

Abstract
Toll-like receptor 2 (TLR2) plays a key role in the host defense against Gram staining positive (Gram+) bacteria and their cell wall envelope components. However, little is known about the expression of TLR2 in the middle ear under otitis media (OM) conditions, and its role in the persistent otitis media with effusion (OME). In this study, we demonstrated that the pneumococcal cell wall component, peptidoglycan-polysaccharides (PGPS), activated the expression of TLR2 in the middle ear epithelial cells through the nuclear factor kappa B (NF-κB)-cytokine signaling pathway while I kappa B alpha mutant (IκBαM), a dominant negative inhibitor of NF-κB, abrogated the expression of TLR2 induced by PGPS. This study suggests that the existence of residual PGPS may maintain a low profile of cytokine production in the middle ear mucosa and thus contribute to the pathogenesis of OME.
Peptidoglycan-polysaccharides (PGPS) are cell envelope components of Gram staining positive (Gram+) bacteria including Streptococcus pneumoniae, a frequent cause of otitis media (OM) in young children (1) and pneumonia in the elderly (2). Antibiotic application kills S. pneumoniae and leads to release of PGPS, which causes otitis media with effusion (OME), a common condition without active bacterial growth but has a low profile of inflammation in the middle ear cleft on a long-term basis. PGPS is resistant to host enzyme digestion by DNases, RNases, and proteases during purification (3), suggesting durability, and it may serve as a persistent stimulus factor in the middle ear cleft. Consistent with the biochemical property, PGPS is long present in middle ear effusion in humans (3). Other components such as proteins, enzymes, and polysaccharides are present in the middle ear cavity for a limited period of time because they are susceptible to host enzyme digestion. Therefore, PGPS-induced immune and inflammatory responses are thought as persistent events in OME, which causes a low profile of chronic inflammation in the middle ear mucosa.
Toll-like receptors (TLRs) are present on the surface of many cell types including epithelial cells, macrophages, monocytes, dendritic cells, lymphocytes, vascular endothelial cells, cardiac myocytes, and adipocytes (4). There are 13 TLR paralogs in mammals that have been found, but TLR2 is the one that recognizes a variety of Gram+ bacterial products such as peptidoglycan, lipoteichoic acid, and lipoarabinomannan, which responds to factors released by Gram staining negative (Gram−) bacteria including nontypeable Hemophilus influenzae (NTHi) (5), coccobacilli, N. meningitidis (6), and the Mycoplasma lipopeptides (7), so-called pathogen-associated molecular patterns (PAMPs). Knockout of TLR2 in mice decreased survival of animals against Gram+ bacterium Staphylococcus aureus (7,8), suggesting that TLR2 plays an important role in the host defense system by activating immune and inflammatory cells. TLR2 is, therefore, recognized as a cell surface receptor for mediating inflammatory responses in the middle ear in the presence of Gram+ metabolites such as cell wall components.
It is known that TLR2 mediates immune and inflammatory reactions through NF-κB in host cells (9). However, it is not clear whether it plays a role in the pathogenic continuum of OM through a self-regulatory mechanism. In this study, we demonstrated that pneumococcal PGPS activates NF-κB via the TLR2 receptor in middle ear epithelial cells (MEECs), and then NF-κB, in turn, up-regulated the expression of TLR2 in a positive feedback manner. This perhaps provides an experimental model for OME in the middle ear mucosa in response to bacterial residual challenges.
MATERIALS AND METHODS
Materials
All human and animal specimens used in this study were procured, handled, and maintained according to the protocols approved by the Institutional Review Board (IRB) and the Institutional Animal Care and Use Committee (IACUC) at the University of Minneosta. The NF-κB subunit (p65) cDNA was a gift from Dr. Frank G. Ondrey at the University of Minnesota, Minneapolis, MN. I kappa B alpha mutant (IκBαM), a gift from Dr. Inder Verma at Salk Institute, was dominant negative NF-κB (10).
Cell culture
The rat and mouse MEEC lines, established in our laboratory from normal rat and mouse middle ear mucosal epithelia with temperature-sensitive simian vacuolating virus 40 (SV40), were used in this study. Cells were maintained in Ham’s F12 medium (ATCC) and supplements (hereafter referred to as full growth medium, FGM) as previously described (11).
q-PCR
RNA was isolated using the Mini-prep RNA isolation kit (Stratagene). Quantitative PCR (qPCR) was performed as previously described (12). Primers pairs used were as follows: mouse TLR2 (5′-gaatcacagtagagaa-cagca a-3′/5′-gcaagaacaaagaaa atgagtc-3′, 410 bp).
Immunohistochemistry
Human archival Eustachian tube sections were deparaffinized in xylene and rehydrated in graded ethanol routinely. Cells were cultured on eight-chamber slides, incubated with and without PGPS at 0.5 μg/mL for 30 min, fixed with methanol (100%), and stored at −30°C before use. Tissue sections and cell culture slides were incubated with anti-TLR2 (eBioscience) or an “activated” form of p65 from Chemicon (MAB 3026, 1:10 dilution). Primary antibodies were detected with diaminobenzidine (DAB) tetrahydrochloride or FITC and tetramethylrhodamine isothiocyanate (TRITC).
Western blot
For p65 cDNA transfection, mouse MEEC cells were cultured up to 60% confluence and incubated with transfected with NF-κB subunit p65 and empty vector for 8 h in serum-free Opti-MEM transfection medium (Invitrogen, CA) containing 6 μg/mL of Lipofectamine (hereafter referred to as transfection medium), recovered in FGM for 24 h. For challenging with various factors, cells were incubated with PGPS at 0.5 μg/mL, lipopolysaccharide (LPS) at 0.1 μg/mL, and TNF-α at 100 ng/mL for 6 h and then harvested for protein isolation from the cytosol and membranes. Briefly, 40 μg of total protein were loaded, separated by electrophoresis, probed by a specific TLR antibody (anti-TLR2, 1:1000 dilution, eBioscience, CA) and reprobed by a polyclonal glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody (1:2000 dilution, Abcam). An ECL kit (Amersham Biosciences) was used to detect specific protein bands.
ELISA
Mouse MEEC and human bronchial epithelial cells (NL-20) were treated with a variety of factors as above at different time points and then harvested for protein isolation. ELISA procedure was performed as previously described (13). Briefly, specific antibody (anti-TLR2, 1:500 dilution, eBioscience) was coated onto the 96 well plates (MaxiSorp, Nunc, IL) and blocked with 10% skim milk at room temperature for 2 h. The total protein from MEEC and NL-20 cells was added to the coated wells for 2 h, then incubated with secondary antibody conjugated to biotin (anti-TLR2, 1:500 dilution, eBioscience) for 2 h, and then incubated with streptavidin-HRP (1:2000 dilution, Zymed) for 1 h. Substrate (TMB, Zymed) was added to each well for 15 min. After the addition of H2SO4 (2N), plates were read at 450 nm on a Microplate ELISA reader (Spectra Classic, TECAN U.S. Inc, NC). Similarly, TLR2 protein expression by PGPS and LPS in the presence of NF-κB dominant negative inhibitor (IκBαM) was performed.
Luciferase assays
Cells at 60% confluence were transfected with NF-κB luciferase reporters at 1.4 μg/mL and cotransfected with β-gal reporters (internal controls) at 0.6 μg/mL for 8 h and recovered in the complete medium for 24 h. After that, cells were incubated with PGPS from 0.125 to 0.5 μg/mL for 6 h and cells with PGPS without incubation served as controls. To study whether PGPS is mediated through NF-κB, IκBαM, a dominant negative NF-κB, was used for canceling out the NF-κB activity induced by PGPS at 0.5 μg/mL. Cells were treated with PGPS at 0.5 μg/mL for 8 h after reporter gene transfection and recovery. Luciferase activity was measured, as previously described (14). The activities of luciferase reporters are presented by a relative luciferase-to-β-galactosidase reporter activity (e.g., relative luciferase activity, RLA). In a similar way, TLR2 luciferase assays were performed as above. The TLR2 promoter sequence from −5660 to 5400 for luciferase assays was constructed as previously described (12) by using the following compound primers (5′-atggccggtaccccgcccgcaagc accatc-3′/5′-gtacgggtattgt-tcttcgaaccgctc-3′), which contain the both KpnI and HindIII endonuclease restriction sites for subcloning into pGL4 plasmids.
Fluorescent activated cell sorting
Cell cultures (60% confluence) were challenged with PGPS at 0.5 μg/mL for 24 h and harvested for the evaluation of positive cells, as previously described. Briefly, cells were washed with PBS, harvested by trypsinization, preincubated with 0.3% saponin in PBS for 10 min, and then incubated the following primary and secondary antibodies sequentially: an “activated” form of p65 from Chemicon (MAB 3026, 1:100 dilution). Nonspecific IgG was used as an antibody control. Cells without any stain served as blank controls. Results are presented as a % of positive cells in a total of 10,000 cells counted per sample.
Microarray data analysis
Affymetrix microarrays on the middle ear mucosa of rats with and without Pneumococci (Pn6A), NTHi, and Eustachian tube obstruction (ETO) were performed similarly as previously described (15). Briefly, cDNA was prepared from 20 μg of total RNA using a T7-dT24 primer. cRNA was synthesized from cDNA and biotinylated using the BioArray High Yield RNA Transcript Labeling Kit (Enzo Diagnostics, Farmingdale, NY) and hybridized with the Rat U34 arrays (Affymetrix). The intensity of each gene expression for the U34 arrays was determined by GeneSifter after normalization and is presented as RLU. A pathway analysis tool module (PEGG) was used for the evaluation of signaling pathways. The results were presented as “Z-scores” that weigh the gene activity of the entire pathway (how many genes changed and to what extent they have changed in a specific pathway) and give scores for up-regulated and down-regulated genes separately. Once the entire pathway was analyzed, important genes involved in TLR signaling were individually analyzed using t test embedded in GeneSifter software, and data are presented as individual heat maps or merged heat maps with P values. Similarly, human NL-20 was incubated with PGPS at 0.5 μg/mL for 8 h, harvested for Affymetrix microarrays (human U34), and evaluated for changes in the expression of TLRs using the same method as above.
Statistical analysis
The t test was used for evaluation of differences between experiments and controls. ANOVA was used when there is a multiple sample comparison. P values <0.05 were considered significant.
RESULTS
TLR2 is expressed in mouse and human middle ear epithelia
To study the expression profile of TLRs in the upper respiratory tract epithelial cells, we examined the expression of TLRs in the human tracheal epithelial cells (NL-20) by Affymetrix microarrays. As expected, TLR1–TLR9 were detected in the NL-20 cells (Fig. 1A). To investigate which TLRs respond to pneumococcal cell wall component PGPS, cells were challenged with PGPS at 0.5 μg/mL for 8 h and harvested to evaluate the TLR expression by microarray as described above. It was found that TLR2, but not others, was significantly up-regulated by PGPS. We therefore focused on the regulation of TLR2 in this study. To verify the expression of TLR2 in OM, the middle ear mucosa with chronic OM, Eustachian tube tissue exposed to pathogens, and cultured mouse MEEC were stained with TLR2 antibody. It was found that the human Eustachian tube and middle ear with OM as well as mouse MEEC were positive for TLR2 protein expression (Fig. 1B). The expression of TLR2 was not detectable in the normal human middle ear epithelium by immunohistochemistry (data not shown).
Figure 1.

Expression of the TLR profile in the human and mouse MEECs. Affymetrix microarray showed the expression of TLR1–TLR9 in the human NL-20 cells and TLR2 but not other TLRs significantly responded to the challenge of PGPS (gray bar) at 0.5 μg/mL for 8 h compared with PBS, black bar (A). Immunohistochemistry demonstrated that TLR2 was expressed in the human Eustachian tube epithelium (left, B, arrows pointing to the cellular membrane and the cytosol being full of mucous granules which are unstained and white in color), diseased middle ear mucosa (middle, B), and cultured mouse MEEC cells. Upper row in B, nonspecific IgG stain as controls for the lower row TLR2 stain; RLU, relative light unit; Scale bar = 20 μM applying to all panels in B; *p < 0.05, n = 3.
PGPS regulates the expression of TLR2 in mouse MEEC cells
The expression of TLR2 was detected in cultured mouse MEEC cells by qPCR (Fig. 2A, left). However, the level of TLR2 protein is fairly low, not detectable by Western blot (Fig. 2B). It is questionable whether such a low level of TLR2 is sufficient for mediating immune and inflammatory reactions. Therefore, we studied the regulation of TLR2. As shown in Fig. 2A, PGPS increased the expression of TLR2 mRNA transcripts at 24 h in a dose-dependent manner. The data showed that PGPS increased the expression of TLR2 ~24 h after the incubation (Fig. 2A, right) or at ~30 h after transfection with p65 (Fig. 2B, lane 3). Cytokine incubation for 6 h showed little increased expression of TLR2 (Fig. 2B, lanes 4 – 6). To study whether this is true to animal models, Affymetrix microarray data were retrieved from our previous studies. It was found that Pneumococci (Pn6A) increased the expression of TLR2 mRNA transcripts in the middle ear mucosa (Fig. 2C). ELISA analysis confirmed that TLR2 was significantly up-regulated by bacterial metabolites such as PGPS and LPS and inflammatory mediators such as TNF-α and NF-κB subunit p65 (Fig. 2D). Similarly, TLR2 was significantly up-regulated by pneumococcal cell wall component PGPS and corresponding cytokines in human tracheal epithelial cells (NL-20, Fig. 2E). To study whether the TLR2 regulation is via NF-κB, cells were incubated with PGPS (0.5 μg/mL) in the presence of NF-κB dominant negative inhibitor (IκBαM) for 6 h and harvested for ELISA. It was found that IκBαM significantly inhibited the TLR2 protein expression compared with its empty vector control (Fig. 2F). To study whether PGPS regulates TLR2, luciferase assays were performed. It was demonstrated that PGPS significantly increased the promoter activity of TLR2 compared with its control (Fig. 2G).
Figure 2.

Pneumococci and their metabolites increased the expression of TLRs in vitro and in vivo. qPCR demonstrated that PGPS at 0.5 μg/mL significantly up-regulated the expression of TLR2 mRNA transcripts in mouse MEEC cells compared with control (A, p < 0.05, n = 3). Western blot analysis demonstrated that PGPS at 0.5 μg/mL, LPS at 0.1 μg/mL, TNF-α at 100 ng/mL, and p65 at 1.4 μg/mL up-regulated the expression of TLR2 at 6 h after recovery (B). Experimental OM verified that Pn6A significantly increased the expression level of TLR2 mRNA transcripts 3–14 d after bullar infection with Pneumococci (Pn6A) compared with those in controls using Affymetrix microarrays (C). ELISA demonstrated that bacterial products (PGPS and LPS) and inflammatory mediators (TNF-α and p65) at the same concentrations as above significantly up-regulated the expression of TLR2 protein in mouse MEEC (D, *p < 0.05, **p < 0.01, n = 3) and NL-20 human bronchial epithelial cells (E, *p < 0.05, **p < 0.01, §p < 0.001, n = 3). IκBαM (gray bar) significantly inhibited the TLR2 protein expression induced by PGPS and LPS compared with control, black bar (F, *p < 0.05, n = 3). PGPS at 1.25 μg/mL significantly increased the luciferase activity of TLR2 compared with its control PBS (G, p < 0.01, n = 9). ev, empty vector; Ctrl, control.
PGPS regulates the activity of NF-κB in Mouse MEEC cells
To study which transcriptional factor regulates the expression of TLR2, mouse MEEC cells were treated with and without PGPS for 6 h in a dose-dependent manner from 0.125 to 0.5 μg/mL and harvested for analyses of luciferase assays. It was demonstrated that PGPS increased the promoter activity of NF-κB in a dose-dependent manner and PGPS at 0.5 μg/mL significantly increased the promoter activity of NF-κB by luciferase assays (Fig. 3A). To study whether PGPS activate the NF-κB protein and translocate into cellular nuclei, fluorescent activated cell sorting (FACS) and immunohistochemistry were performed using an antibody against “activated” NF-κB subunit p65. As expected, PGPS at 0.5 μg/mL for 6 h significantly increased the cell numbers with “activated” p65 by FACS (Fig. 3B) and increased the translocation of “activated” NF-κB into the nuclei by immunohistochemistry (Fig. 3D, arrows) compared with no PGPS (Fig. 3C). IκBαM, a dominant negative NF-κB, significantly reduced the promoter activity of NF-κB induced by PGPS (Fig. 3E). Affymetrix microarray data demonstrated that Pn6A significantly increased the expression of NF-κB mRNA transcripts in the middle ear mucosa of rats (Fig. 3F). This suggests that Pneumococci not only increase the activity of NF-κB but also increase the transcripts of NF-κB.
Figure 3.
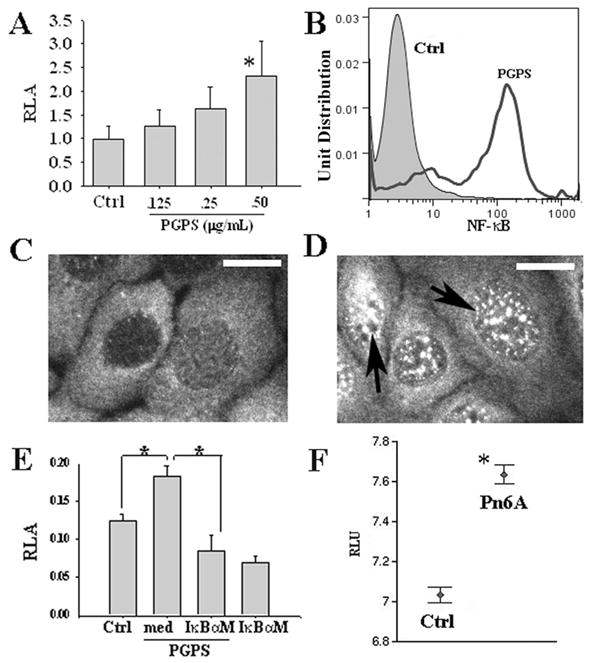
PGPS induced the activity of NF-κB in mouse MEEC cells. PGPS at 0.5 μg/mL significantly increased the promoter activity of NF-κB compared with control (Ctrl) by luciferase assays (A, p < 0.05, n = 6). PGPS at 0.5 μg/mL significantly increased the percentage of activated NF-κB positive cells compared with PBS by FACS (B). PGPS incubation for 30 min increased the “activated” NF-κB translocation into the nuclei (D, black arrows) compared with control (C, bar = 5 μm) by immunohistochemistry. IκBαM significantly inhibited the promoter activity of NF-κB by PGPS at 0.5 μg/mL (E, *p < 0.05, n = 6). Note that IκBαM alone reduced the luciferase activity of NF-κB in nonPGPS treated cells. Pn6A infection in the middle ear of rats increased the expression of NF-κB mRNA transcripts 3–14 d after Pn6A infection in the middle ear of rats (F). *p < 0.05.
Pn6A regulates the TLR signaling pathway
To study the influence of Pn6A on immune and inflammatory responses, the entire TLR signaling pathway activity in the middle ear mucosa with and without Pn6A challenging was evaluated by using Affymetrix microarrays (Pn6A versus PBS from 3 to 14 d). Overall, Pn6A increased the TLR signaling pathway activity at a Z-score of 3.85 (Fig. 4A), which is the highest one in all the pathways studied. There were some genes whose activity was down-regulated in Pn6A-treated ears (scored at 0.37) when compared with PBS. Specifically, among the 33 genes listed in this array (U34A), there were nine genes up-regulated (Fig. 4B: D50558, J04423, L26267, M26744, M98820, AA800678, AI171959, U22414, and X63594, standing for membrane protein, NF-κB p105, IL-1β, IL-6, MIP-1 α, and IκBα) and two genes down-regulated (D64046, PI3 K beta unit, and MAPK14). In contrast, Pn6A did not up-regulate the MAPK signaling pathway much (Z-scores = 1.07 for up-regulation and −0.08 for down-regulation). The data are presented in Fig. 4B.
Figure 4.
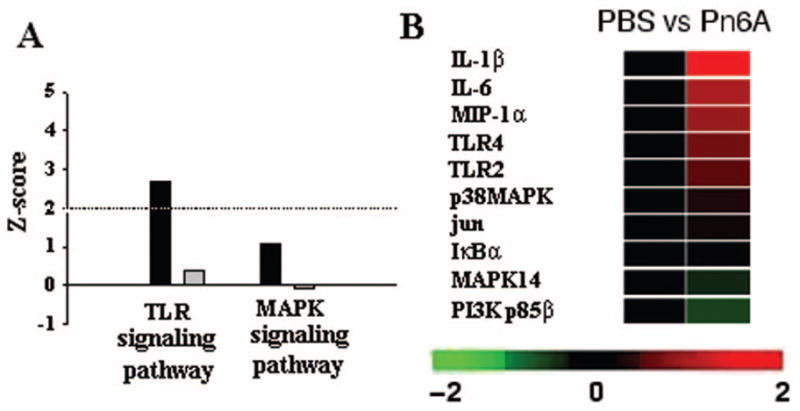
TLR signaling pathway in pneumococcal otitis media of rats. All genes involved in the TLR signaling pathway were evaluated by GeneSifter using Z-score as a parameter. Pn6A vs. PBS control had Z-scores (2.41 for up-regulated genes and −1.85 for down-regulated genes) of TLR signaling pathway (A, black bar, up-regulated; gray bar, down-regulated). Similar changes were observed in the MAPK signaling pathway. Dotted line in A indicates a normal range for Z-scores (A). Microarray heat maps demonstrated that the TLR signaling pathway genes and related genes were up-regulated (IL-1β, IL-6, MIP-1α, TLR4, TLR2, p38MAPK, jun, IκBα) or down-regulated (MAPK14 and PI3 K p85β) (B) in the rat middle ear mucosa after Pn6A challenge for 3–14 d. Upward and downward rows indicate up-regulated and down-regulated genes, respectively.
DISCUSSION
Pneumococcus has been shown to bind to polymeric immunoglobulin A receptor (pIgAR) or platelet-activating factor receptor (PAFR) and enters nasopharyngeal epithelial cells through pIgAR (16) and lung epithelial cells through PAFR (17), but these receptors do not seem to activate host cells and mediate immune and inflammatory responses. It suggests that pIgAR and PAFR are not appropriate receptors for signaling immune and inflammatory reactions. Recent studies indicate that TLR2 is the one that activates the host cells through activation of NF-κB (9), a central molecule for immune and inflammatory reactions. As exemplified in this study, PGPS, a cell wall component that harbors PAMP for Pneumococci, regulates the expression of TLR2 but not others.
TLR2 is barely detected in autopsied specimens from the MEECs but highly expressed in the adult human Eustachian tube epithelium, which has apparently been exposed to pathogens (as judged by a degree of mucous cell metaplasia in the epithelium) and middle ear mucosal epithelial cells with chronic OM, suggesting that TLR2 is up-regulated under the inflammatory conditions or exposure to pathogens in the upper respiratory tract.
Consistent with the findings in the in vivo human data, the in vitro studies demonstrated that the expression of TLR2 in cultured mouse MEEC cells is detected but its expression level is limited. The same applies to the human middle ear epithelium. This suggests that under normal physiological conditions, the expression of TLR2 is limited, which may not be sufficient for mediating immune and inflammatory reactions. It is known that TLRs are involved in antifungal peptide and drosomycin in Drosophila (18). Similarly, TLRs are involved in immune reactions by specifically recognizing bacterial PAMPs (4) and subsequent specific antibody production that yields specific immune responses. Without the expression, an immune response is severely compromised and therefore leads to chronic inflammation or prolongs the infectious disease process. Knockout of TLRs in mice that compromises survival of animals from infection is a good piece of evidence. However, elevated TLR2 in the middle ear mucosa may increase the sensitivity or susceptibility to residual PGPS that is released from killed Pneumococci by antibiotics or immune responses. This may create a scenario in which there is no more bacterial growth but a low profile of inflammation sustains.
It is well known that TLR2 is responsible for the response to peptidoglycan and lipoprotein, whereas TLR4 responds to LPS and lipoteichoic acid (4). It is noted in this study that that both LPS and PGPS up-regulated the expression of TLR2 although LPS is less potent than PGPS in terms of inducing capability. This suggests that there is a cross-regulation between TLR2 and TLR4 in the middle ear epithelium.
One thing clear in this study is that constitutional expression of NF-κB subunit p65 is more potent than the activation of NF-κB (by addition of LPS and PGPS to cells) in terms of TLR2 induction. However, why this occurs in mouse MEEC is not yet understood. Perhaps, it is involved in cellular proliferation and survival because up-regulated NF-κB correlates to the cellular survival of cancer cells because NF-κB also regulates the expression of survival-related genes called inhibitors of apoptosis protein (IAPs) (19), which in turn suppresses caspase activation (20). Perhaps, up-regulated NF-κB in the middle ear after infection with Pneumococci reflect the fact that cells are highly activated and participate in varying immune and inflammatory responses. We know that LPS causes cellular apoptosis in epithelial cells (21,22), including mouse MEEC cells. Unlike LPS, PGPS is involved in more activation than apoptosis in cultured MEECs (data not published). We have observed that NF-κB subunit p65 increases the proliferation of mouse MEEC cells and extends the lifespan of cells by increasing the expression of Bcl-2 and epidermal growth factor receptor (EGFR). Although cells treated with LPS have an increase of TLR2, cells die because LPS is a ligand for death receptors such as CD95 (21). This suggests that PGPS-induced TLR2 expression is more durable than LPS-induced TLR2 expression because cells survive after challenge.
The expression of TLR2 is up-regulated for the enhancement of immune and inflammatory actions in the host under pathological conditions. On one hand, microbial pathogens act although existing TLRs for the production of proinflammatory cytokines such as TNF-α, IL-1β (23), and IL-8 (24), which sustains inflammation. It is well known that NF-κB plays an important role in immune and inflammatory reactions, linking to many immune and inflammatory related genes such as TNF-α and IFN-γ (24 –26). In our earlier study, we showed that PGPS up-regulates the expression of NF-κB, and NF-κB in turn regulates the expression of IL-8 (3), an important chemokine expressed in inflamed middle ear mucosa. However, the expression of TLR2 is up-regulated upon infection in a positive feedback manner on the expression of TLR2 that again extends immune and inflammatory reactions in the middle ear. It constitutes a model specifically for OME in which bacterial culture is sterile but inflammatory reactions persist in the middle ear cavity because residual PGPS in the middle ear cavity and up-regulated TLR2 expression in the host epithelial cells maintain a low profile of inflammation, especially cells that are active and not apoptotic.
To summarize, our data indicate that TLRs regulate the activity of NF-κB, resulting in the production of several cytokines, chemokines, or both, which are long present in the middle ear effusion. At the same time, host epithelial cells that have survived and sensitized with up-regulated NF-κB and TLR2 respond to residual pneumococcal cell wall components such as PGPS in the middle ear cavity and maintain a low profile of inflammation in the middle ear mucosa. The existence of such a feedback regulation of TLR2 in response to PGPS in the MEECs provides a possibility for self-augmentation or self-regulation of cytokine production and thus persistent inflammation in the middle ear.
Acknowledgments
Supported, in part, by the NIDCD Grant R01DC008165, Supplement No. 00010055, and by the Lions 5M Multiple District Hearing Foundation.
Abbreviations
- FACS
fluorescent activated cell sorting
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- IκBαM
I kappa B alpha mutant
- LPS
lipopolysaccharide
- MEEC
middle ear epithelial cell
- NF-κB
nuclear factor kappa B
- PAMP
pathogen-associated molecular pattern
- OM
otitis media
- OME
otitis media with effusion
- PAFR
platelet-activating factor receptor
- PGPS
pneumococcal peptidoglycan-polysaccharides
- TLR
toll-like receptor
References
- 1.Broome CV, Facklam RR, Allen JR, Fraser DW, Austrian R. From the center for disease control. Epidemiology of pneumococcal serotypes in the United States, 1978–1979. J Infect Dis. 1980;141:119–123. doi: 10.1093/infdis/141.1.119. [DOI] [PubMed] [Google Scholar]
- 2.Musher DM. Infections caused by Streptococcus pneumoniae: clinical spectrum, pathogenesis, immunity, and treatment. Clin Infect Dis. 1992;14:801–807. doi: 10.1093/clinids/14.4.801. [DOI] [PubMed] [Google Scholar]
- 3.Tsuchiya K, Toyama K, Hamajima Y, Kim Y, Ondrey FG, Lin J. Pneumococcal peptidoglycan-polysaccharide induces the production of interleukin-8 by way of nuclear factor kappa B, nuclear factor interleukin-6, and action protein 1 dependent mechanisms in the airway epithelial cells. Laryngoscope. 2007;117:86–91. doi: 10.1097/01.mlg.0000244182.81768.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 5.Shuto T, Xu H, Wang B, Han J, Kai H, Gu XX, Murphy TF, Lim DJ, Li JD. Activation of NF-kappa B by nontypeable Hemophilus influenzae is mediated by toll-like receptor 2-TAK1-dependent NIK-IKK alpha/beta-I kappa B alpha and MKK3/6-p38 MAP kinase signaling pathways in epithelial cells. Proc Natl Acad Sci USA. 2001;98:8774–8779. doi: 10.1073/pnas.151236098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wyllie DH, Kiss-Toth E, Visintin A, Smith SC, Boussouf S, Segal DM, Duff GW, Dower SK. Evidence for an accessory protein function for Toll-like receptor 1 in anti-bacterial responses. J Immunol. 2000;165:7125–7132. doi: 10.4049/jimmunol.165.12.7125. [DOI] [PubMed] [Google Scholar]
- 7.Takeuchi O, Kaufmann A, Grote K, Kawai T, Hoshino K, Morr M, Muhlradt PF, Akira S. Cutting edge: preferentially the R-stereoisomer of the mycoplasmal lipopeptide macrophage-activating lipopeptide-2 activates immune cells through a toll-like receptor 2- and MyD88-dependent signaling pathway. J Immunol. 2000;164:554–557. doi: 10.4049/jimmunol.164.2.554. [DOI] [PubMed] [Google Scholar]
- 8.Han F, Yu H, Tian C, Li S, Jacobs MR, Benedict-Alderfer C, Zheng QY. Role for Toll-like receptor 2 in the immune response to Streptococcus pneumoniae infection in mouse otitis media. Infect Immun. 2009;77:3100–3108. doi: 10.1128/IAI.00204-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoshimura A, Lien E, Ingalls RR, Tuomanen E, Dziarski R, Golenbock D. Cutting edge: recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J Immunol. 1999;163:1–5. [PubMed] [Google Scholar]
- 10.Gilmore TD, Koedood M, Piffat KA, White DW. Rel/NF-kappaB/IkappaB proteins and cancer. Oncogene. 1996;13:1367–1378. [PubMed] [Google Scholar]
- 11.Tsuchiya K, Kim Y, Ondrey FG, Lin J. Characterization of a temperature-sensitive mouse middle ear epithelial cell line. Acta Otolaryngol. 2005;125:823–829. doi: 10.1080/00016480510031533. [DOI] [PubMed] [Google Scholar]
- 12.Hu X, Huang J, Feng L, Fukudome S, Harmajima Y, Lin J. Sonic Hedgehog (SHH) promotes the differentiation of mouse cochlear neural progenitors via the Math1–Brn3.1 signaling pathway in vitro. J Neurosci Res. 2010;88:927–935. doi: 10.1002/jnr.22286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin J, Tsprun V, Kawano H, Paparella MM, Zhang Z, Andway R, Ho SB. Characterization of mucins in human middle ear and eustachian tube. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1157–L1167. doi: 10.1152/ajplung.2001.280.6.L1157. [DOI] [PubMed] [Google Scholar]
- 14.Ozeki M, Hamajima Y, Feng L, Ondrey FG, Zheng M, Schlentz EP, Lin J. Id1 induces the proliferation of cochlear sensorineural epithelial cells via the NF-kB/cyclin D1 pathway in vitro. J Neurosci Res. 2007;85:515–524. doi: 10.1002/jnr.21133. [DOI] [PubMed] [Google Scholar]
- 15.Lin J, Tsuboi Y, Pan W, Giebink SG, Adams GL, Kim Y. Analysis by cDNA microarrays of altered gene expression in middle ears of rats following pneumococcal infection. Int J Pediatr Otorhinolaryngol. 2002;65:203–211. doi: 10.1016/s0165-5876(02)00130-1. [DOI] [PubMed] [Google Scholar]
- 16.Zhang JR, Mostov KE, Lamm ME, Nanno M, Shimida S, Ohwaki M, Tuomanen E. The polymeric immunoglobulin receptor translocates Pneumococci across human nasopharyngeal epithelial cells. Cell. 2000;102:827–837. doi: 10.1016/s0092-8674(00)00071-4. [DOI] [PubMed] [Google Scholar]
- 17.Ishizuka S, Yamaya M, Suzuki T, Nakayama K, Kamanaka M, Ida S, Sekizawa K, Sasaki H. Acid exposure stimulates the adherence of Streptococcus pneumoniae to cultured human airway epithelial cells: effects on platelet-activating factor receptor expression. Am J Respir Cell Mol Biol. 2001;24:459–468. doi: 10.1165/ajrcmb.24.4.4248. [DOI] [PubMed] [Google Scholar]
- 18.Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorso-ventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86:973–983. doi: 10.1016/s0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- 19.Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J. NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol. 2001;21:5299–5305. doi: 10.1128/MCB.21.16.5299-5305.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 21.Grassmé H, Kirschnek S, Riethmueller J, Riehle A, von Kurthy G, Lang F, Weller M, Gulbins E. CD95/CD95 ligand interactions on epithelial cells in host defense to Pseudomonas aeruginosa. Science. 2000;290:527–530. doi: 10.1126/science.290.5491.527. [DOI] [PubMed] [Google Scholar]
- 22.Matute-Bello G, Winn RK, Jonas M, Chi EY, Martin TR, Liles WC. Fas (CD95) induces alveolar epithelial cell apoptosis in vivo: implications for acute pulmonary inflammation. Am J Pathol. 2001;158:153–161. doi: 10.1016/S0002-9440(10)63953-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsuguchi T, Musikacharoen T, Ogawa T, Yoshikai Y. Gene expressions of Toll-like receptor 2, but not Toll-like receptor 4, is induced by LPS and inflammatory cytokines in mouse macrophages. J Immunol. 2000;165:5767–5772. doi: 10.4049/jimmunol.165.10.5767. [DOI] [PubMed] [Google Scholar]
- 24.May MJ, Ghosh S. Signal transduction through NF-kappa B. Immunol Today. 1998;19:80–88. doi: 10.1016/s0167-5699(97)01197-3. [DOI] [PubMed] [Google Scholar]
- 25.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 26.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]