

Abstract
The human leukocyte antigen (HLA) class II genes HLA-DRB1, -DQA1 and -DQB1 are the strongest genetic factors for type 1 diabetes (T1D). Additional loci in the major histocompatibility complex (MHC) are difficult to identify due to the region’s high gene density and complex linkage disequilibrium (LD). To facilitate the association analysis, two novel algorithms were implemented in this study: one for phasing the multi-allelic HLA genotypes in trio families, and one for partitioning the HLA strata in conditional testing. Screening and replication were performed on two large and independent datasets: the Wellcome Trust Case–Control Consortium (WTCCC) dataset of 2,000 cases and 1,504 controls, and the T1D Genetics Consortium (T1DGC) dataset of 2,300 nuclear families. After imputation, the two datasets have 1,941 common SNPs in the MHC, of which 22 were successfully tested and replicated based on the statistical testing stratifying on the detailed DRB1 and DQB1 genotypes. Further conditional tests using the combined dataset confirmed eight novel SNP associations around 31.3 Mb on chromosome 6 (rs3094663, p = 1.66 × 10−11 and rs2523619, p = 2.77 × 10−10 conditional on the DR/DQ genotypes). A subsequent LD analysis established TCF19, POU5F1, CCHCR1 and PSORS1C1 as potential causal genes for the observed association.
Electronic supplementary material
The online version of this article (doi:10.1007/s00439-010-0908-2) contains supplementary material, which is available to authorized users.
Introduction
Type 1 diabetes (T1D) is an autoimmune disease that causes the destruction of insulin-producing β-cells of the pancreas. Candidate gene studies and genome-wide association (GWA) studies have identified over 40 non-HLA risk loci for T1D (Barrett et al. 2009; Wallace et al. 2009). Still, the human leukocyte antigen (HLA) class II genes HLA-DRB1, -DQA1 and -DQB1 remain the strongest genetic factors (Erlich et al. 2008; Koeleman et al. 2004). In particular, the haplotypes DRB1*0401-DQA1*0301-DQB1*0302 and DRB1*0301-DQA1*0501-DQB1*0201 confer the highest risk (OR 8.3, p = 1.7 × 10-42; OR 6.6, p = 7.3 × 10−23, respectively) in most populations, whereas DRB1*15-DQA1*01-DQB1*06 is the most protective (OR 0.0, p = 1.1 × 10−27) in a dominant manner (Koeleman et al. 2004).
The major histocompatibility complex (MHC), home to the HLA class II genes, is a 3.6 Mb region on chromosome 6p21.3 between flanking genes MOG (telomeric) and COL11A2 (centromeric). The region is essential to the immune system, and is known to harbor an exceptionally high density of 224 gene loci (Horton et al. 2004). Some of those genes and certain regions in the extended MHC, which includes also 3.9 Mb telomeric and 0.2 Mb centromeric extensions, have been identified to be involved in the etiology of T1D (Table 1). Although the MHC is generally believed to harbor additional T1D-associated genes, the overshadowing effect of the HLA class II genes and the strong and complicated linkage disequilibrium (LD) within that region (Alper et al. 2006; de Bakker et al. 2006) make such associations hard to uncover by conventional methodologies. The high polymorphism of DRB1 (762 alleles), DQA1 (35 alleles) and DQB1 (107 alleles) (EMBL-EBI 2010), and the complex interactions of genes in the region further complicate the problem.
Table 1.
Previously reported T1D loci in extended MHC
| MHC region | Loci | Position (Mb) | References | Evidence for association | Subjects and methods of analyses |
|---|---|---|---|---|---|
| Class II | DPB1 | 33.16 | Cucca et al. (2001) | DPB1*0402 protective mainly on DR4-negative haplotypes (DPB1*0301 at 5.4-fold greater risk than DPB1*0402 on DR3 haplotypes) | Haplotype method transmission disequilibrium test (HM-TDT) adjusted for DR-DQ: 408 European families |
| Cruz et al. (2004) | DPB1*0301 (p = 0.002), DPB1*0202 (p = 0.027) predisposing; DPB1*0402 (p = 0.012) protective | TDT adjusted for DR-DQ: 84 Mexican-American families | |||
| Howson et al. (2009) | Overall p = 5.1 × 10−8 | Recursive partitioning (RP) and logistic regression (LR) controlled for DR-DQ: proband dataset of 2,300 families (T1DGC) | |||
| DOA | 33.08 | Santin et al. (2009) | SNP rs419434 (unconditional OR 1.93; CI 1.52–2.44; p = 10−8) | 15 DR3 homozygous cases and 39 controls with one copy of B18-DR3 extended haplotype for initial screening. 554 cases and 841 controls of southern European origin for Fisher’s exact test in replication study | |
| Class III | HLA-DRA | 32.52 | Nejentsev et al. (2007) | SNP rs9268831 (p = 6.95 × 10−8) | RP and LR controlled for DR-DQ: 2,484 cases and 2,019 controls, mainly Caucasian |
| Centromeric of NOTCH4 | 32.30 | Valdes et al. (2009) | SNP rs2395106 (p = 3.3 × 10−4) | Overall conditional genotype method (OCGM) controlled for DR-DQ: 886 Caucasian families | |
| CFB | 32.02 | Valdes et al. (2010) | SNP rs4151659 on DR3 (p = 1.2 × 10−9) and DR4 (p = 4 × 10-15) haplotypes | TDT on the DR3 and DQ4 haplotypes: 1,411 Caucasian families (T1DGC) | |
| MSH5 | 31.83 | Valdes et al. (2009) | SNP rs707915 (p = 3.5 × 10−4) | OCGM controlled for DR-DQ: 886 Caucasian families | |
| AIF1 | 31.69 | Eike et al. (2009b) | SNP rs2259571 (p = 0.034) | Logistic regression adjusted for DR-DQ: 434 Norwegian families | |
| TNF (200-kb region) | 31.65 | Hanifi Moghaddam et al. (1998) | Microsatellite D6S273 | 120 cases and 83 controls from Belgium matched for DQA1*0301-DQB1*0302 and DQA1*0501-DQB1*0201 | |
| 5’ of LTA | 31.65 | Valdes et al. (2010) | SNP rs7762619 on DR3 (p = 2 × 10−12) and DR4 (p = 8 × 10-8) haplotypes | TDT on the DR3 and DQ4 haplotypes: 1,411 Caucasian families (T1DGC) | |
| Class I | HLA-B | 31.43 | Nejentsev et al. (2000) | Microsatellites C12A (p = 0.00326), STR MICA (p = 0.00018), C125 (p = 0.00239) and C143 (p = 0.00013) for DR3/404; MIB (p = 0.00463) for DR3/401 | Population differentiation test: 75 cases and 181 controls for DR3/404 genotype, and 241 cases and 354 controls for DR3/401 genotype. All from Finland |
| Valdes et al. (2005) | B*4403 (p = 0.036) protective; B*1801 (p = 0.038), B*3906 (p = 0.002) predisposing | TDT adjusted for DR-DQ: 283 Caucasian multiplex families from US | |||
| Nejentsev et al. (2007) | Overall p = 2.01 × 10−19: B*39 predisposing | Recursive partitioning and logistic regression controlled for DR-DQ: 2,484 cases and 2,019 controls, mainly Caucasian | |||
| Eike et al. (2009a) | Overall p = 5.7 × 10−12: B*44 protective; B*39, B*18 predisposing | LR controlled for DR-DQ: proband dataset of 2,312 families (T1DGC) | |||
| Howson et al. (2009) | Overall p = 6.0 × 10−17: B*38 most protective (OR 0.51; CI 0.14–1.77); B*39 most predisposing (OR 2.53; CI 1.57–4.07) | RP and LR controlled for DR-DQ: proband dataset of 2,300 families (T1DGC) | |||
| HLA-C | 31.35 | Valdes et al. (2005) | C*0802 (p = 0.049), C*1601 (p = 0.049) protective; C*0303 (p = 0.0032) predisposing | TDT adjusted for DR-DQ: 283 Caucasian multiplex families from US | |
| HLA-A | 30.02 | Noble et al. (2002) | A*0101 (p = 0.007) protective; A*2402 (p = 0.026), A*3002 (p = 0.046) predisposing | TDT adjusted for DR-DQ: 283 Caucasian multiplex families from US with two affected siblings and unaffected parents | |
| Nejentsev et al. (2007) | Overall p = 2.35 × 10−13: A*01, A*11, A*31 protective; A*24 predisposing | RP and LR controlled for DR-DQ: 2,484 cases and 2,019 controls, mainly Caucasian. | |||
| Howson et al. (2009) | Overall p = 8.8 × 10−13: A*32 most protective; A*24 most predisposing | RP and LR controlled for DR-DQ: proband dataset of 2,300 families (T1DGC) | |||
| HLA-G | 29.91 | Eike et al. (2009a) | SNPs rs4122198 (p = 1.5 × 10−8), rs2394186 (p = 2.1 × 10−8), rs1619379 (p = 1.1 × 10−7) and rs1611133 (p = 3.2 × 10−6) | LR controlled for DR-DQ: 2,321 families (T1DGC) | |
| Between UBD and MAS1L | 29.63–29.56 | Aly et al. (2008) | SNP rs1233478 (p = 1.4 × 10−12 after chromosomes with the highly conserved MHC 8.1 haplotype were removed) | AFBAC: 237 families from US; 1,240 families from UK, US and Denmark (T1DGC) for replication | |
| Extended Class I | Telomeric of HLA-F | 27.77 | Lie et al. (1999) | Microsatellite D6S2223*3 (p = 0.00004) protective | Homozygous parent TDT (HPTDT) on DRB1*03-DQA1*0501-DQB1*0201: 116 families from Norway, Denmark and UK |
| Johansson et al. (2003) | D6S2223*3 (p = 0.03) protective | HPTDT on DRB1*03-DQA1*0501-DQB1*0301: 43 families from Sweden and Southern France | |||
| Santin et al. (2009) | D6S2223*3 (p = 0.001) protective | HPTDT on DRB1*03-DQA1*0501-DQB1*0201: 111 Caucasian parents | |||
| PRSS16 | 27.33 | Santin et al. (2009) | SNPs rs6931711, rs858988, rs9368492, rs9393796, rs9348756, rs9368493 and rs6938397 (p = 0.02, all SNPs in perfect LD) | HPTDT on DRB1*0401-DQA1*03-DQB1*0302: 62 Caucasian parents | |
| BTN3A2 | 26.48 | Viken et al. (2009) | SNP rs9379857 (p = 0.01) | HPTDT on DRB1*03-DQA1*0501-DQB1*0201: 111 Caucasian parents |
There are a handful of statistical techniques for studying the association of markers conditional on certain primary factors (Thomson et al. 2008), to account for any effects due to LD with those factors. One major branch of conditional testing strategies is the stratification approach. Some methods in this family require careful selection of cases and controls that match specific genotypes under consideration. These include matched genotype strategies (Hanifi Moghaddam et al. 1998), and the homozygous parent linkage method (HPLM) for affected sib pairs (Robinson et al. 1993) and the homozygous parent transmission disequilibrium test (HPTDT) (Johansson et al. 2003; Lie et al. 1999; Santin et al. 2009; Viken et al. 2009) for trios. The major drawback of the approach is the reduction in samples available for analysis. Alternative stratification-based methods that use all samples without imposing stringent selection criteria include the conditional haplotype method (CHM), the conditional genotype method (CGM) and the conditional extended transmission disequilibrium test (CETDT) (Cucca et al. 2001; Koeleman et al. 2000a, b). The basic assumption of these three methods is that for no association, the relative allele frequencies of the test locus should be the same in cases and controls in each stratum of primary haplotype or genotype. Both CHM and CETDT, however, require the inference of haplotype phase. As extensions to CHM and CGM, the overall conditional haplotype method (OCHM) (Thomson 1984) and the overall conditional genotype method (OCGM) (Valdes et al. 2009) define a test statistic that measure the additional genetic effect summed over all haplotype or genotype strata. Besides stratification, other conditional testing techniques include conditional logistic regression (CLR) (Eike et al. 2009a; Howson et al. 2009; Nejentsev et al. 2007) and LD-adjusted association tests (Cruz et al. 2004; Noble et al. 2002; Valdes et al. 2005).
In this study, we applied conditional tests on each single nucleotide polymorphism (SNP), stratified by the DR/DQ alleles in a manner similar to the OCHM and OCGM methods described above. The effects of individual SNPs in addition to the DR/DQ genotypes are measured by their respective overall p values obtained through stratified analysis, which does not assume any particular model or involve any likelihood estimations. Thus, the results were expected to be more reliable (Huang 1999). This study was carried out in two stages: an initial screening stage and a replication stage that verifies the associations of the significant SNPs emerged from the screening. First, the case–control dataset from the Wellcome Trust Case–Control Consortium (WTCCC) (WTCCC 2007) was used for screening followed by replication based on an independent trio dataset from the T1D Genetics Consortium (T1DGC) (Brown et al. 2009). Then, the same two-stage procedure was repeated with the roles of the two datasets switched. LD analysis was next performed on the replicated SNPs, and the novelty of each locus was established through conditional tests that adjust, respectively, for the HLA-A, -B, -C and -DPB1 genes, in addition to the DR/DQ genotypes. Finally, the possible causal genes were located if they have one or more SNPs, preferably non-synonymous ones, in reasonably strong LD with any of the SNPs with novel associations. A summary of our analytical plan is shown in Fig. 1. A T1D locus in the MHC class I region was identified by our two-stage study.
Fig. 1.
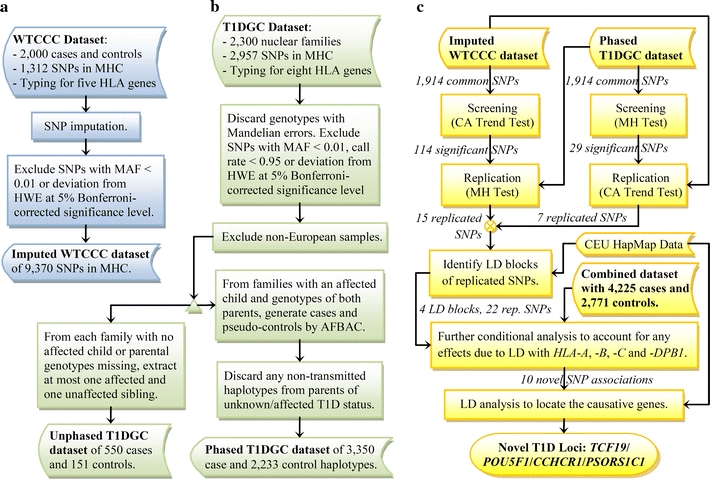
Summary of analytical plan. a Processing of WTCCC dataset. b Processing of T1DGC dataset. c Procedures of statistical and LD analyses
Results
Data preparation
The WTCCC dataset consists of 2,000 T1D cases and 1,504 controls, all from the British population. Samples were genotyped on the Affymetrix500K platform, with 1,312 SNPs across the MHC, of which 330 were also in the T1DGC dataset. Genotype information is available for five HLA loci: HLA-A, -B, -C, -DRB1 and -DQB1. Owing to the small number of common SNPs in the two datasets, imputation of genotypes was necessary using the CEU HapMap (The International HapMap Consortium 2005) samples as a reference. After imputation and imposing quality control by excluding SNPs with minor allele frequency (MAF) < 0.01 or Hardy–Weinberg equilibrium (HWE) p value below the Bonferroni’s-corrected 5% significance level, the dataset was expanded to 9,370 SNPs in the MHC.
The T1DGC MHC Fine Mapping Project is targeted at providing a dense SNP map to ensure comprehensive coverage in the MHC region. When compared with the WTCCC, the T1DGC dataset has a higher SNP density in the MHC. It includes 2,300 nuclear families with 9,749 individuals from nine cohorts, mainly of Caucasian ethnicity. It consists of genotypes for 2,957 SNPs as well as eight HLA genes, namely HLA-A, -B, -C, -DPA1, -DPB1, -DRB1, -DQA1 and -DQB1, in the classical MHC. Any Mendelian errors were set as missing values. Then, SNPs with MAF < 0.01 or call rate < 0.95 were removed, and the remaining SNPs were tested for HWE at 5% significance level after Bonferroni’s correction, leaving a total of 2,259 SNPs. To avoid any inflation of test statistics that could be introduced by population structure, 281 samples of non-European origin were discarded, leaving behind 2,240 families. Families without any affected child (15) or with parental genotypes missing (550) were then removed. This led to 1,675 trio families that have genotypes for both parents and at least one affected child. It is worth noting that parents of affected or unknown T1D status were kept at this point so that their affected child and unaffected spouse could be properly phased. In families with more than one affected child, the one with the lowest count of missing genotypes was selected. Using the affected family-based controls (AFBAC) approach (Thomson 1995), both the SNP and HLA genotypes of each trio were transformed into a case and a pseudo-control, which is composed of the non-transmitted alleles from the parents. Trio phasing and imputation of the SNP genotypes were performed by Beagle v.3.0 (Browning and Browning 2009). The multi-allelic HLA genotypes were phased by a novel algorithm. Details of the algorithm are discussed in the “Materials and methods”. After removing 146 and 971 haplotypes of pseudo-controls, respectively, from parents of affected and unknown T1D status, we arrived at a dataset of 3,350 and 2,233 phased case and control haplotypes, respectively, with a coverage of 2,259 SNPs in the MHC.
Screening and replication study conditional on DR/DQ
We first used the WTCCC dataset for initial screening. With phases unknown, we applied the Cochran–Armitage (CA) trend test (Armitage 1955; Freidlin et al. 2002) on SNP genotypes with subjects stratified by their DRB1 and DQB1 genotypes. By assuming a co-dominant mode of inheritance for the trend test, the dosage effect of an allele can be accounted. As DQA1 is in strong LD with DRB1 and DQB1, it is sufficient to condition only on the two genes when the typing of DQA1 is unavailable. This subject-based approach of stratification, similar to OCGM, has the advantage of capturing both the cis- and trans-effect of the tested loci on the DR/DQ genotypes. When compared with the conventional logistic regression technique for conditional analysis, which involves maximum likelihood estimations of multiple parameters, obtaining p values by exact conditional tests based on the stratification is a more direct approach to identify the positions of associated loci in the gene-dense MHC region with complicated LD. In this stage, only the 1,941 SNPs common to both dataset were tested. Out of these, 114 SNPs passed the two-tailed tests at 5% significance level with Bonferroni’s correction (p < 0.05/1941 ≈ 2.58 × 10−5).
In the next stage, a replication study was done using the T1DGC dataset. With haplotype phases inferred from the trio data, the Mantel–Haenszel (MH) test (Mantel and Haenszel 1959; Yu and Schaid 2007) was applied on each SNP by counting the number of transmitted and non-transmitted alleles stratified by their DRB1-DQA1-DQB1 haplotypes. The MH test has the advantage of being more sensitive when the effects across strata are generally in the same direction. The approach is similar in concept to OCHM, which tests for an additional effect over all haplotypes, in this case, the DR/DQ haplotypes. Haplotype-based matching is more specific at ruling out any effect due to LD on the same chromosome than the subject-based approach which stratifies on genotypes. Of the 114 SNPs which passed the initial screening, 15 were successfully replicated based on the false discovery rate (FDR) approach (Benjamini and Hochberg 1995) (p < 15 × 0.05/114 ≈ 6.58 × 10−3).
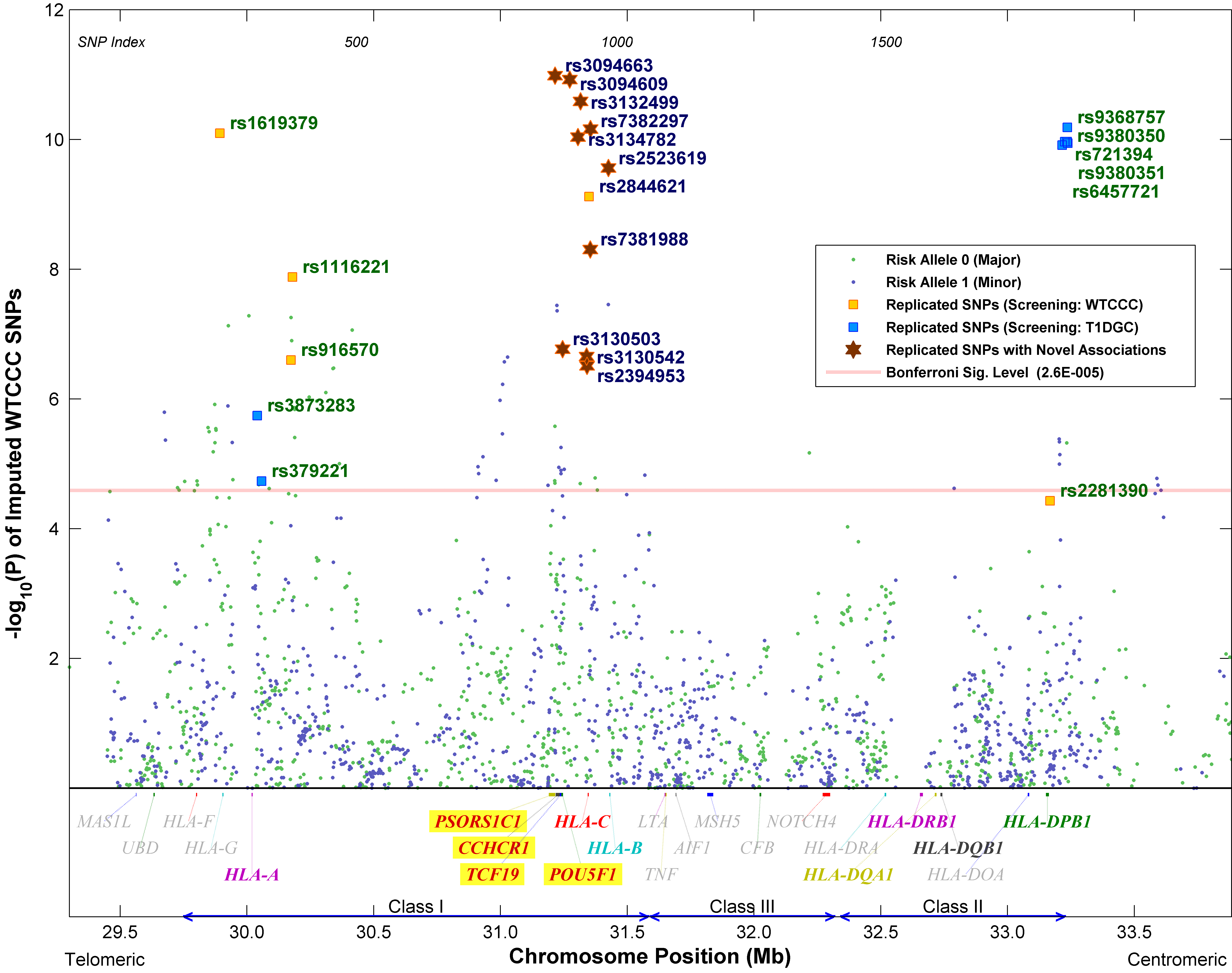
To capture those SNPs showing stronger associations in the T1DGC dataset than in the WTCCC dataset, the conditional analysis was carried out again with the roles of the two datasets switched: T1DGC dataset for initial screening and WTCCC dataset for replication. Out of the 1,941 common SNPs under test, 29 were able to pass the Bonferroni’s-corrected 5% significance level, of which 7 were successfully replicated at 5% FDR (p < 7 × 0.05/29 ≈ 0.0121). It was expected that a fewer number of SNPs passed the screening, since the T1DGC dataset is smaller in size and hence less powerful statistically. The seven newly replicated SNPs were all different from the 15 obtained in the previous round of analysis, making a total of 22 replicated SNPs. These SNPs, together with their conditional p values for the two datasets, are listed in Table 2. Plots of the conditional p values of all SNPs with respect to their chromosome positions are shown in Figs. 2 and 3, respectively, for the two datasets.
Table 2.
22 replicated SNPs with significant T1D associations in both T1DGC and WTCCC datasets
| SNPa | WTCCCb | T1DGCc | Combinedd | Remarks | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Pos (Chr. 6) | Allelese (Maj./Min.) | Control MAF | Case MAF | p f | Control MAF | Case MAF | p g | Control MAF | Case MAF | p f | |
| rs1619379i | 29893214 | CT | 0.422 | 0.394 | 3.98 × 10−11 | 0.393 | 0.393 | 3.89 × 10−4 | 0.410 | 0.394 | 8.00 × 10−11 | LD with HLA-A |
| rs3873283h | 30040979 | AG | 0.146 | 0.098 | 2.90 × 10−3 | 0.164 | 0.104 | 9.51 × 10−6 | 0.154 | 0.101 | 1.81 × 10−6 | |
| rs379221h | 30058119 | AG | 0.177 | 0.133 | 6.37 × 10−3 | 0.168 | 0.112 | 5.91 × 10−6 | 0.173 | 0.123 | 1.85 × 10−5 | |
| rs916570i | 30174010 | AG | 0.272 | 0.302 | 9.94 × 10−7 | 0.261 | 0.299 | 5.95 × 10−3 | 0.267 | 0.301 | 2.51 × 10−7 | |
| rs1116221i | 30179309 | CT | 0.283 | 0.291 | 1.14 × 10−10 | 0.241 | 0.268 | 6.38 × 10−3 | 0.265 | 0.280 | 1.32 × 10−8 | |
| rs3094663 i | 31215066 | CT | 0.280 | 0.288 | 8.48 × 10−10 | 0.283 | 0.254 | 5.30 × 10−3 | 0.281 | 0.272 | 1.04 × 10−11 | Blocks 1 –3: TCF19, POU5F1 and CCHCR1 |
| rs3130503 i | 31245144 | GA | 0.218 | 0.185 | 1.85 × 10−6 | 0.199 | 0.174 | 2.33 × 10−3 | 0.210 | 0.180 | 1.71 × 10−7 | |
| rs3094609 i | 31273545 | CT | 0.148 | 0.119 | 3.79 × 10−11 | 0.140 | 0.112 | 7.36 × 10−5 | 0.144 | 0.116 | 1.20 × 10−11 | |
| rs3134782 i | 31305612 | AG | 0.157 | 0.121 | 3.56 × 10−10 | 0.144 | 0.110 | 1.13 × 10−4 | 0.151 | 0.116 | 9.24 × 10−11 | |
| rs3132499 i | 31315899 | TC | 0.148 | 0.118 | 2.28 × 10−10 | 0.138 | 0.110 | 9.09 × 10−5 | 0.144 | 0.114 | 2.60 × 10−11 | |
| rs3130542 i | 31340090 | GA | 0.195 | 0.157 | 1.84 × 10−6 | 0.202 | 0.163 | 1.48 × 10−3 | 0.198 | 0.160 | 2.23 × 10−7 | |
| rs2394953 i | 31341332 | AG | 0.195 | 0.157 | 1.84 × 10−6 | 0.200 | 0.163 | 5.44 × 10−4 | 0.197 | 0.160 | 3.10 × 10−7 | |
| rs2844621i | 31349516 | TC | 0.305 | 0.281 | 4.05 × 10−12 | 0.278 | 0.253 | 2.40 × 10−3 | 0.294 | 0.268 | 7.62 × 10−10 | |
| rs7381988 i | 31354682 | AG | 0.166 | 0.131 | 1.76 × 10−8 | 0.151 | 0.119 | 2.65 × 10−4 | 0.160 | 0.125 | 4.98 × 10−9 | |
| rs7382297 i | 31355046 | GT | 0.149 | 0.117 | 4.31 × 10−10 | 0.137 | 0.110 | 8.59 × 10−5 | 0.144 | 0.114 | 6.94 × 10−11 | |
| rs2523619 i | 31426123 | TC | 0.203 | 0.195 | 7.07 × 10−9 | 0.204 | 0.160 | 9.81 × 10−4 | 0.204 | 0.179 | 2.77 × 10−10 | PSORS1C1 |
| rs2281390i | 33167647 | GT | 0.113 | 0.065 | 3.72 × 10−6 | 0.170 | 0.110 | 3.43 × 10−3 | 0.137 | 0.085 | 3.72 × 10−5 | LD with HLA-B |
| rs6457721h | 33215729 | TG | 0.181 | 0.101 | 6.26 × 10−5 | 0.170 | 0.089 | 1.50 × 10−8 | 0.176 | 0.096 | 1.22 × 10−10 | Block 4: LD with HLA-DPB1 |
| rs721394h | 33225796 | CT | 0.172 | 0.095 | 3.81 × 10−5 | 0.168 | 0.084 | 2.91 × 10−9 | 0.170 | 0.090 | 1.09 × 10−10 | |
| rs9380350h | 33234843 | CT | 0.172 | 0.096 | 4.28 × 10−5 | 0.169 | 0.084 | 2.23 × 10−9 | 0.171 | 0.091 | 1.08 × 10−10 | |
| rs9368757h | 33236355 | GA | 0.170 | 0.097 | 9.15 × 10−5 | 0.163 | 0.081 | 2.04 × 10−9 | 0.167 | 0.090 | 6.55 × 10−11 | |
| rs9380351h | 33237724 | GA | 0.171 | 0.098 | 1.53 × 10−4 | 0.163 | 0.081 | 2.62 × 10−9 | 0.168 | 0.091 | 1.15 × 10−10 | |
aSNP Information based on NCBI genome build 36.3
b2,000 cases and 1,504 controls from the British population
c3,350 case and 2,233 control haplotypes of European origin
d4,225 cases and 2,771 controls combined from the WTCCC and T1DGC datasets
eAlleles in reference assembly strand orientation with risk alleles underlined
fCochran–Armitage trend test based on subject counts stratified by their DRB1 and DQB1 genotypes
gMantel–Haenszel test based on chromosome counts stratified by their DRB-DQA1-DQB1 haplotypes
hSNPs that pass screening with WTCCC dataset and replication with T1DGC dataset
iSNPs that pass screening with T1DGC dataset and replication with WTCCC dataset. All SNPs showing novel associations are highlighted in bold
Fig. 2.
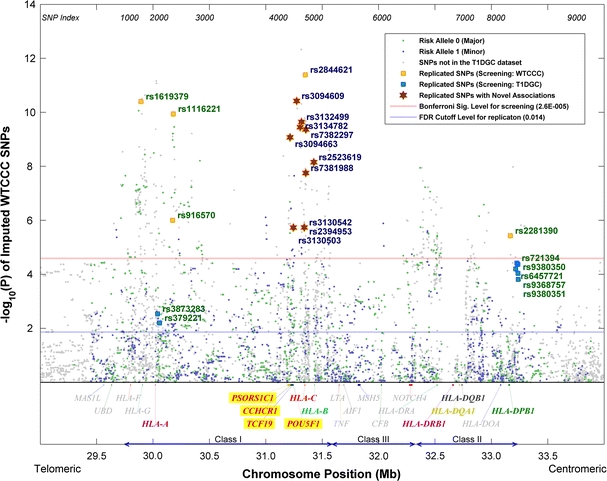
p values of the WTCCC dataset conditional on the HLA-DRB1-DQB1 genotypes based on the stratified Cochran-Armitage trend test. The classical MHC subregions and some relevant genes are included for reference. Position is along chromosome 6, NCBI genome build 36.3
Fig. 3.
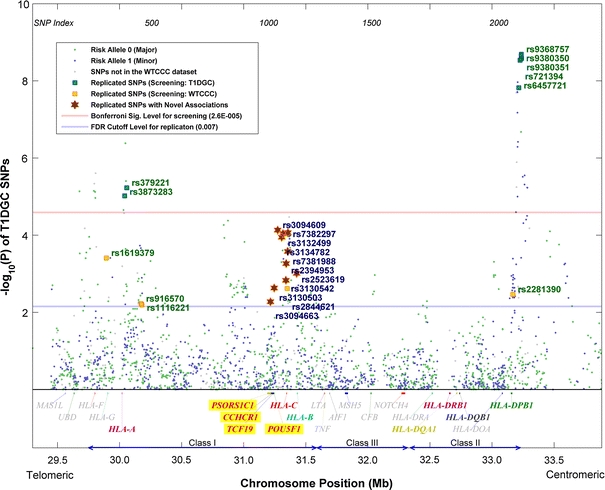
p values of the T1DGC dataset conditional on the HLA-DRB1-DQA1-DQB1 haplotypes based on Mantel–Haenszel test. The classical MHC subregions and some relevant genes are included for reference. Position is along chromosome 6, NCBI genome build 36.3
LD analysis was performed on the replicated SNPs using Haploview (Barrett et al. 2005) based on the CEU HapMap Phase II haplotypes (Fig. 4). Four LD blocks were identified by the confidence interval method (Gabriel et al. 2002) using the default settings, i.e. a block is created if 95% of informative comparisons are strong LD, with the upper and lower 95% confidence bounds for D′ > 0.98 and > 0.7, respectively.
Fig. 4.
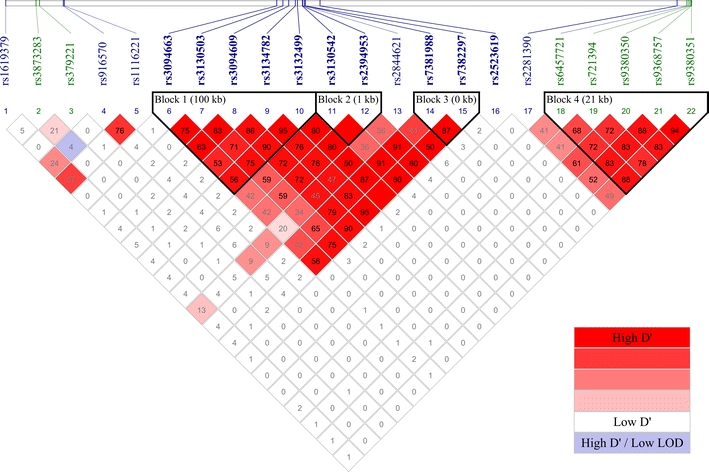
LD between the 22 replicated SNPs. The figure in each box gives the first two decimal places of the r 2 value. LD blocks identified by the confidence interval method were outlined. IDs in blue indicate SNPs that pass screening with WTCCC dataset, whereas IDs in green indicate SNPs that pass screening with T1DGC dataset. All SNPs showing novel associations are highlighted in bold (color figure online)
Further conditional analysis adjusted for HLA-A, -B, -C and -DPB1
To verify whether the associations of any of these loci were the results of LD with some known T1D-associated HLA genes in their neighborhood, more conditional tests were applied. This was done by including the gene to be controlled for as a stratification factor in addition to the DRB1 and DQB1 genes in the CA trend test. The five replicated SNPs within 200 kb from HLA-A were tested for T1D association conditional on the HLA-A, -DQB1 and -DRB1 genotypes. Likewise, tests on Blocks 1–3, rs2844621 and rs2523619 were controlled for HLA-B and -C, and tests on Block 4 and rs2281390 controlled for HLA-DPB1 and -DPA1, due to their proximities with the respective genes on the chromosome. If the p value of the tested locus then becomes insignificant, we can conclude that it is in LD with one or more alleles of the additional stratifying gene which exhibit similar confounding effects on the DR/DQ genotypes.
In general, the overall statistical power of the tests decreases with the introduction of an additional stratification factor due to a fewer number of samples in each stratum. Therefore, we used all samples combined from the two datasets when adjusting for the HLA-A, -B and -C genotypes in the conditional analysis based on the CA trend test. However, since the genotypes of DPB1 and DPA1 are not available in the WTCCC dataset, we could only apply the MH test on the T1DGC dataset to control for the two genes. By counting the number of haplotypes, the approach can help in improving the statistical power, but it is also prone to underestimate the trans-effect of an SNP on the confounding haplotype, leading to more conservative results in general.
The combined dataset has a total of 4,225 cases and 2,771 controls. Apart from the 3,675 cases and 2,620 controls previously utilized in screening and replication, it includes an additional 550 cases and 151 controls with unknown haplotype phases. These samples come from families in the T1DGC dataset with parental genotypes missing, and at most one affected and one unaffected siblings were taken from each family (Curtis 1997). In combining the two datasets, the alleles of all common SNPs were made consistent based on their strand orientation information. HLA genotypes of high resolution were preferred, but those of low resolution were also used.
To further enhance the statistical power, we implemented a partitioning algorithm that reduces the number of HLA strata without compromising the confounding effects of the factors. For each unique stratum, the algorithm progressively grows a subset of the HLA alleles until there is not a remaining allele in the stratum that can exert an additional effect of p < 0.5. The consolidated set of alleles is then adopted as the new stratum. The conservative choice of p < 0.5 is to ensure that even any weak effects of the stratification factors are retained after the partitioning process. For instance, there are altogether 4,011 strata for the HLA-DRB1, -DQB1 and -A genotypes, resulting in an effective sample size of 1,133 cases and 453 controls, after those strata consisting of all controls or all cases were eliminated. Our partitioning algorithm reduces the number of strata to 1,408 and hence increases the effective sample size to 2,302 cases and 745 controls.
After controlling for the HLA-DRB1-DQB1-A genotypes, the five replicated SNPs near HLA-A became insignificant, indicating their effects were mostly due to LD with the gene. Similarly, Blocks 1–3 and SNPs rs2844621 and rs2523619 were tested for association conditional, respectively, on the HLA-DRB1-DQB1-B and HLA-DRB1-DQB1-C genotypes. With five independent loci under test, the 5% significance level with LD-adjusted Bonferroni’s correction (Duggal et al. 2008) becomes 0.05/5 or 0.01. Of the 11 replicated SNPs under investigation, SNPs rs3094663 from Block 1, rs7381988 from Block 3 and rs2523619 were able to pass both conditional tests with p < 0.01. If the 5% FDR was adopted instead, seven more SNPs could pass both tests, with two of them from Block 2. The results show that Blocks 1–3 and SNP rs2523619 harbor additional effects on T1D not due to LD with HLA-B and -C. Finally, conditional tests stratified, respectively by the HLA-DRB1-DQB1-DPB1 and HLA-DRB1-DQB1-DPA1 genotypes were performed on Block 4 and SNP rs2281390. While Block 4 was disproved after adjusting also for DPB1, rs2281390 was able to pass the tests with p < 0.05/2 = 0.025. p values of the conditional tests are summarized in Table 3.
Table 3.
p values of replicated SNPs conditional on nearby T1D-associated HLA genes in addition to DRB1 and DQB1
| Block | SNP ID | SNP Pos | p | |
|---|---|---|---|---|
| CA trend test conditional ona: | DR - DQ - A | |||
| rs1619379 | 29893214 | 0.4107 | ||
| rs3873283 | 30040979 | 0.5397 | ||
| rs379221 | 30058119 | 0.1085 | ||
| rs916570 | 30174010 | 0.4594 | ||
| rs1116221 | 30179309 | 0.4687 | ||
| Block | SNP ID | SNP Pos | p | |
|---|---|---|---|---|
| CA trend test conditional ona: | DR - DQ - B | DR - DQ - C | ||
| 1 | rs3094663 b | 31215066 | 3.18 × 10−3b | 2.57 × 10−3b |
| rs3130503 c | 31245144 | 0.0199c | 0.0120 | |
| rs3094609 c | 31273545 | 0.0246c | 3.13 × 10−3b | |
| rs3134782 c | 31305612 | 0.0259c | 2.52 × 10−3b | |
| rs3132499 c | 31315899 | 0.0388c | 5.88 × 10−3b | |
| 2 | rs3130542 c | 31340090 | 7.68 × 10−3b | 0.0135c |
| rs2394953 c | 31341332 | 0.0107c | 0.0217c | |
| rs2844621 | 31349516 | 7.05 × 10−3b | 0.0515c | |
| 3 | rs7381988 b | 31354682 | 9.84 × 10−3b | 2.36 × 10−3b |
| rs7382297 c | 31355046 | 0.0480c | 7.69 × 10−3b | |
| rs2523619 b | 31426123 | 5.74 × 10−4 | 3.21 × 10−5b | |
| Block | SNP ID | SNP Pos | p | |
|---|---|---|---|---|
| MH test conditional ond | DR - DQ - DPB1 | DR - DQ - DPA1 | ||
| 4 | rs2281390 | 33167647 | 0.0108b | 3.08 × 10−3b |
| rs6457721 | 33215729 | 0.3820 | 4.12 × 10−8 | |
| rs721394 | 33225796 | 0.2711 | 1.20 × 10−8 | |
| rs9380350 | 33234843 | 0.2349 | 9.80 × 10−9 | |
| rs9368757 | 33236355 | 0.2741 | 8.86 × 10−9 | |
| rs9380351 | 33237724 | 0.2937 | 1.11 × 10−8 | |
aUsing combined dataset of 4,225 cases and 2,771 controls
bSNPs or p values that pass the LD-adjusted 5% Bonferroni’s significance level. SNPs showing novel associations are highlighted in bold
cSNPs or p values that pass the 5% FDR criterion
dUsing T1DGC dataset of 3,350 case and 2,233 control haplotypes
The p values of all SNPs conditional on the different HLA genes were plotted in Figs. 5a–d. In Fig. 5a, the stratification on HLA-A almost completely nullified all SNP associations in the neighborhood, while the associations of SNPs farther away remained prominent. This proves the robustness and effectiveness of our partitioning algorithm in reducing the number of HLA strata while retaining most of their confounding effects. Upon conditioning on HLA-B, the associations of rs2523619 and rs3094663 were more outstanding than other replicated SNPs in that region. A considerable level of LD was observed between Block 4 and HLA-B, even though they are about 1.8 Mb apart. In particular, the association of rs2281390 was totally gone after conditioning on HLA-B. Therefore, the SNP was not considered a novel association, despite its passing of the tests conditioned on HLA-DPB1 and -DPA1. When compared with HLA-B, conditioning on HLA-C had smaller impact on the p values and the LD with HLA-DPB1 was seemingly confined to a shorter range.
Fig. 5.
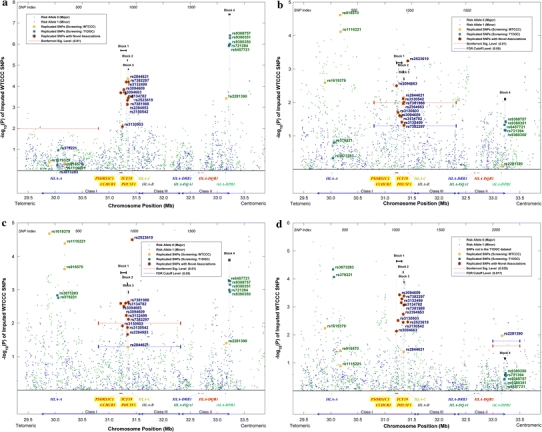
p values conditional on HLA-DRB1-DQB1 together with HLA- a A, b B, c C and d DPB1, respectively. p values of a–c are generated by applying stratified Cochran–Armitage trend test on the combined dataset, whereas p values in d are generated by applying Mantel–Haenszel test on the phased T1DGC haplotypes. Significance levels were indicated in the appropriate regions under test. The classical MHC subregions and some relevant genes are included for reference. Position is along chromosome 6, NCBI genome build 36.3
p values and odds ratios (OR) of each novel SNP association in individual HLA haplotype strata are available in Supplementary Tables 2–7. The statistics were obtained by applying the Fisher’s exact test on the number of major and minor alleles on each HLA haplotype based on the phased T1DGC dataset. For example, with regard to DRB1-DQB1-DQA1, rs2523619 has p = 0.011 and OR 0.3 and OR 0.43, respectively, for the DRB1*802-DQB1*402-DQA1*401 and DRB1*101-DQB1*501-DQA1*101 haplotypes. If HLA-C is taken into consideration as well, we found that the effect of the SNP is particularly strong on C*702, with p = 0.0002, OR 0.07 and p = 0.0264, OR 0.08, respectively, for the DRB1*101-DQB1*501-C*702 and DRB1*802-DQB1*402-C*702 haplotypes. The minor allele of the SNP also tends to exhibit susceptibility across strata.
Locating causal genes through detailed LD analysis
To further track down the causal genes, detailed LD analysis was carried out and the results were summarized in Supplementary Table 1. Of the ten novel SNP associations, rs2394953 and rs7382297 were in perfect LD (r 2 = 1) with rs3130542 and rs3132499, respectively, and can thus be excluded. For the remaining eight SNPs, we examined their LD with SNPs residing in the functional areas of some nearby genes. A list of gene-affiliated SNPs was retrieved through NCBI Entrez.
The 30.7–32 Mb region on chromosome 6 was considered. The range was chosen to encompass the SNPs under study with flanking margins of 500 kb. Of the over 10,000 SNPs obtained, only a small fraction was available in the CEU HapMap haplotypes. After removing the redundant SNPs, i.e. those from the same gene and in perfect LD, we had 115 non-synonymous and 502 synonymous SNPs, covering about 50 and 84% of the genes in that region.
For the 7 SNPs in Blocks 1–3, rs2073721 of gene TCF19 (31.237 Mb) emerged consistently as the one showing the strongest LD (in terms of r 2) among all the non-synonymous SNPs, despite the fact that two of the SNPs in Block 1, rs3094663 and rs3130503, are in the introns of PSORS1C1 (31.2 Mb) and POU5F1 (31.243 Mb). In particular, D′ = 0.959 and r 2 = 0.818 between rs3094663 and rs2073721, and D′ = 1 and r 2 = 0.848 between rs3130503 and rs2073721. In addition, synonymous SNPs rs1065461 and rs2073722 of TCF19 showed moderate to strong LD with the SNPs in Blocks 1–3, with r 2 ranging from 0.59 to 1 proving further the relevance of the gene.
Apart from TCF19, POU5F1 and CCHCR1 (31.22 Mb) also have several SNPs in considerable LD with Blocks 1–3. For instance, the non-synonymous SNP rs3130932 in POU5F1 has D′ = 1 and r 2 = 0.675 with rs3130503. For CCHCR1, two non-synonymous SNPs, rs130066 and rs3130453, have, respectively, an LD of D′ = 1, r 2 = 0.28 and D′ = 1, r 2 = 0.31 with rs3130503.
Replicated SNP rs2523619 has weak LD (r 2 < 0.3) with all the non-synonymous SNPs in the region. Nevertheless, among the ten synonymous SNPs having the strongest LD with rs2523619, six were from PSORS1C1, of which three were at the top and the highest LD being D′ = 0.753 and r 2 = 0.478 with rs3815087. The LD between the eight replicated SNPs with novel associations and the missense SNPs in three of the genes were shown in Fig. 6a–c. The risk alleles of the replicated SNPs in Blocks 1–3, which happened to be all minor alleles, are consistently in stronger LD with the major alleles of the missense SNPs of the four genes under study. However, the risk allele of rs2523619 does not always map to the same missense allele as the other replicated SNPs. It is only in unison with others for PSORS1C1.
Fig. 6.
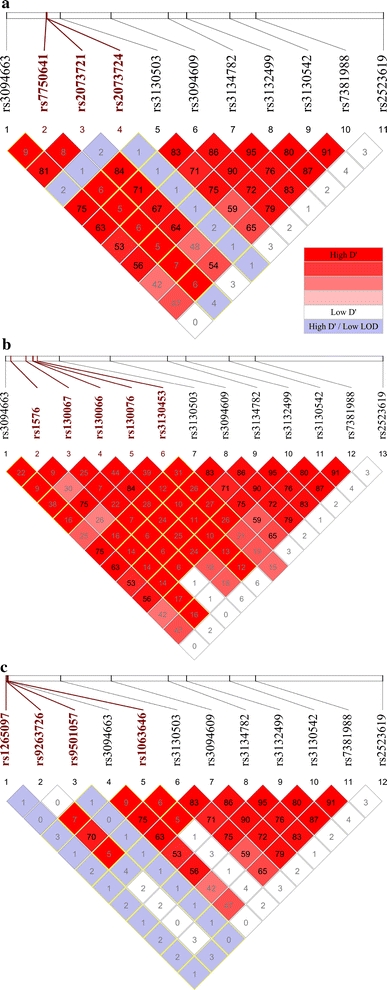
LD between the eight replicated SNPs with novel T1D associations and the missense SNPs within a TCF19, b CCHCR1 and c PSORS1C1. Only SNPs available in the CEU HapMap dataset were studied and any redundant SNPs were removed. All missense SNPs are highlighted in bold and red. Boxes of D′ = 1 are outlined in yellow (color figure online)
The effects of the missense alleles on the amino acids and their side chain properties, which may in turn influence the protein structures, are summarized in Table 4. The results of LD analysis hint at the potential roles of TCF19, POU5F1, CCHCR1 and PSORS1C1 in the etiology of T1D, with TCF19 supported by the strongest r 2 measure.
Table 4.
Protein-coding information for selected missense SNPs of TCF19, POU5F1, CCHCR1 and PSORS1C1
| Gene | No. of exons | No. of isoforms | Missense SNP | Allele changea | Residue change | Residue polarity and charge change | ||
|---|---|---|---|---|---|---|---|---|
| ID | Position | Exon No. | ||||||
| TCF19 | 4 | 2 | rs7750641 | 31237289 | 3 | C→T | P→S | np→p |
| rs2073721 | 31237595 | 3 | G→A | V→M | np→np | |||
| rs2073724 | 31237686 | 3 | C→T | P→L | np→np | |||
| POU5F1 | 4 | 6 | rs3130932 | 31241922 | 1 | G→T | R→M | p+→np |
| CCHCR1 | 18 | 14 | rs1576 | 31218370 | 18 | C→G | S→C | p→np |
| rs130067 | 31226490 | 6 | A→C | E→D | p−→p− | |||
| rs130066 | 31230294 | 4 | C→G | S→R | p→p+ | |||
| rs130076 | 31230461 | 4 | C→T | R→W | p+→np | |||
| rs3130453 | 31232828 | 2 | C→T | W→X | np→(N/A) | |||
| PSORS1C1 | 6 | 2 | rs1265097 | 31214438 | 5 | G→T | P→T | np→p |
| rs9263726 | 31214478 | 5 | G→A | R→H | p+→p+ | |||
| rs9501057 | 31214495 | 5 | C→T | P→S | np→p | |||
| rs1063646 | 31215627 | 6 | C→T | P→L | np→np | |||
Abbreviations for amino acid side chain properties: p polar, np, non-polar; +, positively charged; −, negatively charged
aFrom major to minor alleles
Discussion
TCF19 (Transcription Factor 19, previously known as SC1) is a late growth-regulated gene with a putative amino acid sequence that bears the characteristics of a trans-activating factor; hence, implying its role in the regulation of expression of other genes necessary for the later stages of cell cycle progression (Ku et al. 1991). TCF19 is one of the transcription factors involved in cell proliferation or differentiation being up-regulated in human pro-B and pre-B cells (Hystad et al. 2007). It is also highly expressed in germinal center cells. A non-synonymous SNP in TCF19, rs7750641, was found to be associated with lymphocyte count, mean cell hemoglobin, white blood cell count, hematocrit count, and eosinophil count (p = 2.3 × 10−7) in a multivariate analysis (Ferreira et al. 2009). Although the cause is largely unknown, there is evidence that the destruction of islet β-cells in T1D is the result of a disorder of immunoregulation: islet β-cell autoreactive T cells dominate over protective regulatory T cells, which develop inadequately due to some genetic or environmental factors. TCF19 may be one of the key players in maintaining such immunological balance.
POU5F1 (POU class 5 Homeobox 1, also known as OCT3 or OCT4) encodes a transcription factor containing a POU homeodomain. This transcription factor is involved in the embryonic development, especially during early embryogenesis, and is necessary for embryonic stem cell pluripotency. It is also essential for endoderm formation (Lunde et al. 2004), which is a precursor to pancreas development. Specifically, the gene is found to be expressed in pancreatic endocrine progenitor cells.
CCHCR1 (coiled-coil alpha-helical rod protein 1, formerly known as HCR), a plausible candidate gene for psoriasis (Asumalahti et al. 2002), was shown to promote steroidogenesis by interacting with the steroidogenic acute regulator protein (StAR) (Sugawara et al. 2003). The protein is abundantly expressed in heart, liver, skeletal muscle, kidney and pancreas. Steroid hormones mediate various vital physiological functions, such as the regulation of metabolism and immune response. In some T1D patients, steroids appear to lower the number of islet cell antibodies and prolong the time between the development of the antibodies and the onset of symptoms. On the other hand, steroids can increase insulin resistance in liver and skeletal muscle, thus reducing glucose uptake in cells. The changes in the insulin sensitivity are known to have impact on the T1D process (Greenbaum 2002).
Two synonymous SNPs in exon 2 of PSORS1C1 (psoriasis susceptibility 1 candidate 1, earlier known as SEEK1) were reported to be associated with psoriasis in the Swedish population even upon stratification for the predisposing HLA-Cw*0602 allele (Holm et al. 2003). The gene has two protein isoforms, and is expressed in pancreas and kidney among other tissues. However, the function of the protein in biological processes remains largely unknown. The two missense SNPs, rs1265097 and rs9501057, in complete LD (D′ = 1) with all the eight novel SNP associations, are both located in exon 5 of PSORS1C1, which is translated in both isoforms.
In summary, we performed a detailed stratified analysis in search of additional T1D loci in the MHC using the large and independent WTCCC and T1DGC datasets. To facilitate the analysis, two novel algorithms were implemented: one for phasing the multi-allelic HLA genotypes in trio families, and the other one for partitioning the HLA strata, while retaining their confounding effects in conditional testing, with the goal to enhance the statistical power. When compared with the likelihood-based approaches, our proposed model-free approach can more accurately determine the strength of association of each SNP over other potential factors, and is computationally feasible for large datasets. Therefore, we believe it is a promising framework for future conditional analyses in the MHC. Our study has identified eight novel SNP associations which established the evidence for an additional T1D locus in the MHC class I region at around 31.3 Mb on chromosome 6. Detailed LD analysis has singled out four potential causal genes responsible for the observed SNP associations. They include TCF19, POU5F1, CCHCR1 and PSORS1C1, all within a 56 kb region. Owing to high gene density and complicated LD, further fine mapping and analyses are necessary for pinpointing the exact causal polymorphism in that region.
Materials and methods
Wellcome Trust Case–Control Consortium (WTCCC) genotype dataset for initial screening
The dataset has 2,000 T1D cases and 1,504 controls (from the 1958 British Birth Cohort). All samples are from the British population and genotyped on the Affymetrix500K platform. HLA typing information is available for five loci: HLA-A, -B, -C, -DRB1 and -DQB1. To improve SNP density in the MHC, the IMPUTE (Howie et al. 2009) software was run on the dataset based on the CEU HapMap (The International HapMap Consortium 2005) haplotypes with ancestry from northern to western Europe, expanding the coverage to 9,878 SNPs and at the same time inferring any missing genotypes. The threshold for calling genotypes in IMPUTE was set to be 0.9, i.e. for each SNP of each individual, the genotype will be treated as missing if the maximum genotype probability is < 0.9. Furthermore, SNPs with MAF < 0.01 in all samples or p < 0.05/9878 ≈ 5.06 × 10−6 for HWE tests in the controls were excluded, resulting in a total of 9,370 SNPs. Information on the access to the WTCCC T1D dataset is available at http://www.wtccc.org.uk/info/access_to_data_samples.shtml.
Type 1 diabetes genetics consortium (T1DGC) genotype dataset for replication study
The dataset consists of 2,300 trio families with 9,749 individuals from 9 cohorts; namely, Asia Pacific (AP; 191), British Diabetic Association (BDA; 418), Danish (DAN; 147), European (EUR; 475), Human Biological Data Interchange (HBDI; 431), Joslin Diabetes Center (JOS; 112), North American (NA; 334), Sardinian (SAR; 78) and United Kingdom (UK; 114). Based on the primary, secondary and tertiary ethnic groups of each subject, 281 individuals of non-European origin from 80 families (AP: 21; EUR: 14; NA: 40; UK: 5) were removed, to avoid any statistical bias due to the existence of population structure. After excluding parent samples with missing genotypes or unknown/affected T1D status, only 1,675 families remained eligible for AFBAC.
The fine mapping data include 2,957 SNPs in the MHC genotyped on two oligonucleotide pool assays (OPA) using the Illumina Golden Gate platform at the Wellcome Trust Sanger Institute. HLA typing information is available for eight loci: HLA-A, -B, -C, -DPA1, -DPB1, -DRB1, -DQA1 and -DQB1. Quality control on the SNP genotypes was performed using PLINK (Purcell et al. 2007). First, any Mendelian errors were set as missing. Then, SNPs with call rate < 0.95 and MAF < 0.01 were discarded. The remaining 2,338 SNPs were tested for HWE and those with p < 0.05/2338 ≈ 2.14 × 10−5 were removed, resulting in a total of 2,259 SNPs. Information on access to data and samples is available at http://www.t1dgc.org/views/vw_access.cfm, and details of the dataset can be found in (Brown et al. 2009).
Affected family-based controls
Families from the T1DGC dataset were converted into cases and pseudo-controls using the AFBAC method (Thomson 1995). Samples of any affected parents were removed. In families with multiple children, all but the child with the least missing genotypes were discarded. The affected child became the case, and the two non-transmitted haplotypes from the unaffected parents were merged into a pseudo control. For the SNP genotypes, the AFBAC procedure was done by Beagle v.3.0 (Browning and Browning 2009), which at the same time performed haplotype phase inference and imputation for missing values. For the multiallelic HLA genotypes, AFBAC was implemented using Matlab.
Phasing of multi-allelic HLA genotypes in trio families
In trio families, haplotype phases can be unambiguously determined, except for the case when both parents and the child are heterozygous for the same genotype, say AB. With highly diversified HLA alleles, only about 1.8% of the genotypes fall into that category. In those situations, the phase can be inferred from the haplotypes of the nearby HLA genes. In this work, we considered all HLA genes within 1 Mb of the target whose phase was being inferred. However, if there were fewer than two genes within that distance, the closest two were picked, even though they might be outside the range or both lie on the same side of the target. Those nearby genes with directly determined phases made up four known haplotypes: one transmitted and one non-transmitted haplotype from each parent. Based on the other known haplotypes in the dataset, we had counts n Ai, n Bi of the number of A alleles and B alleles, respectively, with haplotype i. A one-sided p value was then obtained using the MH test statistics, with each end of the p value in support of one of the two possible solutions for phasing. The inferred phase was adopted when the two-sided p value was < 0.05. Otherwise, the algorithm was run again with reference to one fewer gene, excluding the most distant one first. The process was repeated until a two-sided p < 0.05 was reached using the greatest number of closest genes in computing the statistics. If none of the gene combinations could result in p < 0.05, the solution supported by the smallest p value was adopted. Overall, the phase could be inferred at an average p value of 0.0034.
p value-based partitioning of HLA strata
The objective of the algorithm was to improve the statistical power of stratified analysis by reducing the number of HLA strata without compromising their confounding effects in the conditional analysis. Each stratum consisted of a set of genotypes G i, where i = 1,…,n. We first selected the minimum set of G i that gave a T1D association with p < threshold, with preference given to primary factors HLA-DRB1 and -DQB1. In this work, we chose a conservative threshold of 0.5 to ensure that even factors with weak confounding effects are retained. Then, we progressively grew the set of genotypes by including the next one that shows the strongest additional association with p < 0.5. The process continued until there were no more genotypes in the stratum that could further differentiate the cases and controls with p < 0.5. This effectively eliminated any uninformative factors in a stratum. The power of the conditional analysis was thus improved with more samples per stratum. This approach also works for haplotype strata.
Haplotype-based MH test for conditional analysis
The method was used to test the T1D association of a locus, an SNP in this case, conditional on the DR/DQ haplotypes, the primary genetic factors of the disease. It is an extension to the conditional haplotype method (CHM), such that an overall p value across all DR/DQ haplotype strata is obtained for each SNP by applying the MH test statistic (Mantel and Haenszel 1959; Yu and Schaid 2007). The only assumption of the statistic is the randomization of subjects into the levels of the factor. Let there be m cases and n controls for the DR/DQ haplotype stratum i. Suppose N out of the (m + n) haplotypes carry allele 1 at the test SNP with x of them being controls. Then, variable x follows the hypergeometric distribution with mean and variance given by
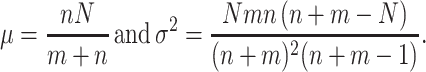 |
To test whether an SNP is associated with the disease conditional on the primary haplotypes, statistics of individual strata can be combined into one MH statistic
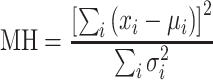 |
Under the null hypothesis that there is no association in any stratum, the MH statistic has a χ2 distribution with one degree of freedom. For the asymptotic distribution to hold, it only requires the overall sample size be reasonably large. Since the MH method is targeted at detecting average effects across strata, it is a common method for assessing average partial association. All haplotype counts were determined using Matlab, and the conditional p values of each SNP were calculated by the mantelhaen.test function in R’s stats library.
Genotype-based CA trend test with stratification for conditional analysis
The CA trend test (Armitage 1955) is typically used in the analysis of ordered categorical data and is a common genotype-based test for association studies (Armitage 1955; Freidlin et al. 2002). For a biallelic marker with alleles denoted by a and A, there are three possible genotypes {aa, aA and AA} indexed by k = 0, 1, 2. The categories are ordered by an increasing number of allele A in the genotypes. For each DR/DQ genotype i, let m k, n k be the number of cases and controls of genotype k, and M, N be the total number of cases and controls within the stratum. Then, the CA statistic is given by
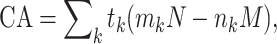 |
where t k are weights, and the expression can be viewed as the sum of differences between the fraction of cases and the fraction of controls across the genotype categories. Under the null hypothesis that there is no association in any category, the CA statistic is a zero-mean normal distribution at large sample size. The choice of weights t k for optimum statistical power is dependent on the type of associations. For instance, the optimum t values for testing whether allele A is dominant or recessive over allele a are (0, 1, 1) and (1, 1, 0), respectively. In this study, we chose t = (0, 1, 2), which is optimum for testing codominant or additive inheritance and often used in genome-wide association studies. The independence_test function in R’s coin library was used to perform the trend tests with stratification by DR/DQ genotypes. Details of the conceptual framework and implementation of the conditional inference procedures can be found in (Hothorn et al. 2008).
Linkage disequilibrium analysis
The LD among the 22 replicated SNPs was analyzed using Haploview v.4.1 (Barrett et al. 2005). The D′ and r 2 measure was generated for each SNP pair based on the HapMap CEU haplotypes for data release #22, with a total of 120 haplotypes of the northern and western European ancestry. The dataset consists of three different files: data, sample and legend, which can be downloaded from http://hapmap.ncbi.nlm.nih.gov/downloads/phasing/2007-08_rel22/phased/. Only SNPs under investigation were extracted from the haplotype files for input to Haploview.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
p values of the combined dataset conditional on HLA-DRB1-DQB1 genotypes (PNG 310 kb)
Results of LD analysis for locating the causal genes (XLS 44 kb)
p values of each novel SNP association in individual HLA haplotype strata (XLS 153 kb)
Supplementary materials are available at Human Genetics’ website.
Acknowledgments
This study utilizes data generated by the Wellcome Trust Case–Control Consortium and also resources provided by the Type 1 Diabetes Genetics Consortium, a collaborative clinical study sponsored by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institute of Allergy and Infectious Diseases (NIAID), National Human Genome Research Institute (NHGRI), National Institute of Child Health and Human Development (NICHD), and Juvenile Diabetes Research Foundation International (JDRF) and supported by U01 DK062418. A full list of the investigators who contributed to the generation of the data is available from http://www.wtccc.org.uk. Funding for the project was provided by the Wellcome Trust under award 076113. We thank Prof. Tian Zheng, Department of Statistics, Columbia University, for helpful discussions.
Conflict of interest
The authors declare no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Alper CA, Larsen CE, Dubey DP, Awdeh ZL, Fici DA, Yunis EJ. The haplotype structure of the human major histocompatibility complex. Hum Immunol. 2006;67:73–84. doi: 10.1016/j.humimm.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Aly TA, Baschal EE, Jahromi MM, Fernando MS, Babu SR, Fingerlin TE, Kretowski A, Erlich HA, Fain PR, Rewers MJ, Eisenbarth GS. Analysis of single nucleotide polymorphisms identifies major type 1A diabetes locus telomeric of the major histocompatibility complex. Diabetes. 2008;57:770–776. doi: 10.2337/db07-0900. [DOI] [PubMed] [Google Scholar]
- Armitage P. Tests for linear trends in proportions and frequencies. Biometrics. 1955;11:375–386. doi: 10.2307/3001775. [DOI] [Google Scholar]
- Asumalahti K, Veal C, Laitinen T, Suomela S, Allen M, Elomaa O, Moser M, de Cid R, Ripatti S, Vorechovsky I, Marcusson JA, Nakagawa H, Lazaro C, Estivill X, Capon F, Novelli G, Saarialho-Kere U, Barker J, Trembath R, Kere J. Coding haplotype analysis supports HCR as the putative susceptibility gene for psoriasis at the MHC PSORS1 locus. Hum Mol Genet. 2002;11:589–597. doi: 10.1093/hmg/11.5.589. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C, Plagnol V, Pociot F, Schuilenburg H, Smyth DJ, Stevens H, Todd JA, Walker NM, Rich SS. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41:703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57:289–300. [Google Scholar]
- Brown WM, Pierce J, Hilner JE, Perdue LH, Lohman K, Li L, Venkatesh RB, Hunt S, Mychaleckyj JC, Deloukas P (2009) Overview of the MHC fine mapping data. Diabetes Obes Metab 11(Suppl 1):2–7 [DOI] [PMC free article] [PubMed]
- Browning BL, Browning SR. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009;84:210–223. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz TD, Valdes AM, Santiago A, Frazer de Llado T, Raffel LJ, Zeidler A, Rotter JI, Erlich HA, Rewers M, Bugawan T, Noble JA. DPB1 alleles are associated with type 1 diabetes susceptibility in multiple ethnic groups. Diabetes. 2004;53:2158–2163. doi: 10.2337/diabetes.53.8.2158. [DOI] [PubMed] [Google Scholar]
- Cucca F, Dudbridge F, Loddo M, Mulargia AP, Lampis R, Angius E, De Virgiliis S, Koeleman BP, Bain SC, Barnett AH, Gilchrist F, Cordell H, Welsh K, Todd JA. The HLA-DPB1–associated component of the IDDM1 and its relationship to the major loci HLA-DQB1, -DQA1, and -DRB1. Diabetes. 2001;50:1200–1205. doi: 10.2337/diabetes.50.5.1200. [DOI] [PubMed] [Google Scholar]
- Curtis D. Use of siblings as controls in case-control association studies. Ann Hum Genet. 1997;61:319–333. doi: 10.1017/S000348009700626X. [DOI] [PubMed] [Google Scholar]
- de Bakker PI, McVean G, Sabeti PC, Miretti MM, Green T, Marchini J, Ke X, Monsuur AJ, Whittaker P, Delgado M, Morrison J, Richardson A, Walsh EC, Gao X, Galver L, Hart J, Hafler DA, Pericak-Vance M, Todd JA, Daly MJ, Trowsdale J, Wijmenga C, Vyse TJ, Beck S, Murray SS, Carrington M, Gregory S, Deloukas P, Rioux JD. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat Genet. 2006;38:1166–1172. doi: 10.1038/ng1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggal P, Gillanders EM, Holmes TN, Bailey-Wilson JE. Establishing an adjusted p-value threshold to control the family-wide type 1 error in genome wide association studies. BMC Genomics. 2008;9:516. doi: 10.1186/1471-2164-9-516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eike MC, Becker T, Humphreys K, Olsson M, Lie BA. Conditional analyses on the T1DGC MHC dataset: novel associations with type 1 diabetes around HLA-G and confirmation of HLA-B. Genes Immun. 2009;10:56–67. doi: 10.1038/gene.2008.74. [DOI] [PubMed] [Google Scholar]
- Eike MC, Olsson M, Undlien DE, Dahl-Jorgensen K, Joner G, Ronningen KS, Thorsby E, Lie BA. Genetic variants of the HLA-A, HLA-B and AIF1 loci show independent associations with type 1 diabetes in Norwegian families. Genes Immun. 2009;10:141–150. doi: 10.1038/gene.2008.88. [DOI] [PubMed] [Google Scholar]
- EMBL-EBI (2010) The International Immunogenetics Information System (IMGT) HLA Database. http://www.ebi.ac.uk/imgt/hla/stats.html
- Erlich H, Valdes AM, Noble J, Carlson JA, Varney M, Concannon P, Mychaleckyj JC, Todd JA, Bonella P, Fear AL, Lavant E, Louey A, Moonsamy P. HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes. 2008;57:1084–1092. doi: 10.2337/db07-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira MA, Hottenga JJ, Warrington NM, Medland SE, Willemsen G, Lawrence RW, Gordon S, de Geus EJ, Henders AK, Smit JH, Campbell MJ, Wallace L, Evans DM, Wright MJ, Nyholt DR, James AL, Beilby JP, Penninx BW, Palmer LJ, Frazer IH, Montgomery GW, Martin NG, Boomsma DI. Sequence variants in three loci influence monocyte counts and erythrocyte volume. Am J Hum Genet. 2009;85:745–749. doi: 10.1016/j.ajhg.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freidlin B, Zheng G, Li Z, Gastwirth JL. Trend tests for case-control studies of genetic markers: power, sample size and robustness. Hum Hered. 2002;53:146–152. doi: 10.1159/000064976. [DOI] [PubMed] [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- Greenbaum CJ. Insulin resistance in type 1 diabetes. Diabetes Metab Res Rev. 2002;18:192–200. doi: 10.1002/dmrr.291. [DOI] [PubMed] [Google Scholar]
- Hanifi Moghaddam P, de Knijf P, Roep BO, Van der Auwera B, Naipal A, Gorus F, Schuit F, Giphart MJ. Genetic structure of IDDM1: two separate regions in the major histocompatibility complex contribute to susceptibility or protection. Belgian Diabetes Registry. Diabetes. 1998;47:263–269. doi: 10.2337/diabetes.47.2.263. [DOI] [PubMed] [Google Scholar]
- Holm SJ, Carlen LM, Mallbris L, Stahle-Backdahl M, O’Brien KP. Polymorphisms in the SEEK1 and SPR1 genes on 6p21.3 associate with psoriasis in the Swedish population. Exp Dermatol. 2003;12:435–444. doi: 10.1034/j.1600-0625.2003.00048.x. [DOI] [PubMed] [Google Scholar]
- Horton R, Wilming L, Rand V, Lovering RC, Bruford EA, Khodiyar VK, Lush MJ, Povey S, Talbot CC, Jr, Wright MW, Wain HM, Trowsdale J, Ziegler A, Beck S. Gene map of the extended human MHC. Nat Rev Genet. 2004;5:889–899. doi: 10.1038/nrg1489. [DOI] [PubMed] [Google Scholar]
- Hothorn T, Hornik K, van de Wiel MAV, Zeileis A. Implementing a class of permutation tests: the coin package. J Stat Softw. 2008;28:1–23. [Google Scholar]
- Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howson JMM, Walker NM, Clayton D, Todd JA, Consortium TDG. Confirmation of HLA class II independent type 1 diabetes associations in the major histocompatibility complex including HLA-B and HLA-A. Diabetes Obes Metab. 2009;11:31–45. doi: 10.1111/j.1463-1326.2008.01001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C-Y (1999) Factors influencing the reliability of DIF detection methods. Dissertation Abstracts International 59:2944
- Hystad ME, Myklebust JH, Bo TH, Sivertsen EA, Rian E, Forfang L, Munthe E, Rosenwald A, Chiorazzi M, Jonassen I, Staudt LM, Smeland EB. Characterization of early stages of human B cell development by gene expression profiling. J Immunol. 2007;179:3662–3671. doi: 10.4049/jimmunol.179.6.3662. [DOI] [PubMed] [Google Scholar]
- Johansson S, Lie BA, Todd JA, Pociot F, Nerup J, Cambon-Thomsen A, Kockum I, Akselsen HE, Thorsby E, Undlien DE. Evidence of at least two type 1 diabetes susceptibility genes in the HLA complex distinct from HLA-DQB1,-DQA1 and-DRB1. Genes Immun. 2003;4:46–53. doi: 10.1038/sj.gene.6363917. [DOI] [PubMed] [Google Scholar]
- Koeleman BP, Dudbridge F, Cordell HJ, Todd JA. Adaptation of the extended transmission/disequilibrium test to distinguish disease associations of multiple loci: the conditional extended transmission/disequilibrium test. Ann Hum Genet. 2000;64:207–213. doi: 10.1046/j.1469-1809.2000.6430207.x. [DOI] [PubMed] [Google Scholar]
- Koeleman BP, Herr MH, Zavattari P, Dudbridge F, March R, Campbell D, Barnett AH, Bain SC, Mulargia AP, Loddo M, Amos W, Cucca F, Todd JA. Conditional ETDT analysis of the human leukocyte antigen region in type 1 diabetes. Ann Hum Genet. 2000;64:215–221. doi: 10.1046/j.1469-1809.2000.6430215.x. [DOI] [PubMed] [Google Scholar]
- Koeleman BP, Lie BA, Undlien DE, Dudbridge F, Thorsby E, de Vries RR, Cucca F, Roep BO, Giphart MJ, Todd JA. Genotype effects and epistasis in type 1 diabetes and HLA-DQ trans dimer associations with disease. Genes Immun. 2004;5:381–388. doi: 10.1038/sj.gene.6364106. [DOI] [PubMed] [Google Scholar]
- Ku DH, Chang CD, Koniecki J, Cannizzaro LA, Boghosian-Sell L, Alder H, Baserga R. A new growth-regulated complementary DNA with the sequence of a putative trans-activating factor. Cell Growth Differ. 1991;2:179–186. [PubMed] [Google Scholar]
- Lie BA, Todd JA, Pociot F, Nerup J, Akselsen HE, Joner G, Dahl-Jorgensen K, Ronningen KS, Thorsby E, Undlien DE. The predisposition to type 1 diabetes linked to the human leukocyte antigen complex includes at least one non-class II gene. Am J Hum Genet. 1999;64:793–800. doi: 10.1086/302283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunde K, Belting HG, Driever W. Zebrafish pou5f1/pou2, homolog of mammalian Oct4, functions in the endoderm specification cascade. Curr Biol. 2004;14:48–55. doi: 10.1016/j.cub.2003.11.022. [DOI] [PubMed] [Google Scholar]
- Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst. 1959;22:719–748. [PubMed] [Google Scholar]
- Nejentsev S, Gombos Z, Laine AP, Veijola R, Knip M, Simell O, Vaarala O, Akerblom HK, Ilonen J. Non-class II HLA gene associated with type 1 diabetes maps to the 240-kb region near HLA-B. Diabetes. 2000;49:2217–2221. doi: 10.2337/diabetes.49.12.2217. [DOI] [PubMed] [Google Scholar]
- Nejentsev S, Howson JM, Walker NM, Szeszko J, Field SF, Stevens HE, Reynolds P, Hardy M, King E, Masters J, Hulme J, Maier LM, Smyth D, Bailey R, Cooper JD, Ribas G, Campbell RD, Clayton DG, Todd JA. Localization of type 1 diabetes susceptibility to the MHC class I genes HLA-B and HLA-A. Nature. 2007;450:887–892. doi: 10.1038/nature06406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble JA, Valdes AM, Bugawan TL, Apple RJ, Thomson G, Erlich HA. The HLA class I A locus affects susceptibility to type 1 diabetes. Hum Immunol. 2002;63:657–664. doi: 10.1016/S0198-8859(02)00421-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson WP, Barbosa J, Rich SS, Thomson G. Homozygous parent affected sib pair method for detecting disease predisposing variants: application to insulin dependent diabetes mellitus. Genet Epidemiol. 1993;10:273–288. doi: 10.1002/gepi.1370100502. [DOI] [PubMed] [Google Scholar]
- Santin I, Castellanos-Rubio A, Aransay AM, Gutierrez G, Gaztambide S, Rica I, Vicario JL, Noble JA, Castano L, Bilbao JR. Exploring the diabetogenicity of the HLA-B18-DR3 CEH: independent association with T1D genetic risk close to HLA-DOA. Genes Immun. 2009;10:596–600. doi: 10.1038/gene.2009.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara T, Shimizu H, Hoshi N, Nakajima A, Fujimoto S. Steroidogenic acute regulatory protein-binding protein cloned by a yeast two-hybrid system. J Biol Chem. 2003;278:42487–42494. doi: 10.1074/jbc.M302291200. [DOI] [PubMed] [Google Scholar]
- The International HapMap Consortium (2005) A haplotype map of the human genome. Nature 437:1299–1320 [DOI] [PMC free article] [PubMed]
- Thomson G. HLA DR antigens and susceptibility to insulin-dependent diabetes mellitus. Am J Hum Genet. 1984;36:1309–1317. [PMC free article] [PubMed] [Google Scholar]
- Thomson G. Mapping disease genes: family-based association studies. Am J Hum Genet. 1995;57:487–498. [PMC free article] [PubMed] [Google Scholar]
- Thomson G, Barcellos LF, Valdes AM. Searching for additional disease loci in a genomic region. Adv Genet. 2008;60:253–292. doi: 10.1016/S0065-2660(07)00411-7. [DOI] [PubMed] [Google Scholar]
- Valdes AM, Erlich HA, Noble JA. Human leukocyte antigen class I B and C loci contribute to type 1 diabetes (T1D) susceptibility and age at T1D onset. Hum Immunol. 2005;66:301–313. doi: 10.1016/j.humimm.2004.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdes AM, Thomson G, Consortium TDG. Several loci in the HLA class III region are associated with T1D risk after adjusting for DRB1-DQB1. Diabetes Obes Metab. 2009;11:46–52. doi: 10.1111/j.1463-1326.2008.01002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdes AM, Thomson G, Barcellos LF. Genetic variation within the HLA class III influences T1D susceptibility conferred by high-risk HLA haplotypes. Genes Immun. 2010;11:209–218. doi: 10.1038/gene.2009.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viken MK, Blomhoff A, Olsson M, Akselsen HE, Pociot F, Nerup J, Kockum I, Cambon-Thomsen A, Thorsby E, Undlien DE, Lie BA. Reproducible association with type 1 diabetes in the extended class I region of the major histocompatibility complex. Genes Immun. 2009;10:323–333. doi: 10.1038/gene.2009.13. [DOI] [PubMed] [Google Scholar]
- Wallace C, Smyth DJ, Maisuria-Armer M, Walker NM, Todd JA, Clayton DG. The imprinted DLK1-MEG3 gene region on chromosome 14q32.2 alters susceptibility to type 1 diabetes. Nat Genet. 2009;42:68–71. doi: 10.1038/ng.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WTCCC Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Schaid DJ. Sequential haplotype scan methods for association analysis. Genet Epidemiol. 2007;31:553–564. doi: 10.1002/gepi.20228. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
p values of the combined dataset conditional on HLA-DRB1-DQB1 genotypes (PNG 310 kb)
Results of LD analysis for locating the causal genes (XLS 44 kb)
p values of each novel SNP association in individual HLA haplotype strata (XLS 153 kb)