of this finding was demonstrated by experiments in vivo showing that a VCAM-1 blocking antibody reduced leukocyte-endothelial cell adhesion in EIRAKO mice to below control levels. Epidemiological evidence supports that upregulation of VCAM-1 in association with insulin resistance is clinically important, because in patients with type 2 diabetes, soluble VCAM-1 concentrations are independently associated with cardiovascular mortality (Jager et al., 2000). However, previously published mechanistic studies on the effect of insulin on the expression of factors that promote adhesion have been contradictory. Several studies, among them work by De Catarina et al. (Madonna et al., 2004), have shown that insulin can increase the expression of ICAM-1 and VCAM-1 in endothelial cells, although other studies have shown that insulin inhibits expression of ICAM-1 ((Aljada et al., 2000)) and P-selectin (Booth et al., 2001). We are not aware of previously published evidence demonstrating regulation of VCAM-1 by insulin in vivo.
In the current study, NO-mediated vasodilator function was impaired in EIRAKO mice, and loss of insulin-stimulated phosphorylation of Akt and eNOS was clearly demonstrated in the aorta in vivo and in isolated endothelial cells. NO synthesized by eNOS decreases both leukocyte rolling and adhesion (Lefer et al., 1999), and NO has been shown to inhibit expression of VCAM-1 and other factors promoting leukocyte adhesion through deactivation of NFκB (De Caterina et al., 1995; Khan et al., 1996). Therefore, decreased endothelium-derived NO could result in increased expression of adhesion molecules, or promote atherosclerotic plaque development and complexity through other mechanisms. For example, impaired NO production could increase the number of intraplaque vascular smooth muscle cells, as shown by our data from brachial artery immunohistochemistry, because NO inhibits vascular smooth muscle cell migration (Sarkar et al., 1996). However, NO production could not account for insulin-stimulated downregulation of VCAM-1 in our cell culture studies, as L-NAME was unable to block these effects of insulin. Therefore, it is likely that the regulation of VCAM-1 by insulin is mediated by another mechanism, perhaps through the nuclear factor forkhead box O1 (FoxO1/FKHR). Transcriptional activity of FoxO1 is suppressed by insulin (Nakae et al., 1999; Tang et al., 1999) and FoxO1 activation upregulates VCAM-1 expression (Abid et al., 2006). Such mechanisms may be similar to or separate from the pro-inflammatory and pro-atherosclerotic changes observed in whole-body Akt1−/− mice (Fernandez-Hernando et al., 2007). These questions are currently being investigated in our laboratory.
We observed a dramatic increase in mononuclear cell adhesion when mononuclear cells from wild-type mice were injected into EIRAKO mice. In contrast, no abnormality of mononuclear cell adhesion to endothelial cells was observed when mononuclear cells from EIRAKO mice were injected into control littermates or wild-type mice. These data show that increased leukocyte-endothelial cell interaction in EIRAKO mice is caused by endothelial cell dysfunction rather than changes in monocyte function. Similarly, replacement of bone marrow in apoE null mice with bone marrow from EIRAKO mice or their controls showed that insulin receptor downregulation in bone marrow-derived cells was not sufficient to accelerate atherosclerosis. We therefore conclude that the vascular inflammation and accelerated atherosclerosis observed in EIRAKO mice is a result of the loss of insulin signaling in endothelial cells, with no significant contribution from the downregulation of insulin signaling in leukocytes.
A previous study has shown that the loss of insulin receptors in macrophages or leukocytes may decrease atherosclerosis in apoE null mice (Baumgartl et al., 2006). Furthermore, replacing the bone marrow in apoE null mice with transplants from insulin receptor substrate-2 (IRS-2)/apoE double knockout mice decreased atherosclerosis even though IRS-2/apoE double knockout mice had increased atherosclerosis compared to apoE null controls (Baumgartl et al., 2006; Gonzalez-Navarro et al., 2008). Another study showed that transplantation of bone marrow from insulin receptor null mice to lethally irradiated LDL receptor null mice had no effect after 8 weeks of an atherogenic diet, and demonstrated a modest increase after 12 weeks (Han et al., 2006), as well as increased apoptosis of macrophages and an increased rate of necrotic core formation in atherosclerotic plaques. Therefore, even though decreased expression of insulin receptor in mononuclear cells in EIRAKO mice did not appear to affect increased leukocyte-endothelial interaction or atherosclerotic development, complete loss of insulin signaling in leukocytes, or loss of insulin action in certain subsets of bone-marrow derived cells, may decrease the progression of atherosclerosis while promoting necrotic core formation in more advanced plaques.
In vascular tissue from obese rats and patients with diabetes, the major insulin signaling pathway represented by PI3K and Akt is affected selectively by insulin resistance, whereas the pathway represented by mitogen-activated protein kinase (MAPK) is intact (Jiang et al., 1999). We have therefore proposed that in the normal, insulin-sensitive state, insulin action on the endothelium is mainly mediating anti-atherogenic effects by activating the PI3K/Akt pathway (Jiang et al., 1999). However, in the insulin resistant state, often associated with hyperinsulinemia, pro-atherogenic insulin action on endothelium may be mediated by MAPK pathways (Rask-Madsen and King, 2007). By using a mouse model with a null mutation of the insulin receptor gene in endothelial cells, our study may underestimate the contribution of selective insulin resistance in the endothelium in disease states because potential pro-atherosclerotic insulin signaling mediated by MAPK is lost in the mouse model, while it may be functional in human disease. Future studies will be needed to determine whether enhancement of the insulin receptor substrate/PI3K/Akt signaling pathway in endothelium can delay atherosclerosis development.
In summary, we have demonstrated that insulin action on the endothelium is mainly anti-atherogenic and that loss of insulin signaling in endothelial cells promotes initiation of atherosclerosis and its progression, and increases the complexity of advanced atherosclerotic lesions. The findings further indicate that insulin plays a quantitatively prominent role in maintaining normal endothelial function, and that endothelial insulin resistance may accelerate atherosclerosis by causing impaired activation of eNOS, increased endothelial expression of VCAM-1, and increased leukocyte interaction with endothelial cells. These results provide the rationale for identifying interventions which can decrease the risk of atherosclerotic disease in patients with insulin resistance or diabetes by alleviating insulin resistance in the endothelium.
Experimental Procedures
Additional details of the experimental procedures are included in the Online Supplemental Information.
Animals
Vascular endothelial insulin receptor knockout (VENIRKO) mice (Vicent et al., 2003) were cross-bred with apoE null mice using breeding of Cre-positive males and Cre-negative females. Male Cre+ Insrflox/flox Apoe−/− (EIRAKO) mice and littermate Cre− Insr flox/flox Apoe−/− (control) mice were used for experiments. Mice were fed a a regular chow diet with 9.0% (w/w) fat and 0.221 ppm cholesterol. All protocols for animal use and euthanasia were reviewed and approved by the Animal Care Committee of the Joslin Diabetes Center and were in accordance with NIH guidelines.
Cell culture
Cell culture was obtained from lung tissue or aorta after digestion with collagenase. Endothelial cells were isolated during the first two passages with the use of magnetic beads complexed to ICAM-2 antibody as described previously (Hartwell et al., 1998). Aortic smooth muscle cells were obtained as the ICAM-2-negative fraction after immunoselection of cells cultured from aortic digest. MS1 endothelial cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and cultured in DMEM containing 5% FBS.
Carotid vasodilation
Concentration-response studies of vasodilation stimulated by acetylcholine and sodium nitroprusside were performed as described previously (Atochin et al., 2007).
Intravital microscopy and quantitation of leukocyte-endothelial cell interaction
Leukocyte-endothelial cell interaction was observed in post-capillary venules of the peri-intestinal microcirculation as described (Ouedraogo et al., 2007).
Analysis of atherosclerotic plaques in the aorta, the brachiocephalic artery, and the aortic sinus
The whole aorta, from just distal to the aortic sinus to the iliac bifurcation, was stained with Sudan IV. The stained aortas were placed between two glass slides and photographed under polarized light through a dissection microscope. Lipids were extracted from the brachiocephalic artery and analyzed for cholesteryl ester abundance by mass spectrometry as previously described (Kuo et al., 2008). Cross-sections of paraffin-embedded brachiocephalic artery and cryosections of the aortic sinus were stained as described previously (Kuo et al., 2008).
Statistical analysis
Comparisons were made using paired or unpaired t-test, as appropriate, with p<0.05 considered statistically significant. In text and graphs, data are presented as the mean ± standard error of the mean.
Supplementary Material
Acknowledgments
We are grateful for expert technical assistance provided by Alevtina Pinkhasov of the Advanced Microscopy Core at Joslin Diabetes Center, supported by NIH grant 5P30DK036836; and by Michael Kalbfleisch for work using mass spectrometry, and Donetta Gifford-Moore for work on brachiocepahlic artery histology. Measurement of serum insulin was performed by the Joslin Diabetes Center Specialized Assay Core which is supported by NIH grant 5P30DK036836. Plasma lipid profiles were measured by Dr. Vladimir Babaev at the Lipid, Lipoprotein and Atherosclerosis Core of the Vanderbilt Mouse Metabolic Phenotype Centers with support from NIH grant DK59637. This work was funded in part by support to Dr. Rask-Madsen from the Danish Medical Research Council fellowship (grant 22-01-0498), the Danish Heart Foundation (grant 01-2-2-79-22946), and by a Mary K. Iacocca Fellowship provided by the Iacocca Foundation. The study was also supported by NIH R01 grants DK064344-05 to Dr. Scalia, DK31036 to Dr. Kahn, and DK053105-09 to Dr. King. The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies. None of the authors declare any conflict of interest in reporting this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abid MR, Shih SC, Otu HH, Spokes KC, Okada Y, Curiel DT, Minami T, Aird WC. A novel class of vascular endothelial growth factor-responsive genes that require forkhead activity for expression. J Biol Chem. 2006;281:35544–35553. doi: 10.1074/jbc.M608620200. [DOI] [PubMed] [Google Scholar]
- Aljada A, Saadeh R, Assian E, Ghanim H, Dandona P. Insulin inhibits the expression of intercellular adhesion molecule-1 by human aortic endothelial cells through stimulation of nitric oxide. J Clin Endocrinol Metab. 2000;85:2572–2575. doi: 10.1210/jcem.85.7.6677. [DOI] [PubMed] [Google Scholar]
- Atochin DN, Wang A, Liu VW, Critchlow JD, Dantas AP, Looft-Wilson R, Murata T, Salomone S, Shin HK, Ayata C, Moskowitz MA, Michel T, Sessa WC, Huang PL. The phosphorylation state of eNOS modulates vascular reactivity and outcome of cerebral ischemia in vivo. J Clin Invest. 2007;117:1961–1967. doi: 10.1172/JCI29877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartl J, Baudler S, Scherner M, Babaev V, Makowski L, Suttles J, McDuffie M, Tobe K, Kadowaki T, Fazio S, Kahn CR, Hotamisligil GS, Krone W, Linton M, Bruning JC. Myeloid lineage cell-restricted insulin resistance protects apolipoproteinE-deficient mice against atherosclerosis. Cell Metab. 2006;3:247–256. doi: 10.1016/j.cmet.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth G, Stalker TJ, Lefer AM, Scalia R. Elevated ambient glucose induces acute inflammatory events in the microvasculature: effects of insulin. Am J Physiol Endocrinol Metab. 2001;280:E848–856. doi: 10.1152/ajpendo.2001.280.6.E848. [DOI] [PubMed] [Google Scholar]
- Cardillo C, Nambi SS, Kilcoyne CM, Choucair WK, Katz A, Quon MJ, Panza JA. Insulin stimulates both endothelin and nitric oxide activity in the human forearm. Circulation. 1999;100:820–825. doi: 10.1161/01.cir.100.8.820. [DOI] [PubMed] [Google Scholar]
- De Caterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA, Jr, Shin WS, Liao JK. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest. 1995;96:60–68. doi: 10.1172/JCI118074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despres JP, Lamarche B, Mauriege P, Cantin B, Dagenais GR, Moorjani S, Lupien PJ. Hyperinsulinemia as an independent risk factor for ischemic heart disease. N Engl J Med. 1996;334:952–957. doi: 10.1056/NEJM199604113341504. [DOI] [PubMed] [Google Scholar]
- Drummond GR, Cai H, Davis ME, Ramasamy S, Harrison DG. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression by hydrogen peroxide. Circ Res. 2000;86:347–354. doi: 10.1161/01.res.86.3.347. [DOI] [PubMed] [Google Scholar]
- Du X, Edelstein D, Obici S, Higham N, Zou MH, Brownlee M. Insulin resistance reduces arterial prostacyclin synthase and eNOS activities by increasing endothelial fatty acid oxidation. J Clin Invest. 2006;116:1071–1080. doi: 10.1172/JCI23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan ER, Crossey PA, Walker S, Anilkumar N, Poston L, Douglas G, Ezzat VA, Wheatcroft SB, Shah AM, Kearney MI. Effect of endothelium-specific insulin resistance on endothelial function in vivo. Diabetes. 2008;57:3307–3314. doi: 10.2337/db07-1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Hernando C, Ackah E, Yu J, Suarez Y, Murata T, Iwakiri Y, Prendergast J, Miao RQ, Birnbaum MJ, Sessa WC. Loss of Akt1 leads to severe atherosclerosis and occlusive coronary artery disease. Cell Metab. 2007;6:446–457. doi: 10.1016/j.cmet.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geraldes P, Yagi K, Ohshiro Y, He Z, Maeno Y, Yamamoto-Hiraoka J, Rask-Madsen C, Chung SW, Perrella MA, King GL. Selective regulation of heme oxygenase-1 expression and function by insulin through IRS1/phosphoinositide 3-kinase/Akt2 pathway. J Biol Chem. 2008;283:34327–34336. doi: 10.1074/jbc.M807036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Navarro H, Vinue A, Vila-Caballer M, Fortuno A, Beloqui O, Zalba G, Burks D, Diez J, Andres V. Molecular mechanisms of atherosclerosis in metabolic syndrome: role of reduced IRS2-dependent signaling. Arterioscler Thromb Vasc Biol. 2008;28:2187–2194. doi: 10.1161/ATVBAHA.108.175299. [DOI] [PubMed] [Google Scholar]
- Grenett HE, Benza RL, Fless GM, Li XN, Davis GC, Booyse FM. Genotype-specific transcriptional regulation of PAI-1 gene by insulin, hypertriglyceridemic VLDL, and Lp(a) in transfected, cultured human endothelial cells. Arterioscler Thromb Vasc Biol. 1998;18:1803–1809. doi: 10.1161/01.atv.18.11.1803. [DOI] [PubMed] [Google Scholar]
- Han S, Liang CP, DeVries-Seimon T, Ranalletta M, Welch CL, Collins-Fletcher K, Accili D, Tabas I, Tall AR. Macrophage insulin receptor deficiency increases ER stress-induced apoptosis and necrotic core formation in advanced atherosclerotic lesions. Cell Metab. 2006;3:257–266. doi: 10.1016/j.cmet.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Hartwell DW, Mayadas TN, Berger G, Frenette PS, Rayburn H, Hynes RO, Wagner DD. Role of P-selectin cytoplasmic domain in granular targeting in vivo and in early inflammatory responses. J Cell Biol. 1998;143:1129–1141. doi: 10.1083/jcb.143.4.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann C, Assmus B, Urbich C, Zeiher AM, Dimmeler S. Insulin-mediated stimulation of protein kinase Akt: A potent survival signaling cascade for endothelial cells. Arterioscler Thromb Vasc Biol. 2000;20:402–409. doi: 10.1161/01.atv.20.2.402. [DOI] [PubMed] [Google Scholar]
- Howard G, O'Leary DH, Zaccaro D, Haffner S, Rewers M, Hamman R, Selby JV, Saad MF, Savage P, Bergman R. Insulin sensitivity and atherosclerosis. The Insulin Resistance Atherosclerosis Study (IRAS) Investigators. Circulation. 1996;93:1809–1817. doi: 10.1161/01.cir.93.10.1809. [DOI] [PubMed] [Google Scholar]
- Jager A, van Hinsbergh VW, Kostense PJ, Emeis JJ, Nijpels G, Dekker JM, Heine RJ, Bouter LM, Stehouwer CD. Increased levels of soluble vascular cell adhesion molecule 1 are associated with risk of cardiovascular mortality in type 2 diabetes: the Hoorn study. Diabetes. 2000;49:485–491. doi: 10.2337/diabetes.49.3.485. [DOI] [PubMed] [Google Scholar]
- Jiang ZY, Lin YW, Clemont A, Feener EP, Hein KD, Igarashi M, Yamauchi T, White MF, King GL. Characterization of selective resistance to insulin signaling in the vasculature of obese Zucker (fa/fa) rats. J Clin Invest. 1999;104:447–457. doi: 10.1172/JCI5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan BV, Harrison DG, Olbrych MT, Alexander RW, Medford RM. Nitric oxide regulates vascular cell adhesion molecule 1 gene expression and redox-sensitive transcriptional events in human vascular endothelial cells. Proc Natl Acad Sci U S A. 1996;93:9114–9119. doi: 10.1073/pnas.93.17.9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- Kuboki K, Jiang ZY, Takahara N, Ha SW, Igarashi M, Yamauchi T, Feener EP, Herbert TP, Rhodes CJ, King GL. Regulation of endothelial constitutive nitric oxide synthase gene expression in endothelial cells and in vivo : a specific vascular action of insulin. Circulation. 2000;101:676–681. doi: 10.1161/01.cir.101.6.676. [DOI] [PubMed] [Google Scholar]
- Kuo MS, Kalbfleisch JM, Rutherford P, Gifford-Moore D, Huang XD, Christie R, Hui K, Gould K, Rekhter M. Chemical analysis of atherosclerotic plaque cholesterol combined with histology of the same tissue. J Lipid Res. 2008;49:1353–1363. doi: 10.1194/jlr.D700037-JLR200. [DOI] [PubMed] [Google Scholar]
- Lefer DJ, Jones SP, Girod WG, Baines A, Grisham MB, Cockrell AS, Huang PL, Scalia R. Leukocyte-endothelial cell interactions in nitric oxide synthase-deficient mice. Am J Physiol. 1999;276:H1943–1950. doi: 10.1152/ajpheart.1999.276.6.H1943. [DOI] [PubMed] [Google Scholar]
- Madonna R, Pandolfi A, Massaro M, Consoli A, De Caterina R. Insulin enhances vascular cell adhesion molecule-1 expression in human cultured endothelial cells through a pro-atherogenic pathway mediated by p38 mitogen-activated protein-kinase. Diabetologia. 2004;47:532–536. doi: 10.1007/s00125-004-1330-x. [DOI] [PubMed] [Google Scholar]
- Murdoch C, Tazzyman S, Webster S, Lewis CE. Expression of Tie-2 by human monocytes and their responses to angiopoietin-2. J Immunol. 2007;178:7405–7411. doi: 10.4049/jimmunol.178.11.7405. [DOI] [PubMed] [Google Scholar]
- Nakae J, Park BC, Accili D. Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a Wortmannin-sensitive pathway. J Biol Chem. 1999;274:15982–15985. doi: 10.1074/jbc.274.23.15982. [DOI] [PubMed] [Google Scholar]
- Naruse K, Rask-Madsen C, Takahara N, Ha SW, Suzuma K, Way KJ, Jacobs JR, Clermont AC, Ueki K, Ohshiro Y, Zhang J, Goldfine AB, King GL. Activation of vascular protein kinase C-beta inhibits Akt-dependent endothelial nitric oxide synthase function in obesity-associated insulin resistance. Diabetes. 2006;55:691–698. doi: 10.2337/diabetes.55.03.06.db05-0771. [DOI] [PubMed] [Google Scholar]
- Nordt TK, Sawa H, Fujii S, Bode C, Sobel BE. Augmentation of arterial endothelial cell expression of the plasminogen activator inhibitor type-1 (PAI-1) gene by proinsulin and insulin in vivo. J Mol Cell Cardiol. 1998;30:1535–1543. doi: 10.1006/jmcc.1998.0719. [DOI] [PubMed] [Google Scholar]
- Okouchi M, Okayama N, Shimizu M, Omi H, Fukutomi T, Itoh M. High insulin exacerbates neutrophil-endothelial cell adhesion through endothelial surface expression of intercellular adhesion molecule-1 via activation of protein kinase C and mitogen-activated protein kinase. Diabetologia. 2002;45:556–559. doi: 10.1007/s00125-001-0773-6. [DOI] [PubMed] [Google Scholar]
- Oliver FJ, de la Rubia G, Feener EP, Lee ME, Loeken MR, Shiba T, Quertermous T, King GL. Stimulation of endothelin-1 gene expression by insulin in endothelial cells. J Biol Chem. 1991;266:23251–23256. [PubMed] [Google Scholar]
- Ouedraogo R, Gong Y, Berzins B, Wu X, Mahadev K, Hough K, Chan L, Goldstein BJ, Scalia R. Adiponectin deficiency increases leukocyte-endothelium interactions via upregulation of endothelial cell adhesion molecules in vivo. J Clin Invest. 2007;117:1718–1726. doi: 10.1172/JCI29623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rask-Madsen C, Ihlemann N, Krarup T, Christiansen E, Kober L, Nervil Kistorp C, Torp-Pedersen C. Insulin therapy improves insulin-stimulated endothelial function in patients with type 2 diabetes and ischemic heart disease. Diabetes. 2001;50:2611–2618. doi: 10.2337/diabetes.50.11.2611. [DOI] [PubMed] [Google Scholar]
- Rask-Madsen C, King GL. Mechanisms of Disease: endothelial dysfunction in insulin resistance and diabetes. Nat Clin Pract Endocrinol Metab. 2007;3:46–56. doi: 10.1038/ncpendmet0366. [DOI] [PubMed] [Google Scholar]
- Reusch JE, Draznin BB. Atherosclerosis in diabetes and insulin resistance. Diabetes Obes Metab. 2007;9:455–463. doi: 10.1111/j.1463-1326.2006.00620.x. [DOI] [PubMed] [Google Scholar]
- Sarkar R, Meinberg EG, Stanley JC, Gordon D, Webb RC. Nitric oxide reversibly inhibits the migration of cultured vascular smooth muscle cells. Circ Res. 1996;78:225–230. doi: 10.1161/01.res.78.2.225. [DOI] [PubMed] [Google Scholar]
- Scalia R, Gooszen ME, Jones SP, Hoffmeyer M, Rimmer DM, 3rd, Trocha SD, Huang PL, Smith MB, Lefer AM, Lefer DJ. Simvastatin exerts both anti-inflammatory and cardioprotective effects in apolipoprotein E-deficient mice. Circulation. 2001;103:2598–2603. doi: 10.1161/01.cir.103.21.2598. [DOI] [PubMed] [Google Scholar]
- Steinberg HO, Brechtel G, Johnson A, Fineberg N, Baron AD. Insulin-mediated skeletal muscle vasodilation is nitric oxide dependent. A novel action of insulin to increase nitric oxide release. J Clin Invest. 1994;94:1172–1179. doi: 10.1172/JCI117433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg HO, Chaker H, Leaming R, Johnson A, Brechtel G, Baron AD. Obesity/insulin resistance is associated with endothelial dysfunction. Implications for the syndrome of insulin resistance. J Clin Invest. 1996;97:2601–2610. doi: 10.1172/JCI118709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symons JD, McMillin SL, Riehle C, Tanner J, Palionyte M, Hillas E, Jones D, Cooksey RC, Birnbaum MJ, McClain DA, Zhang QJ, Gale D, Wilson LJ, Abel ED. Contribution of insulin and Akt1 signaling to endothelial nitric oxide synthase in the regulation of endothelial function and blood pressure. Circ Res. 2009;104:1085–1094. doi: 10.1161/CIRCRESAHA.108.189316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang ED, Nunez G, Barr FG, Guan KL. Negative regulation of the forkhead transcription factor FKHR by Akt. J Biol Chem. 1999;274:16741–16746. doi: 10.1074/jbc.274.24.16741. [DOI] [PubMed] [Google Scholar]
- Vicent D, Ilany J, Kondo T, Naruse K, Fisher SJ, Kisanuki YY, Bursell S, Yanagisawa M, King GL, Kahn CR. The role of endothelial insulin signaling in the regulation of vascular tone and insulin resistance. J Clin Invest. 2003;111:1373–1380. doi: 10.1172/JCI15211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng G, Quon MJ. Insulin-stimulated production of nitric oxide is inhibited by wortmannin. Direct measurement in vascular endothelial cells. J Clin Invest. 1996;98:894–898. doi: 10.1172/JCI118871. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.