

Abstract
Protein kinase RNA-regulated (PKR) is an established component of innate antiviral immunity. Recently, PKR has been shown to be essential for signal transduction in other situations of cellular stress. The relationship between PKR and the stress-activated protein kinases (SAPKs), such as p38 mitogen-activated protein kinase (MAPK), is not clear. Using embryonic fibroblasts from PKR wild-type and null mice, we established a requirement for PKR in the activation of SAPKs by double-stranded RNA, lipopolysaccharide (LPS) and proinflammatory cytokines. This does not reflect a global failure to activate SAPKs in the PKR-null background as these kinases are activated normally by anisomycin and other physicochemical stress. Activation of p38 MAPK was restored in immortalized PKR-null cells by reconstitution with human PKR. We also show that LPS induction of interleukin-6 and interleukin-12 mRNA is defective in PKR-null cells, and that production of these cytokines is impaired in PKR-null mice challenged with LPS. Our findings indicate, for the first time, that PKR is required for p38 MAPK signaling and plays a potentially important role in the innate response against bacterial endotoxin.
Keywords: innate immunity/interleukins/LPS/ p38 MAPK/PKR
Introduction
The innate immune response is initiated by the recognition of invariant molecular components of infectious agents such as lipopolysaccharide (LPS), the signature motif of Gram-negative bacteria and double-stranded RNA (dsRNA), a common intermediate of viral replication (Medzhitov and Janeway, 1997). Binding of these foreign moieties to specific cellular receptors triggers distinct signaling pathways that lead to physiological events designed to contain or eliminate the pathogens. While the receptors for LPS are extracellular and transmembrane proteins (i.e. CD14 and toll-like receptor 4, respectively) (Qureshi et al., 1999), dsRNA is only known to bind intracellular targets, including the enzyme protein kinase RNA-regulated (PKR).
PKR, a ubiquitous serine/threonine kinase that is induced by interferons (IFNs), was identified and characterized initially as a translational inhibitor in an antiviral pathway regulated by IFNs (Stark et al., 1998). Upon binding to dsRNA, PKR is activated by autophosphorylation and subsequently phosphorylates the α-subunit of eukaryotic initiation factor 2 (eIF-2α), thereby inhibiting translation (Clemens and Elia, 1997). This effectively limits virion production and has led to some viruses evolving strategies to inhibit the action of PKR (Gale and Katze, 1998). More recently, PKR has been implicated in other aspects of cellular function, including normal growth control, induction of apoptosis and signal transduction (Williams, 1997). Its role as a signal transducer has been established for dsRNA, IFN-γ and tumor necrosis factor (TNF)-α, where PKR has been shown to mediate the activation of important transcription factors such as nuclear factor κB (NF-κB), activating transcription factor-2 (ATF-2), signal transducer and activator of transcription-1 (STAT1) and interferon regulatory factor-1 (IRF-1) (Yang et al., 1995; Der et al., 1997; Kumar et al., 1997; Bandyopadhyay et al., 2000; Ramana et al., 2000; Zamanian-Daryoush et al., 2000).
In recent years, many mammalian kinases have been shown to mediate cellular stress responses, including the p38 and c-Jun N-terminal kinase (JNK) subgroups of mitogen-activated protein kinases (MAPKs) (Tibbles and Woodgett, 1999). Collectively known as stress-activated protein kinases (SAPKs), p38 and JNK MAPKs are activated by a wide array of stress stimuli in diverse cell types (Widmann et al., 1999). These stimuli include genotoxic agents (UV irradiation and carcinogens), pathogenic signals (LPS and dsRNA) (Iordanov et al., 2000), proinflammatory cytokines [TNF-α and interleukin (IL)-1β], homeostatic perturbations [in temperature, tonicity, pH (Shrode et al., 1997), oxygen partial pressure (Conrad et al., 1999) or intracellular calcium (Kawasaki et al., 1997)] and other chemical insults (arsenite and anisomycin). Activated SAPKs act on downstream targets, including other kinases and many transcription factors, to promote cellular adaptation or apoptosis (Tibbles and Woodgett, 1999). The relationship between PKR and the SAPKs is not well defined.
In the present study, we demonstrate that PKR mediates the activation of p38 and JNK MAPKs by specific proinflammatory stress stimuli. Reconstitution of PKR-null mouse fibroblasts with functional human PKR restores their ability to activate p38 MAPK. We also show that PKR-null mice are impaired in their production of inflammatory cytokines in response to LPS challenge.
Results and discussion
PKR-null mouse embryonic fibroblasts are defective in activating SAPK in response to proinflammatory signals
Mouse embryonic fibroblasts (MEFs) deficient in PKR have been shown previously to be defective in the activation of NF-κB and IRF-1 in response to IFN-γ and poly(IC) (a synthetic form of dsRNA) (Yang et al., 1995; Kumar et al., 1997). Furthermore, they undergo apoptosis less readily than wild-type MEFs in response to TNF-α, LPS and poly(IC) (Der et al., 1997). Since these agents, as well as the proinflammatory cytokine IL-1β, are all known to activate SAPKs (Raingeaud et al., 1995; Goh et al., 1999; Iordanov et al., 2000), we sought to determine whether PKR deficiency has any effects on their activation of SAPKs. Using antibodies that detect either activated, dual-phosphorylated SAPK or total SAPK, we observed that activation of both p38 MAPK (Figure 1A, upper panel) and JNK (Figure 1C, upper panel) was diminished in PKR-null fibroblasts exposed to each of the above stimuli. The PKR genotype did not affect the amount of p38 MAPK (Figure 1A, lower panel) or JNK (Figure 1C, lower panel) relative to total cell protein. The impaired phosphorylation is not an intrinsic defect of SAPKs in PKR-null cells, since various classical SAPK activators (anisomycin, UV irradiation, osmotic shock, sodium arsenite, hydrogen peroxide and heat shock) resulted in comparable activation of these SAPKs in both types of MEFs (Figure 2A and C). IFN-γ or hydrogen peroxide treatment did not activate JNK (Figure 1, lanes 7 and 8, and Figure 2, lanes 9 and 10).

Fig. 1. PKR is required for the activation of p38 and JNK MAPKs by proinflammatory stimuli. Cells were treated with the indicated agonists prior to lysis: lanes 1 and 2, untreated; lanes 3 and 4, 100 µg/ml poly(IC) for 1 h; lanes 5 and 6, 100 ng/ml LPS for 1 h; lanes 7 and 8, 1000 U/ml IFN-γ for 5 min; lanes 9 and 10, 20 ng/ml IL-1β for 15 min; lanes 11 and 12, 10 ng/ml TNF-α for 20 min. Cell lysates (30 µg) were resolved by SDS–PAGE and probed with antibodies as indicated. Results shown are representative of three separate experiments.

Fig. 2. PKR is not required for the activation of p38 and JNK MAPKs by various stress stimuli. Cells were treated with the indicated agonists prior to lysis: lanes 1 and 2, 10 µg/ml anisomycin for 15 min; lanes 3 and 4, 40 J/m2 UV254 followed by 30 min recovery; lanes 5 and 6, 400 mM NaCl for 30 min; lanes 7 and 8, 0.5 mM sodium arsenite for 1 h; lanes 9 and 10, 2 mM H2O2 for 15 min; lanes 11 and 12, 42°C for 45 min; lanes 13 and 14, untreated. Cell lysates (30 µg) were resolved by SDS–PAGE and probed with antibodies as indicated. Results shown are representative of three separate experiments.
The mammalian MAPK is usually activated in a modular three-kinase cascade (Widmann et al., 1999) involving sequentially a MAPK kinase kinase (MKKK), a MAPK kinase (MKK) and the terminal MAPK. While the MKKKs are a heterogeneous group, the MKKs and MAPKs constitute highly homologous families within themselves, and the activator–effector relationships in vivo between each MKK–MAPK couple are better characterized (Widmann et al., 1999). The upstream activators of p38 MAPK are MKK3 and MKK6 (Raingeaud et al., 1996), whose activated forms are recognized by the same phosphospecific antibody due to high homology in their activation segments. Targeted disruption of MKK4 has been shown to obliterate JNK activation (Yang et al., 1997). To determine whether PKR is also a prerequisite for signaling to MKKs, the same lysates from panels A and C in Figures 1 and 2 were used to assess MKK activation. We found that the activation profile for MKK3/6 correlated well with that of p38 MAPK (compare Figure 1A and B), while MKK4 was activated in tandem with JNK (compare Figure 1C and D), consistent with their respective activator–effector relationship. As in the case of JNK, treatment with IFN-γ or hydrogen peroxide did not lead to MKK4 activation. Thus, we conclude that PKR acts upstream of both the MKK3/6–p38 and MKK4–JNK pathways.
Two trends are noteworthy in Figures 1 and 2. First, the PKR-dependent stress stimuli are proinflammatory ligands that bind specifically to distinct receptors for intracellular signaling, i.e. CD14 and toll-like receptor-4 for LPS (Qureshi et al., 1999), PKR for poly(IC) and the respective cytokine receptors for IFN-γ, IL-1β and TNF-α. In contrast, the PKR-independent stress stimuli do not impact solely upon specific receptors, but rather affect cellular components on a global scale. For instance, UV irradiation and hydrogen peroxide cause chemical damage to labile functional groups present on all subcellular components, while heat shock impinges on the conformational stability of most biomolecules. The ‘indiscriminate’ mode of their actions might generate a wide spectrum of downstream effectors such that these stimuli can bypass PKR to activate SAPKs. Thus, the emerging paradigm is that ‘receptor-mediated’ proinflammatory stress stimuli require PKR to activate SAPKs whereas ‘globally acting’ stress stimuli do not. Secondly, while activation of the MKK4–JNK pathway by LPS and poly(IC) is severely compromised in PKR-null MEFs, residual activation is significant with IL-1β or TNF-α (Figure 1C and D). Therefore, additional pathway(s), independent of PKR, are likely to play a major role in cytokine signaling to JNK but a lesser role in the case of poly(IC) or LPS.
Activation of p38 MAPK by poly(IC), LPS and the proinflammatory cytokines in immortalized fibroblast cell lines generated from the MEFs was found to be dependent on PKR, similar to the outcome depicted in Figure 1 (data not shown). However, JNK activation was no longer dependent on PKR in these cell lines regardless of the stress stimuli used (Chu et al., 1999 and data not shown). This suggests that during crisis, molecular changes have occurred in the cells that uncouple MKK4–JNK activation from the PKR regulatory pathway.
Modulation of p38 MAPK activation by the manipulation of PKR levels in immortalized mouse fibroblasts
We reasoned that if PKR plays a direct signaling role, an elevated protein level would lead to enhanced SAPK activation, much as overexpression of a signaling protein can lead to increased reporter activity. When wild-type MEFs were pre-treated overnight with IFN-α, the PKR level was enhanced and this presumably led to a more robust activation of p38 MAPK upon TNF-α treatment (compare lanes 3 and 7 in Figure 3A). However, TNF-α-induced p38 MAPK activation is also enhanced in PKR-null fibroblasts pre-treated with IFN-α (compare lanes 4 and 8 in Figure 3A), implying the presence of other IFN-stimulated gene(s) that can also support p38 MAPK activation. In this regard, immortalized fibroblasts overexpressing RNase L, an IFN-inducible protein, have been shown to exhibit a stronger activation of p38 MAPK in response to viral infection (Iordanov et al., 2000).


Fig. 3. Modulation of p38 MAPK activation through manipulation of the PKR protein level. (A) Wild-type MEFs were pre-treated with 1000 U/ml IFN-α or carrier for 18 h prior to stimulation with 10 ng/ml TNF-α or carrier (Con) for 20 min. Membranes were probed with antibodies as indicated. (B) Immortalized PKR-null fibroblasts, the parental line or expressing either wild-type PKR or K296R mutant, were deprived of tetracycline for 24 h before cells were treated with the indicated agonists: lanes 1–3, 100 ng/ml LPS for 1 h; lanes 4–6, 100 µg/ml poly(IC) for 1 h; lanes 7–9, untreated; lanes 10–12, 10 ng/ml TNF-α for 20 min; lanes 13–15, 10 µg/ml anisomycin for 15 min. Cell lysates (30 µg) were resolved by SDS–PAGE and probed with antibodies as indicated.
To analyze whether the kinase activity of PKR was required for p38 MAPK activation, we engineered immortalized PKR-null fibroblasts to express either wild-type or a kinase-inactive mutant of human PKR (K296R) in a tetracycline-regulated manner. This allowed us to investigate the contribution of the mutant without interference from the endogenous wild-type enzyme. The K296R mutant was confirmed to be devoid of PKR kinase activity by in vitro kinase assay (data not shown). Our results (Figure 3B) suggest that PKR plays a structural role in supporting the activation of p38 MAPK by TNF-α, since the K296R mutant can restore p38 MAPK activation to the same extent as the wild-type enzyme (compare lane 11 with 12). In contrast, poly(IC) and LPS require PKR kinase activity for p38 MAPK activation (Figure 3B, compare lane 2 with 3, and lane 5 with 6). While this is expected for poly(IC), which binds and activates PKR directly, it is surprising for LPS, which does not activate PKR in these cells (data not shown). It is not understood why LPS requires the kinase activity of PKR for p38 MAPK signaling, yet does not result in PKR activation. Anisomycin activates p38 MAPK similarly in all three clones (Figure 3B, lanes 13–15).
Impaired production of proinflammatory cytokines in PKR-null fibroblasts and mice
p38 MAPK was discovered by virtue of its role in mediating the production of proinflammatory cytokines in response to endotoxin (Han et al., 1994; Lee et al., 1994). Subsequently, it was found to regulate the expression of many other proinflammatory genes (Ono and Han, 2000), including other interleukins, chemokines, cell adhesion molecules, matrix metalloproteinases, nitric oxide synthase 2 (NOS2) and cyclooxygenase-2 (COX-2). The pivotal role of p38 MAPK in inflammation suggests that PKR may have a direct impact on the inflammatory response.
The induction of IL-6 and IL-12 p40 mRNA in response to LPS, poly(IC) and encephalomyocarditis virus (EMCV) infection was examined in immortalized murine fibroblasts since these cytokines are inducible in fibroblasts (Chu et al., 1999) and their transcription is, in part, regulated by the p38 MAPK pathway (Lu et al., 1999; Iordanov et al., 2000). IL-6 is a pleiotropic cytokine produced by a wide range of cell types, and is considered a major regulator of the acute phase protein response in the liver (van der Poll and van Deventer, 1998). IL-12, a potent activator of cytotoxic T cells and natural killer (NK) cells, is highly inducible by LPS and serves as an important bridge connecting innate immunity to adaptive immunity (Trinchieri, 1995). IL-6 mRNA expression was attenuated in PKR-null fibroblasts upon LPS, poly(IC) and EMCV treatment (Figure 4A, lanes 1–8), while IL-12 p40 mRNA expression was severely impaired in LPS-treated PKR-null fibroblasts (lanes 9–12). No induction of IL-12 p40 mRNA by poly(IC) or EMCV treatment was observed (data not shown). The reduced IL-6 mRNA levels in PKR-null fibroblasts led to reduced cytokine production in culture supernatants in response to LPS and poly(IC) treatment (Figure 4B). Despite induction of its mRNA, IL-12 p40 was not detected in culture supernatants, probably due to the inability of fibroblasts to process and secrete this protein efficiently compared with immune effectors such as phagocytic cells and antigen-presenting cells (Trinchieri, 1995). Nevertheless, it is evident that the innate immune response in fibroblasts is impaired when PKR is absent.
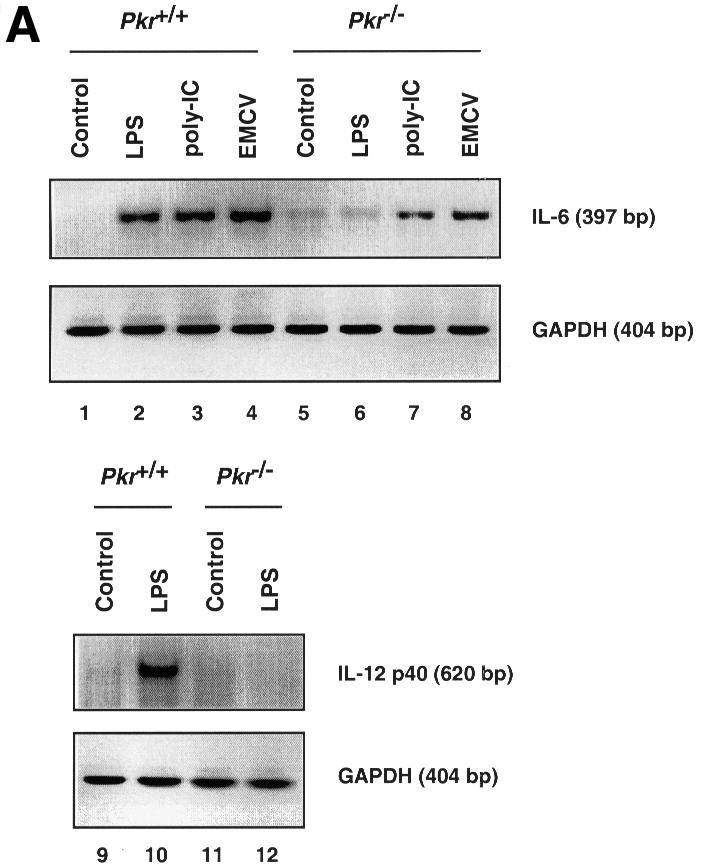



Fig. 4. Differential mRNA expression and cytokine production in PKR wild-type versus PKR-null fibroblasts and mice. (A) Cells in 6-well plates were treated with the indicated agonists prior to RNA extraction: lanes 1, 5, 9 and 11, untreated; lanes 2, 6, 10 and 12, 100 ng/ml LPS for 5 h; lanes 3 and 7, 100 µg/ml poly(IC) for 5 h; lanes 4 and 8, EMCV at an m.o.i. of 5 for 5 h. RT–PCR was performed and the UV-visualized image was inverted after scanning. (B) Cells (1 × 106) in 2 ml of medium in 6-well plates were treated with the indicated agonists for 9 h before culture supernatant was collected by centrifugation. Results shown are representative of two experiments. (C) Mice were injected with 100 µg of LPS and bled at 2 h interval (n = 2). Results shown are representative of two experiments. (D) Mice were injected with 10 µg of LPS and bled at 2 h interval (n = 2). Results shown are representative of two experiments.
Although fibroblasts participate in inflammatory processes and non-specific immunity by producing cytokines and mediators in response to LPS, their phenotype may not be representative of specific immune cell types as reflected by the lack of IL-12 secretion. Furthermore, the absence of CD14 in fibroblasts implies an LPS signaling pathway independent of this co-receptor (Tominaga et al., 1997). To determine the effects of PKR deletion at the systemic level, mice were challenged with i.p. injection of LPS and monitored for serum levels of IL-6 and IL-12 p40. The production of both cytokines was reduced in PKR-null mice compared with wild-type animals (Figure 4C and D). Consistent with reports in the literature (Engelberts et al., 1991), IL-6 production peaked at the 2 h time point while that of IL-12 p40 peaked 2 h later. We conclude that PKR is required for the maximal expression of these interleukins in a physiological context of endotoxin challenge.
Recent work has demonstrated an essential role for PKR in mediating the activation of NF-κB [by poly(IC)] (Zamanian-Daryoush et al., 2000), STAT1 (by IFN-γ) (Ramana et al., 2000) and ATF-2 [by poly(IC) and TNF-α] (Bandyopadhyay et al., 2000), which are transcription factors important for an inflammatory response to the respective agents. For example, TNF-α and poly(IC) induction of E-selectin, an adhesion molecule that facilitates leukocyte rolling during inflammation, is impaired in PKR-null endothelial cells presumably due to the defective activation of NF-κB and ATF-2 (Bandyopadhyay et al., 2000). We can thus formulate a model whereby PKR signals downstream to p38 MAPK to regulate the activity of these transcription factors (Figure 5). In accord with this model, ATF-2 is an established substrate of p38 MAPK (Widmann et al., 1999), STAT1 transcriptional activity is attenuated by the inhibition of p38 MAPK (Goh et al., 1999) and p38 MAPK activity has been demonstrated to be required for NF-κB-dependent transcription (Schulze-Osthoff et al., 1997). While the role of JNK in these pathways remains to be established, its activation was not impaired in immortalized PKR-null fibroblasts (Chu et al., 1999 and data not shown) despite their defect in LPS response. In conclusion, our findings reveal p38 MAPK as a critical link between PKR and its recently recognized proinflammatory properties, suggesting a potentially important role for PKR in the innate antibacterial response.

Fig. 5. A role for PKR in regulating p38 MAPK, its downstream targets and the induction of proinflammatory genes.
Materials and methods
Reagents and antibodies
LPS (from Escherichia coli serotype 0111:B4), poly(IC), anisomycin, sodium arsenite and hydrogen peroxide were obtained from Sigma Chemical Co. (St Louis, MO). Murine IFN-α, TNF-α, IL-1β and IFN-γ were obtained from R&D Systems (Minneapolis, MN). Antibodies were obtained from Santa Cruz Biotechnology (p38 MAPK, MKK6, JNK and MKK4) and New England Biolabs (phos-p38, phos-MKK3/6, phos-JNK and phos-MKK4). PKR antibody has been described previously (Zamanian-Daryoush et al., 2000).
Cell culture
MEFs were isolated from genotyped littermates as described previously (Der et al., 1997) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal calf serum (FCS; Gibco-BRL), 100 U/ml penicillin and 100 µg/ml streptomycin. Immortalized fibroblasts were generated from MEFs according to a previously described protocol (Yang et al., 1995). Unless otherwise specified, cells were grown in 10 cm dishes to ∼80% confluence and used in stress treatment without prior serum starvation.
Cell lysis, SDS–PAGE and immunoblotting
Cell lysates were prepared as previously described (Goh et al., 1999). A 30 µg aliquot of protein was resolved by 10% SDS–PAGE and electroblotted onto a PVDF membrane. Incubation with antibodies and immunodetection (Amersham ECL system) were performed according to the manufacturer’s instructions.
Vector construction
The ptTA-GPT vector was obtained from Dr M.S.Schlissel (Sheehy and Schlissel, 1999). The ptV5 vector was constructed by excising the pEF promoter from pEF6/His C (Invitrogen) by digestion with MluI and EcoRI. The ptetO promoter was excised from the pTet-Splice vector (Gibco-BRL) by digestion with BssHII and BamHI. This fragment containing the tetO promoter was gel purified. The V5 epitope was added by annealing two oligonucleotides: V5BamHI (GATCTCGCCCACCATGGCAGCTGGTAAGCCTATCCCTAACCCTCTCCTCGGTCTCGATTCTACGGGATCCG) and V5EcoR1 (AATTCGGATCCCGTAGAATCGATACCGAGGAGAGGGTTAGGGATAGGCTTACCAGCTGCCATGGTGGGCGA). The oligonucleotides were annealed in 1× ligation buffer (Gibco-BRL) and then ligated to the purified tetO fragment from above. This was then ligated to the pEF6/His C plasmid digested with MluI and EcoRI. The full coding region from both the wild-type and mutant PKR cDNA templates was amplified using the PCR with the following primer pair: PKRBamFWD (AACTACTGGATCCATGGCTGGTGATCTTTCAGCAGGTTTC) and PKRNotREV (AATACATCTGCGGCCGCGGTAAATATCTATTGATATTCCCTAGC). The PCR was performed with the elongase kit according to the manufacturer’s instructions (Gibco-BRL). Amplified DNA was digested with BamHI and cloned into dephosphorylated BamHI-digested ptV5 vector. The final plasmid construct containing the wild-type PKR cDNA was designated ptV5PKR and the mutant construct was designated ptV5K296R. All positive clones were identified by restriction digestion and confirmed by DNA sequencing.
Generation of stable cell lines expressing PKR
Immortalized fibroblasts derived from PKR-null mice were transfected with the ptTA-GPT plasmid using Lipofectamine (Gibco-BRL) and stable cell lines were selected using the selection strategy outlined in Mulligan and Berg (1981). Once cell lines expressing the tTA protein were successfully established, two of these lines were transfected with either ptV5PKR or ptV5K296R and stable cell lines were selected in the presence of 2 µg/ml blasticidin (Invitrogen). Individual cell clones were isolated by limited dilution and selection of individual blasticidin-resistant colonies using sterilized glass cloning rings (Bellco Glass, Vineland, NJ). Expression of PKR was analyzed from clones grown in medium containing either 1 µg/ml tetracycline (Sigma) or no tetracycline. Inducible expression was confirmed by western blotting (data not shown) using the V5 epitope antibody (Invitrogen). Cell lines expressing either wild-type or mutant PKR in a tetracycline-inducible fashion were used for further analysis.
RNA preparation and RT–PCR
Experiments were done in 6-well plates. Total RNA was prepared using the High Pure RNA isolation kit (Boehringer Mannheim) according to the manufacturer’s instructions. A 10 ng aliquot of total RNA was used as template in a 20 µl reaction using the SuperScript One-Step RT–PCR System (Gibco-BRL). The RT–PCR conditions are: for GAPDH, forward primer 5′-CCTCAACTACATGGTCTAC-3′, reverse primer 5′-CCTTCCACAATGCCAAAGT-3′, 30 cycles at 94°C for 30 s, 52°C for 30 s and 72°C for 1 min; for IL-6, forward primer 5′-TTGCCTTCTTGG GACTGATG-3′, reverse primer 5′-CTGAAGGACTCTGGCTTTGT-3′, 30 cycles at 94°C for 30 s, 56°C for 30 s and 72°C for 1 min; for IL-12 p40, forward primer 5′-GAGGTGGACTGGACTCCCGA-3′, reverse primer 5′-CAAGTTCTTGGGCGGGTCTG-3′, 35 cycles at 94°C for 30 s, 60°C for 30 s and 72°C for 1 min.
Animals and treatments
Homozygous PKR-null mice (Yang et al., 1995) were backcrossed 10 generations onto the C57BL/6 background. Control C57BL/6 mice were obtained from Taconic Farms, Inc. Seven- to nine-week-old sex-matched mice were studied. Mice were given i.p. injections of 100 µl of LPS [either 0.1 or 1 mg/ml in phosphate-buffered saline (PBS)] or vehicle. Approximately 200 µl of blood was obtained by retro-orbital puncture at each time point. Two animals were sampled per time point per treatment. Animal handling protocols conform to guidelines of the NIH.
Cytokine measurements
The levels of murine IL-6 and IL-12 p40 present in sera and culture supernatants were determined using ELISA kits from R&D Systems as per the manufacturer’s instructions.
Acknowledgments
Acknowledgements
We are grateful to Drs Robert Silverman and Andrew Larner for helpful comments on the manuscript. This work was supported by grants to B.R.G.W. from the National Institutes of Health (P01-CA62220 and AI34039).
References
- Bandyopadhyay S.K., de la Motte,C.A. and Williams,B.R.G. (2000) Induction of E-selectin expression by dsRNA and TNF-α is attenuated in murine aortic endothelial cells derived from PKR-null mice. J. Immunol., 164, 2077–2083. [DOI] [PubMed] [Google Scholar]
- Chu W.M., Ostertag,D., Li,Z.W., Chang,L., Chen,Y., Hu,Y., Williams,B., Perrault,J. and Karin,M. (1999) JNK2 and IKKβ are required for activating the innate response to viral infection. Immunity, 11, 721–731. [DOI] [PubMed] [Google Scholar]
- Clemens M.J. and Elia,A. (1997) The double-stranded RNA-dependent protein kinase PKR: structure and function. J. Interferon Cytokine Res., 17, 503–524. [DOI] [PubMed] [Google Scholar]
- Conrad P.W., Rust,R.T., Han,J., Millhorn,D.E. and Beitner-Johnson,D. (1999) Selective activation of p38α and p38γ by hypoxia. Role in regulation of cyclin D1 by hypoxia in PC12 cells. J. Biol. Chem., 274, 23570–23576. [DOI] [PubMed] [Google Scholar]
- Der S.D., Yang,Y.L., Weissmann,C. and Williams,B.R. (1997) A double-stranded RNA-activated protein kinase-dependent pathway mediating stress-induced apoptosis. Proc. Natl Acad. Sci. USA, 94, 3279–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelberts I., von Asmuth,E.J., van der Linden,C.J. and Buurman,W.A. (1991) The interrelation between TNF, IL-6 and PAF secretion induced by LPS in an in vivo and in vitro murine model. Lymphokine Cytokine Res., 10, 127–131. [PubMed] [Google Scholar]
- Gale M. Jr and Katze,M.G. (1998) Molecular mechanisms of interferon resistance mediated by viral-directed inhibition of PKR, the interferon-induced protein kinase. Pharmacol. Ther., 78, 29–46. [DOI] [PubMed] [Google Scholar]
- Goh K.C., Haque,S.J. and Williams,B.R. (1999) p38 MAP kinase is required for STAT1 serine phosphorylation and transcriptional activation induced by interferons. EMBO J., 18, 5601–5608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J., Lee,J.D., Bibbs,L. and Ulevitch,R.J. (1994) A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science, 265, 808–811. [DOI] [PubMed] [Google Scholar]
- Iordanov M.S., Paranjape,J.M., Zhou,A., Wong,J., Williams,B.R., Meurs,E.F., Silverman,R.H. and Magun,B.E. (2000) Activation of p38 mitogen-activated protein kinase and c-Jun N-terminal kinase by double-stranded RNA and encephalomyocarditis virus: involvement of RNase L, protein kinase R and alternative pathways. Mol. Cell. Biol., 20, 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki H., Morooka,T., Shimohama,S., Kimura,J., Hirano,T., Gotoh,Y. and Nishida,E. (1997) Activation and involvement of p38 mitogen-activated protein kinase in glutamate-induced apoptosis in rat cerebellar granule cells. J. Biol. Chem., 272, 18518–18521. [DOI] [PubMed] [Google Scholar]
- Kumar A., Yang,Y.L., Flati,V., Der,S., Kadereit,S., Deb,A., Haque,J., Reis,L., Weissmann,C. and Williams,B.R. (1997) Deficient cytokine signaling in mouse embryo fibroblasts with a targeted deletion in the PKR gene: role of IRF-1 and NF-κB. EMBO J., 16, 406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.C. et al. (1994) A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature, 372, 739–746. [DOI] [PubMed] [Google Scholar]
- Lu H.T., Yang,D.D., Wysk,M., Gatti,E., Mellman,I., Davis,R.J. and Flavell,R.A. (1999) Defective IL-12 production in mitogen-activated protein (MAP) kinase kinase 3 (Mkk3)-deficient mice. EMBO J., 18, 1845–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R. and Janeway,C.A.,Jr (1997) Innate immunity: the virtues of a nonclonal system of recognition. Cell, 91, 295–298. [DOI] [PubMed] [Google Scholar]
- Mulligan R.C. and Berg,P. (1981) Selection for animal cells that express the Escherichia coli gene coding for xanthine–guanine phosphoribosyltransferase. Proc. Natl Acad. Sci. USA, 78, 2072–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono K. and Han,J. (2000) The p38 signal transduction pathway: activation and function. Cell Signal., 12, 1–13. [DOI] [PubMed] [Google Scholar]
- Qureshi S.T., Gros,P. and Malo,D. (1999) Host resistance to infection: genetic control of lipopolysaccharide responsiveness by toll-like receptor genes. Trends Genet., 15, 291–294. [DOI] [PubMed] [Google Scholar]
- Raingeaud J., Gupta,S., Rogers,J.S., Dickens,M., Han,J., Ulevitch,R.J. and Davis,R.J. (1995) Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J. Biol. Chem., 270, 7420–7426. [DOI] [PubMed] [Google Scholar]
- Raingeaud J., Whitmarsh,A.J., Barrett,T., Derijard,B. and Davis,R. (1996) MKK3- and MKK-6-regulated gene expression is mediated by the p38 MAPK signal transduction pathway. Mol. Cell. Biol., 16, 1247–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramana C.V., Grammatikakis,N., Chernov,M., Nguyen,H., Goh,K.C., Williams,B.R. and Stark,G.R. (2000) Regulation of c- myc expression by IFN-γ through Stat1-dependent and -independent pathways. EMBO J., 19, 263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze-Osthoff K., Ferrari,D., Riehemann,K. and Wesselborg,S. (1997) Regulation of NF-κB activation by MAP kinase cascade. Immunobiology, 198, 35–49. [DOI] [PubMed] [Google Scholar]
- Sheehy A.M. and Schlissel,M.S. (1999) Overexpression of RelA causes G1 arrest and apoptosis in a pro-B cell line. J. Biol. Chem., 274, 8708–8716. [DOI] [PubMed] [Google Scholar]
- Shrode L.D., Rubie,E.A., Woodgett,J.R. and Grinstein,S. (1997) Cytosolic alkalinization increases stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) activity and p38 mitogen-activated protein kinase activity by a calcium-independent mechanism. J. Biol. Chem., 272, 13653–13659. [DOI] [PubMed] [Google Scholar]
- Stark G.R., Kerr,I.M., Williams,B.R.G., Silverman,R.H. and Schreiber,R.D. (1998) How cells respond to interferons. Annu. Rev. Biochem., 67, 227–264. [DOI] [PubMed] [Google Scholar]
- Tibbles L.A. and Woodgett,J.R. (1999) The stress-activated protein kinase pathways. Cell. Mol. Life Sci., 55, 1230–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga K., Kirikae,T. and Nakano,M. (1997) Lipopolysaccharide (LPS)-induced IL-6 production by embryonic fibroblasts isolated and cloned from LPS-responsive and LPS-hyporesponsive mice. Mol. Immunol., 34, 1147–1156. [DOI] [PubMed] [Google Scholar]
- Trinchieri G. (1995) Interleukin-12: a proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu. Rev. Immunol., 13, 251–276. [DOI] [PubMed] [Google Scholar]
- van der Poll T. and van Deventer,S.J. (1998) The role of interleukin 6 in endotoxin-induced inflammatory responses. Prog. Clin. Biol. Res., 397, 365–377. [PubMed] [Google Scholar]
- Widmann C., Gibson,S., Jarpe,M.B. and Johnson,G.L. (1999) Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol. Rev., 79, 143–180. [DOI] [PubMed] [Google Scholar]
- Williams B.R.G. (1997) Role of the double-stranded RNA-activated protein kinase in cell regulation. Biochem. Soc. Trans., 25, 509–513. [DOI] [PubMed] [Google Scholar]
- Yang D., Tournier,C., Wysk,M., Lu,H.T., Xu,J., Davis,R.J. and Flavell,R.A. (1997) Targeted disruption of the MKK4 gene causes embryonic death, inhibition of c-Jun NH2-terminal kinase activation and defects in AP-1 transcriptional activity. Proc. Natl Acad. Sci. USA, 94, 3004–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.L., Reis,L.F., Pavlovic,J., Aguzzi,A., Schafer,R., Kumar,A., Williams,B.R., Aguet,M. and Weissmann,C. (1995) Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO J., 14, 6095–7106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian-Daryoush M., Mogensen,T.H., DiDonato,J.A. and Williams,B.R.G. (2000) NF-κB activation by the dsRNA-activated protein PKR is mediated through the NF-κB inducing kinase and IκB kinase. Mol. Cell. Biol., 20, 1278–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]