

Abstract
Background
This study evaluates the relationship between cytochrome P450 (CYP) 3A5 genotype and vincristine-induced peripheral neuropathy in children with precursor B cell acute lymphoblastic leukemia (preB ALL). We have shown in vitro that vincristine is metabolized significantly more efficiently by CYP3A5 than by CYP3A4. We also found that vincristine neurotoxicity is less common in African-Americans (70% express CYP3A5) than in Caucasians. We test the hypothesis that CYP3A5 expressers experience less vincristine neuropathy than do CYP3A5 non-expressers.
Procedure
This study of pharmacogenetics of vincristine neuropathy in children with preB ALL was completed at Indiana University Simon Cancer Center. Whole blood for DNA extraction and genotyping was collected as well as plasma from a single time-point for analysis of vincristine and primary metabolite (M1) concentrations. Vincristine neuropathy was captured via chart review and graded per the National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0.
Results
89% of CYP3A5 expressers experienced neurotoxicity versus 100% of non-expressers (p=0.03). The proportion of treatment months with neurotoxicity was significantly different between the expressers and non-expressers (16% vs. 27%, p=0.0007). Limited pharmacokinetic data suggest different rates of vincristine metabolism between CYP3A5 genotype groups with higher primary metabolite (M1) plasma concentrations (p=0.0004) and lower metabolic ratios ([vincristine]/[M1]) (p=0.036) in the CYP3A5 expressers compared to the CYP3A5 non-expressers. M1 concentration was also inversely related to severity of neuropathy (p=0.0316).
Conclusions
In children with preB ALL, CYP3A5 expressers experience less vincristine-induced peripheral neuropathy, produce more M1, and have lower metabolic ratios compared to CYP3A5 non-expressers.
Keywords: vincristine, pharmacogenetics, acute lymphoblastic leukemia, peripheral neuropathy
INTRODUCTION
Vincristine is one of the mainstay drugs in pediatric oncology used for treatment of a variety of malignancies, including pediatric acute lymphoblastic leukemia (ALL). In the United States, over half of children with cancer who receive chemotherapy are given a treatment regimen that includes vincristine. Furthermore, over 30,000 adults with cancer receive vincristine as part of their therapeutic regimen each year as well. Given the lack of myelosuppression associated with vincristine at its recommended dose and the fact that it is quite inexpensive, it is also used as part of many chemotherapeutic regimens in third world countries. Despite its abundant use, there exists a paucity of information regarding its disposition and its optimal therapeutic dosing strategy [1]. The dose limiting toxicity of vincristine consists of a peripheral neuropathy characterized by progressive motor, sensory, and autonomic involvement in varying combinations [2]. Initial studies in adult patients showed up to 10-fold interpatient variability in pharmacokinetics as well as some degree of intrapatient variability after repeated dosing [3]. More recent pediatric clinical pharmacokinetic studies also reported a 19-fold difference in the dose-corrected area-under-the concentration-time curve (AUC, a measure of drug exposure) for vincristine between patients [4]. Since toxicity is often correlated with drug exposure, one might expect that AUC would correlate with vincristine neurotoxicity; however, Crom et al. [5] found no such association. There are potential explanations for this including: 1) vincristine neurotoxicity may be due to increased sensitivity to vincristine rather than exposure and; 2) existing data in the literature may not be adequate to demonstrate an association due to large variability in vincristine pharmacokinetics coupled with insensitive measures of neuropathy. As such, we do not believe that the potential link between vincristine toxicity and AUC has been adequately explored. Hence, the identification of factors influencing vincristine’s pharmacokinetics may be important in determining appropriate dosing regimens that will provide optimal therapeutic effect while limiting associated neurotoxicity.
Evidence from multiple studies concluded that the CYP3A family of enzymes is responsible for the metabolism of the vinca alkaloids [1,2,6-17]. In vitro and ex vivo data from our laboratory revealed that the metabolic clearance for vincristine is significantly greater with CYP3A5 than with CYP3A4 [6,7]. The predicted intrinsic clearance for vincristine is 5-fold greater in CYP3A5 expressers versus nonexpressers[6,7]. The CYP3A5*1 allele is required for the production of a functional enzyme. In Caucasians, the most common allelic variants include CYP3A5*3, CYP3A5*6, and CYP3A5*7. The CYP3A5*6 and CYP3A5*7 allelic variants have been shown to result in the expression of little to no active CYP3A5 enzyme and the CYP3A5*3 allele possesses a single nucleotide polymorphism in intron 3 leading to a premature termination codon [18]. In contrast, more than 70% of African-Americans have been shown to have at least one CYP3A5*1 allele which allows expression of active CYP3A5 enzyme [19]. Because such a small amount of active CYP3A5 is produced in individuals without the CYP3A5*1 allele, the other genotypes described are effectively void of active CYP3A5 enzyme.
Given the racial differences in CYP3A5 genotype and thus expression, we retrospectively evaluated the correlation between race and the frequency of vincristine-induced peripheral neuropathy (VIPN) in children with preB ALL [20]. This study found that vincristine-related neurotoxicity was much more frequent in Caucasians compared to African-Americans (p=0.007). We also found that Caucasians missed more overall doses (p<0.01) and had more total doses reduced (p<0.0001) due to vincristine-related neurotoxicity than did African-Americans. Furthermore, Caucasians experienced more severe vincristine-associated neurotoxicity than did African-American with average neurotoxicity grades of 2.72 vs. 1.0, respectively (p<0.0001) [20].
These findings support the hypothesis that CYP3A5 genotype resulting in polymorphic expression of CYP3A5 enzyme could account for differences in vincristine-induced neuropathy and metabolism. Polymorphic CYP3A5 expression may be an important contributor to interindividual and interracial differences in CYP3A-mediated vincristine disposition and toxicity [18]. This leads to the specific hypothesis of the study presented here, which is that carriers of one or two copies of the active CYP3A5*1 allele experience less vincristine-induced peripheral neuropathy than do subjects with no CYP3A5*1 alleles. We sought to evaluate this in children with preB ALL by testing for an association between CYP3A5 genotype and vincristine-induced neurotoxicity.
METHODS
This study provides data for pharmacogenetic analysis of vincristine-induced peripheral neuropathy within a pediatric population receiving vincristine as part of preB ALL treatment. This clinical trial was approved by the Indiana University Simon Cancer Center Scientific Review Committee and the Indiana University Purdue University Indianapolis Institutional Review Board. After written informed consent was provided by the subjects’ parents or legal guardians, 130 subjects were enrolled. The 107 subjects who had completed at least one year of ALL therapy were included in this analysis. Subjects were treated on or according to the following treatment protocols for preB ALL: Children Cancer Groups Studies: 1961 and 1991 and Children’s Oncology Group (COG) studies: AALL0331, AALL0232, AALL0434 and AALL01P1. All treatment regimens include a standard vincristine dose of 1.5 mg/m2 with a maximum dose of 2 mg administered weekly during induction chemotherapy. Subsequent dosing intervals varied only slightly by treatment protocol and arm. Clinical assessments of vincristine-related neurotoxicity were evaluated by reviewing patient charts. This was done by a blinded reviewer who was not part of the treatment team for the subjects, but abstracted evidence of VIPN at monthly interval throughout the course of ALL therapy based on documented incidents in each patient’s medical record. Subjects did not undergo any specific testing to evaluate neurotoxicity as part of this study. Severity of neurotoxicity (sensory, motor, and autonomic) observed was graded via National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0 (CTCAE v3.0), assigning a grade of one (lowest) to five (highest) and the highest grade for each month was reported. In addition, a cumulative toxicity grade for the first year of ALL therapy for each subject was calculated by adding the highest grades of neurotoxicity experienced during each month of therapy…
Eligibility Criteria
Subjects ≥1 and ≤18 years of age with a confirmed diagnosis of preB ALL were eligible for this trial. Subjects with baseline peripheral neuropathy greater than grade 1, a history of allergic reactions to vincristine, a history of liver disease, or who were pregnant were excluded from this study.
Genotyping
One whole blood sample for DNA extraction and genotyping was collected from each subject on enrollment in this study. DNA was extracted from whole blood using a Qiagen QIAmp Midi or Mini kit (Qiagen, Inc., Valencia, California, USA). Genotyping for CYP3A5 *3,*6, and *7 (the most common functionally significant polymorphisms in this gene) was performed using Taqman Real-Time polymerase chain reaction assays using previously published methods (Applied Biosystems, Darmstadt, Germany) [21]. A CYP3A5*1 genotype is assigned by default if testing for other alleles (*3, *6, and *7) is negative.
Pharmacokinetic Analysis
Plasma samples were collected one-hour after administration of up to two doses of vincristine from subjects still receiving vincristine (74 of the 107 subjects included in this analysis). Vincristine and its primary metabolite, M1 were quantified in these plasma samples using a previously published liquid chromatography-tandem mass spectrometry assay [22]. Results were analyzed for an association between CYP3A5 genotype and concentrations of vincristine and M1 and the metabolic ratio ([vincristine]/[M1]) as well as between severity of neuropathy and concentrations of vincristine and M1 and the metabolic ratio ([vincristine]/[M1]).
Statistical Analysis
Chi-squared tests and t-tests were used to analyze the differences in demographic data and neurotoxicity between CYP3A5 expressers and non-expressers. Pharmacokinetic data were obtained after multiple doses for some individuals. Therefore, each individual’s median vincristine and M1 concentrations normalized for dose were evaluated using the Wilcoxon Rank Sum approach to determine differences between CYP3A5 expressers and non-expressers. A P-value of less than 0.05 was considered significant. All analyses were performed in R v 2.9.2.
RESULTS
For the purposes of this analysis, data from 107 subjects, all who had completed a minimum of one year of ALL treatment where included. Racial breakdown of this cohort was 105 Caucasians (6 Hispanic or Latino), one African-American, and one Asian. This distribution reflects the population distribution in the state of Indiana. CYP3A5 genotyping revealed 88 subjects (82%) who were CYP3A5*3/*3 (CYP3A5 non-expressers) and 19 subjects (18%) who were CYP3A5*1/*3 (CYP3A5 expressers). The patients were treated for their disease on a number of different cooperative group trials and institutional treatment plans (see Methods section for treatment plans) all of which included vincristine as a core chemotherapeutic agent. The cumulative vincristine doses received, the mean age at diagnosis, and the average number of months of ALL therapy completed at the time of analysis were not statistically different between the CYP3A5 expresser and non-expresser cohorts.
The presence of vincristine neuropathy in CYP3A5 expressers versus CYP3A5 non-expressers was found to be statistically significant (P=0.03) (Table I). The average cumulative neurotoxicity grade was also significantly greater in the CYP3A5 non-expressers compared to the expressers (p=0.035). As measures of burden of neuropathy, the total number of months and the proportion of total treatment months completed in which vincristine neurotoxicity was present were compared between the genotype groups. CYP3A5 non-expressers spent more months on average through their entire course of therapy with toxicity than did CYP3A5 expressers (8.08±5.70 vs. 4.37±3.11 months, p=0.00025) and spent a greater proportion of their therapy with toxicity (p=0.0007). Furthermore, CYP3A5 non-expressers had more doses of vincristine reduced and omitted from their ALL therapy due to neuropathy than did the CYP3A5 expressers on average (p=0.006 and p=0.003, respectively).
Table I.
Neurotoxicity assessment in CYP3A5 expressers versus non expressers
| CYP3A5 Non-expresser (n=88) |
CYP3A5 Expresser (n=19) |
P value | |
|---|---|---|---|
| Experienced vincristine neurotoxicity | 88/88 (100%) | 17/19 (89%) | 0.030 |
| Experienced non-sensory neurotoxicity (motor/autonomic) | 75/88 | 7/19 | 0.04 |
| Maximum grade of neurotoxicity | 2.69 ± 0.93 | 2.16±1.21 | 0.035 |
| Maximum grade of non-sensory neurotoxicity (motor/autonomic) | 2 ± 1.28 | 1.42 ± 1.39 | 0.08 |
| Total number of months with toxicity | 8.08 ± 5.70 | 4.37 ± 3.11 | 0 .00025 |
| Average month of onset of toxicity | 1.59 ± 1.37 | 3.17 ± 4.15 | 0.14 |
| Average number of months with toxicity during the first year of therapy |
4.94 ± 2.66 | 2.90 ± 2.23 | 0.00147 |
| Average number of doses reduced due to vincristine neurotoxicity | 49/3360 | 1/655 | 0.006 |
| Average number of doses missed due to vincristine neurotoxicity | 55/3360 | 3/655 | 0.003 |
| Proportion of months of therapy with toxicity | 711/2622 | 83/509 | 0.0002 |
| Average proportion of months on therapy with toxicity | 27±16% | 16±10% | 0.0007 |
| Proportion of months on therapy with non-sensory toxicity (motor/autonomic) |
331/2622 | 30/509 | 0.00001 |
| Experienced neurotoxicity during the 1st month of therapy | 69/88 (78%) | 10/19 (52%) | 0.040 |
| Experienced neurotoxicity after 1 year of therapy | 61/88 (69%) | 11/19 (58%) | 0.336 |
| Average cumulative neurotoxicity grade at the end of first year of therapy |
13.9±11.2 | 7.21±5.51 | 0.0003 |
| Number of patients with grades 3/4 neuropathy | 50/88 | 7/19 | 0.11 |
| Mean frequency of grades 3/4 neuropathy among patients with grades 3/4 neuropathy during first year of therapy |
1.95 ± 1.29 | 1 ± 0 | 0.001 |
PK samples were collected from 74 of the 107 patients included in this analysis. The genotype distribution of the PK cohort included 64 CYP3A5 non-expressers and 11 CYP3A5 expressers. Figure 1 illustrates the average concentrations of vincristine and M1 (normalized for vincristine dose) one-hour after vincristine dosing in the CYP3A5 expresser and non-expresser cohorts. At that time-point, CYP3A5 expressers had produced significantly more M1 than had the CYP3A5 non-expressers (1286±1068ρg/ml vs. 329±277ρg/ml, p=0.0004). There was also a greater than two-fold difference in metabolic ratio ([vincristine]/ [M1]) between CYP3A5 expressers and non-expressers (p=0.036) (Figure 1C). Evaluation for an association between neuropathy and the PK measures revealed a significant inverse relationship between M1 concentration and severity of neurotoxicity grade (Figure 2, p=0.0316).
Figure 1.

This figure illustrates dose normalized concentrations of vincristine (panel A) and M1 (panel B) and the ratio of vincristine/M1 (panel C) measured one-hour after vincristine administration in CYP3A5 expressers and non-expressers. CYP3A5 expressers produce significantly more M1 by the one-hour time point than do non-expressers (1B, p=0.0004). The metabolic ratio is significantly lower in the CYP3A5 expressers at the one-hour time point compared to the non-expressers (1C, p=0.036). There is no difference between the two groups in vincristine concentration at this time point (1A, p=0.159). CYP3A5*0 designates CYP3A5 non-expressers; CYP3A5*1 designates CYP3A5 expressers.
Figure 2.
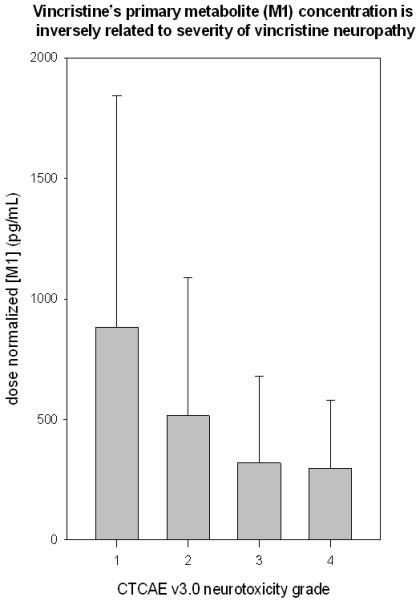
This figure illustrates that M1 plasma concentration (dose normalized) measured one-hour after vincristine administration is inversely related to severity of vincristine-induced neuropathy as measured by CTCAE v3.0 (p=0.0316). These data represent ten patients who had grade 1 neuropathy, 25 patients with grade 2, 24 patients with grade 3 and 15 patients with grade 4.
DISCUSSION
Our results demonstrate significant differences in the amount of vincristine-induced peripheral neuropathy experienced and the amount of M1 produced by CYP3A5 non-expressers and expressers. Expressers of active CYP3A5 enzyme experience less vincristine-induced neuropathy than do CYP3A5 non-expressers. While there was a significant difference in rates of vincristine-induced neuropathy between CYP3A5 expresser and non-expresser groups, the frequencies were very high for both cohorts and were somewhat higher than expected. For example, Aytac et al. reported neuropathy (using toxicity descriptions similar to NCI CTCAE v3.0 grades 3 and 4 (3/4)) in 15% of children treated for ALL on St. Jude Total XI and XIII protocols [23]. In contrast, in one study of 18 patients with lymphoma followed carefully for development of peripheral neuropathy, all subjects were found to have absent ankle reflexes and 75% had sensory signs or symptoms after exposure to three months of vincristine therapy [24]. Furthermore, Crom et al. retrospectively evaluated neurotoxicity in children with ALL who had received vincristine as part of their therapy. Fifty-two out of 64 subjects had evidence of neuropathy: 32 with grade 1 neuropathy [25]. Unlike the many studies that capture only severe neurotoxicity, we adopted a more inclusive approach for collecting VIPN data in this study. Since our ultimate goal is to predict an individual patient’s risk of neuropathy prior to starting therapy, capturing all VIPN was of paramount importance to us. Furthermore, we included motor, sensory, and autonomic neuropathies in an attempt to develop a better understanding of the full spectrum of VIPN in this population. This approach likely contributed to the higher rate of VIPN found in our study compared to others. Given that we found evidence of neuropathy in nearly all patients (98%) enrolled on this study, we looked beyond simply presence or absence of vincristine-induced neuropathy to determine whether there are other differences in the burden or severity of neurotoxicity between the genotype groups. This requires evaluation of severity of side effects these children experienced in an effort to ultimately optimize dosing for individual patients. In fact, we found that CYP3A5 non-expressers experienced more severe neuropathy than did CYP3A5 expressers based on average grade of neurotoxicity. Though the comparison of presence of grade 3/4 neuropathy between the two CYP3A5 genotype groups was not different, the frequencies of episodes of grade 3/4 VIPN in the first year of therapy and after the first year of therapy among children who experienced grade 3/4 neurotoxicity were higher among the nonexpressers compared to the expressers (p=0.001 and p=0.01, respectively). Furthermore, we found that vincristine-induced neuropathy was very common in both genotype groups early in preB ALL therapy when vincristine dose density is greatest. However, vincristine neuropathy in the fourth to twelfth months of ALL therapy (when vincristine dose density decreases significantly compared to induction chemotherapy) was much more common in the CYP3A5 non-expresser cohort. We also found vincristine-induced neuropathy to be an isolated or very short-term (less than five months) side effect in CYP3A5 expressers. In contrast, in many CYP3A5 non-expressers, neuropathy was a longer-term side effect, and lasted throughout the first year of preB ALL therapy. In looking beyond the first year of therapy at the number of children with neuropathy extending farther into their ALL treatment when vincristine dosing is significantly less frequent than it is during year one of treatment, 58% of CYP3A5 expressers continue to experience vincristine-neuropathy compared to 69% of CYP3A5 non-expresser subjects. While this finding is not statistically significant, most cases of toxicity after the first year of therapy among the CYP3A5 expressers where isolated cases in contrast to CYP3A5 non-expressers who experienced protracted courses of toxicity. This indicates a further possibility of how the burden neuropathy may be more significant in terms of severity and duration in the CYP3A5 non-expressers compared to the expressers. Furthermore, given the retrospective nature of this study and the fact that capturing toxicity was completely dependent on care provider documentation, we suspect that we are likely underestimating what many of the patients with mild neuropathy were experiencing.
Patient care providers may decide to omit or reduce scheduled doses of vincristine in patients who are experiencing significant vincristine-neuropathy to allow the patient to recover from the toxicity. As such, we collected data on vincristine dose reductions and omissions for subjects enrolled on this trial. We found a significant difference in both of these surrogate measures between genotype groups, i.e., with CYP3A5 non-expressers having more vincristine doses reduced (p=0.006) and omitted (p=0.003) due to toxicity than did the CYP3A5 expressers. This provides additional evidence of a difference in burden of this side effect between genotype groups.
Limited PK sampling was carried out in this study to look for any evidence of differences in vincristine metabolism between the genotype groups and to evaluate for a correlation with neurotoxicity. Our PK data show that the CYP3A5 expressers produce more M1 and have a lower metabolic ratio ([vincristine]/[M1]) than do CYP3A5 non-expressers one-hour after vincristine dosing. The time-point for collection of a PK sample was selected based on patient convenience with an understanding of its significant limitations. Based on what is known about vincristine PK from the literature [25-27], within minutes after dosing the distribution phase for vincristine should be complete and metabolism will have initiated. As such given that only about 5% of parent drug metabolism that will have occurred at one-hour post-dose, one would only expect to find a difference in metabolic ratio and primary metabolite formation between CYP3A5 expressers and non-expressers if there is a very large difference in vincristine metabolism between based on genotype. In spite of their limitations, these data do reveal significantly more M1 formation and a lower ratio of [vincristine]/[M1] in the CYP3A5 expresser cohort compared to the CYP3A5 non-expresser cohort. At this very early time-point, we did not find a significant difference in average vincristine concentration between the genotype groups. This is not unexpected given that relatively little parent drug metabolism will have actually taken place so early after drug dosing. As such, vincristine concentration at one-hour post-dose is likely to be a better reflection of vincristine distribution throughout the body than it is of metabolism; and thus would likely not be affected by CYP3A5 genotype or expression. In order to evaluate vincristine exposure over time, pharmacokinetic samples would need to be collected over more than one half-life of the drug. The PK evidence provided by this study suggests that CYP3A5 expressers may have faster rates of vincristine conversion to M1 and vincristine clearance and thus potentially less vincristine exposure than do the CYP3A5 non-expressers. This fits well with what is known about vincristine metabolism in vitro [6,7] with vincristine being converted to M1 significantly more efficiently by CYP3A5 expressing compared to non-expressing human liver microsomes. Given the fact that drug toxicity is often directly related to exposure, these findings support the hypothesis that CYP3A5 non-expressers may have higher rates of vincristine-associated toxicity due to slower vincristine clearance and greater vincristine exposure.
Though we have identified a correlation between M1 concentration as well as metabolic ratio and neuropathy, there are other possible explanations for the differences in neurotoxicity between CYP3A5 expressers versus non-expressers in this study. One possibility is that the difference in neurotoxicity may be related to other genetic polymorphisms involved in vincristine disposition such as ABCB1. In fact, we are exploring the potential impact of other pharmacogenetic biomarkers of VIPN in patients enrolled to this study in combination with children with preB ALL enrolled to a second ongoing trial. Another possibility is that interindividual differences in VIPN are due to variability in sensitivity to vincristine (e.g., due to underlying conditions like Charcot Marie Tooth) rather than to exposure to the drug. We did not specifically test these subjects for such genetic disorders but rather excluded potential subjects with any evidence of underlying neuropathies; however, this still could have missed children with subclinical neuropathy at baseline. Alternatively, variability in vincristine-associated neurotoxicity could be due to non-genetic differences in sensitivity to the drug such as environmental differences and/or epigenetic effects. Exposure to heavy metals (e.g., lead or mercury) could result in increased severity of VIPN; however, again our exclusion criteria of evidence of baseline neuropathy should have minimized the risks of these environmental confounders. Furthermore, interactions with current medications (e.g., azoles or nifedipine) may also increase VIPN; but none of the subjects in our cohort received medications known to increase this risk.
Although we observed significant differences in several parameters, there were limitations to this study. One weakness of this research in terms of making specific recommendations for vincristine dosing based on genotype is in the phenotyping of the patients. For this research, we have reviewed patient medical records in order to capture the vincristine neurotoxicity that each patient experienced. As a result, the research is dependent on medical care providers’ reporting of signs and symptoms of this toxicity, which is highly variable. In addition, we have utilized the NCI CTCAE v3.0 which is routinely used to grade toxicities experienced as a result of cancer chemotherapy; however, there is conflicting evidence regarding effectiveness of this tool in the assessment of neurotoxicity [28-30]. These weaknesses could be improved upon in future clinical trials by 1) enrolling patients at the time of diagnosis of ALL and following them prospectively and 2) utilizing additional measurement tools to carefully evaluate neuropathy. This should significantly improve the neuropathy phenotype captured.
An additional weakness is in the very limited PK data available from this study. The single time-point PK sample collection was selected to minimize the burden to the research subjects and their families. However, in order to make definitive conclusions about the relationship between genotype and vincristine metabolism and exposure as well as between vincristine PK parameters and neurotoxicity, more complete PK sampling will be required.
Finally, the ultimate goal of treating a child with ALL is to cure his or her disease. The overarching goal of this type of research is to identify predictors of vincristine neuropathy thus providing the knowledge base needed to determine optimal dosing of vincristine for individual pediatric cancer patients. Once we confirm the genes associated with vincristine neuropathy and disposition, it will be possible to develop improved dosing strategies for this critical agent to optimize therapeutic efficacy while minimizing toxicity. Specifically, our hypothesis is that knowing a patient’s vinca alkaloid pathway genotypes prior to initiating therapy with vincristine will allow us to predict which patients may require a decrease in starting dose to avoid excessive toxicity; and equally importantly, to identify patients who require higher dosing to reach therapeutic exposure to vincristine thereby improving response and overall survival. While it is too early in the course of this study to analyze the data for predictors of disease outcome, Lonnerholm et al. recently reported a small study in which they found that children with standard risk ALL have a faster than average vincristine clearance or less than average vincristine exposure and a higher risk of relapse than do those children with slower clearance and greater exposure to vincristine [27]. Based on the vincristine PK data from this study and what is known about vincristine metabolism in vitro, one could hypothesize that CYP3A5 genotype is an important determinant of vincristine clearance and exposure in patients. This will need to be evaluated prospectively; however, if CYP3A5 genotype is a strong predictor of vincristine toxicity and PK as well as disease outcome, it may provide a starting point for an improved dosing strategy for a drug that is critical in the treatment of multiple curable childhood cancers.
The Children’s Oncology Group has recognized the potential benefits of a more systematic evaluation of vincristine dosing strategies and of possible vincristine dose intensification. The ongoing COG study for patients with intermediate risk ALL is evaluating the potential benefit of vincristine dose intensification by randomizing children to 1.5 mg/m2/dose with a 2 mg cap versus 2 mg/m2/dose with a 2.5 mg cap. However, as evidenced by the recent report of unexpected grade 3 toxicity rates in adolescents randomized to the higher dose arm on this trial, randomization of dose escalation may not be the best approach to take in optimizing vincristine therapy for individual patients. Randomization among just CYP3A5 expressers could be attempted; however, CYP3A5 likely only accounts for a small part of the variability in vincristine response; therefore, additional investigation is needed before embarking on such trials. It is likely that many patients would not tolerate dose intensification and also may not benefit from it. In contrast, there is also likely a group of subjects who could tolerate and may benefit from such dose intensification. However, this type of individualized dosing strategy requires additional data to be put into place. Because of the inherent limitations of this study, our next step is to further evaluate these finding in an ongoing multicenter prospective clinical trial including collection of DNA and pharmacokinetic samples as well as objective assessments of neuropathy throughout therapy for ALL to optimize phenotype data and in order to critically evaluate the best way to assess VIPN in children. This should allow us to identify the most important biomarkers in predicting risk of severe neuropathy in individual patients prior to initiating chemotherapy with vincristine and provide the knowledge-base necessary to make informed decisions about which patients would tolerate and could potentially benefit from vincristine dose-intensification.
Acknowledgments
Funding: This study was funded by K23 RR019956, an award from the Showalter Foundation, Indiana University School of Medicine, Indianapolis, IN, and a Biomedical Research Grant/Elwert Award from Indiana University School of Medicine, Indianapolis, IN, USA.
Footnotes
The authors have no conflicts of interest to disclose.
References
- 1.McCune JS, Lindley C. Appropriateness of maximum-dose guidelines for vincristine. Am J Health Syst Pharm. 1997;54(15):1755–1758. doi: 10.1093/ajhp/54.15.1755. [DOI] [PubMed] [Google Scholar]
- 2.Gidding CE, Boer GJ Meeuwsen-de, Koopmans P, et al. Vincristine pharmacokinetics after repetitive dosing in children. Cancer Chemother Pharmacol. 1999;44(3):203–209. doi: 10.1007/s002800050968. [DOI] [PubMed] [Google Scholar]
- 3.Van den Berg HW, Desai ZR, Wilson R, et al. The pharmacokinetics of vincristine in man: reduced drug clearance associated with raised serum alkaline phosphatase and dose-limited elimination. Cancer Chemother Pharmacol. 1982;8(2):215–219. doi: 10.1007/BF00255487. [DOI] [PubMed] [Google Scholar]
- 4.Frost BM, Lonnerholm G, Koopmans P, et al. Vincristine in childhood leukaemia: no pharmacokinetic rationale for dose reduction in adolescents. Acta Paediatr. 2003;92(5):551–557. [PubMed] [Google Scholar]
- 5.Crom W, de Graaf S, Synold T, et al. Pharmacokinetics of vincristine in children and adolescents with acute lymphoblastic leukemia. Journal of Pediatrics. 1994;125:642–649. doi: 10.1016/s0022-3476(94)70027-3. [DOI] [PubMed] [Google Scholar]
- 6.Dennison JB, Jones DR, Renbarger JL, et al. Effect of CYP3A5 expression on vincristine metabolism with human liver microsomes. The Journal of pharmacology and experimental therapeutics. 2007;321(2):553–563. doi: 10.1124/jpet.106.118471. [DOI] [PubMed] [Google Scholar]
- 7.Dennison JB, Kulanthaivel P, Barbuch RJ, et al. Selective metabolism of vincristine in vitro by CYP3A5. Drug metabolism and disposition: the biological fate of chemicals. 2006;34(8):1317–1327. doi: 10.1124/dmd.106.009902. [DOI] [PubMed] [Google Scholar]
- 8.Gidding CE, Kellie SJ, Kamps WA, et al. Vincristine revisited. Crit Rev Oncol Hematol. 1999;29(3):267–287. doi: 10.1016/s1040-8428(98)00023-7. [DOI] [PubMed] [Google Scholar]
- 9.Kajita J, Kuwabara T, Kobayashi H, et al. CYP3A4 is mainly responsibile for the metabolism of a new vinca alkaloid, vinorelbine, in human liver microsomes. Drug metabolism and disposition: the biological fate of chemicals. 2000;28(9):1121–1127. [PubMed] [Google Scholar]
- 10.Leveque D, Jehl F. Clinical pharmacokinetics of vinorelbine. Clin Pharmacokinet. 1996;31(3):184–197. doi: 10.2165/00003088-199631030-00003. [DOI] [PubMed] [Google Scholar]
- 11.Leveque D, Jehl F, Quoix E, et al. Clinical pharmacokinetics of vinorelbine alone and combined with cisplatin. J Clin Pharmacol. 1992;32(12):1096–1098. [PubMed] [Google Scholar]
- 12.Marquet P, Lachatre G, Debord J, et al. Pharmacokinetics of vinorelbine in man. Eur J Clin Pharmacol. 1992;42(5):545–547. doi: 10.1007/BF00314866. [DOI] [PubMed] [Google Scholar]
- 13.Rahmani R, Bruno R, Iliadis A, et al. Clinical pharmacokinetics of the antitumor drug navelbine (5′-noranhydrovinblastine) Cancer Res. 1987;47(21):5796–5799. [PubMed] [Google Scholar]
- 14.Ratain MJ, Vogelzang NJ, Sinkule JA. Interpatient and intrapatient variability in vinblastine pharmacokinetics. Clin Pharmacol Ther. 1987;41(1):61–67. doi: 10.1038/clpt.1987.9. [DOI] [PubMed] [Google Scholar]
- 15.Sethi VS, Jackson DV, Jr., White DR, et al. Pharmacokinetics of vincristine sulfate in adult cancer patients. Cancer Res. 1981;41(9 Pt 1):3551–3555. [PubMed] [Google Scholar]
- 16.Wargin WA, Lucas VS. The clinical pharmacokinetics of vinorelbine (Navelbine) Semin Oncol. 1994;21(5 Suppl 10):21–27. [PubMed] [Google Scholar]
- 17.Zhou XJ, Placidi M, Rahmani R. Uptake and metabolism of vinca alkaloids by freshly isolated human hepatocytes in suspension. Anticancer Res. 1994;14(3A):1017–1022. [PubMed] [Google Scholar]
- 18.Kuehl P, Zhang J, Lin Y, et al. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat Genet. 2001;27(4):383–391. doi: 10.1038/86882. [DOI] [PubMed] [Google Scholar]
- 19.Xie HG, Wood AJ, Kim RB, et al. Genetic variability in CYP3A5 and its possible consequences. Pharmacogenomics. 2004;5(3):243–272. doi: 10.1517/phgs.5.3.243.29833. [DOI] [PubMed] [Google Scholar]
- 20.Renbarger JL, McCammack KC, Rouse CE, et al. Effect of race on vincristine-associated neurotoxicity in pediatric acute lymphoblastic leukemia patients. Pediatr Blood Cancer. 2007 doi: 10.1002/pbc.21435. [DOI] [PubMed] [Google Scholar]
- 21.Eap CB, Buclin T, Hustert E, et al. Pharmacokinetics of midazolam in CYP3A4- and CYP3A5-genotyped subjects. Eur J Clin Pharmacol. 2004;60(4):231–236. doi: 10.1007/s00228-004-0767-7. [DOI] [PubMed] [Google Scholar]
- 22.Dennison JB, Renbarger JL, Walterhouse DO, et al. Quantification of vincristine and its major metabolite in human plasma by high-performance liquid chromatography/tandem mass spectrometry. Therapeutic drug monitoring. 2008;30(3):357–364. doi: 10.1097/FTD.0b013e31816b92c9. [DOI] [PubMed] [Google Scholar]
- 23.Aytac S, Yetgin S, Tavil B. Acute and long-term neurologic complications in children with acute lymphoblastic leukemia. Turk J Pediatr. 2006;48(1):1–7. [PubMed] [Google Scholar]
- 24.Pal PK. Clinical and electrophysiological studies in vincristine induced neuropathy. Electromyogr Clin Neurophysiol. 1999;39(6):323–330. [PubMed] [Google Scholar]
- 25.Crom WR, de Graaf SS, Synold T, et al. Pharmacokinetics of vincristine in children and adolescents with acute lymphocytic leukemia. J Pediatr. 1994;125(4):642–649. doi: 10.1016/s0022-3476(94)70027-3. [DOI] [PubMed] [Google Scholar]
- 26.de Graaf SS, Bloemhof H, Vendrig DE, et al. Vincristine disposition in children with acute lymphoblastic leukemia. Med Pediatr Oncol. 1995;24(4):235–240. doi: 10.1002/mpo.2950240405. [DOI] [PubMed] [Google Scholar]
- 27.Lonnerholm G, Frost BM, Abrahamsson J, et al. Vincristine pharmacokinetics is related to clinical outcome in children with standard risk acute lymphoblastic leukemia. Br J Haematol. 2008;142(4):616–621. doi: 10.1111/j.1365-2141.2008.07235.x. [DOI] [PubMed] [Google Scholar]
- 28.Cavaletti G, Bogliun G, Marzorati L, et al. Grading of chemotherapy-induced peripheral neurotoxicity using the Total Neuropathy Scale. Neurology. 2003;61(9):1297–1300. doi: 10.1212/01.wnl.0000092015.03923.19. [DOI] [PubMed] [Google Scholar]
- 29.Cavaletti G, Jann S, Pace A, et al. Multi-center assessment of the Total Neuropathy Score for chemotherapy-induced peripheral neurotoxicity. J Peripher Nerv Syst. 2006;11(2):135–141. doi: 10.1111/j.1085-9489.2006.00078.x. [DOI] [PubMed] [Google Scholar]
- 30.Smith E. Clinimetric evaluation of peripheral neuropathy measurement approaches. University of Utah; Salt Lake City: 2008. [Google Scholar]