

Abstract
RDCs for the 14 kDa protein hen egg-white lysozyme (HEWL) have been measured in eight different alignment media. The elongated shape and strongly positively charged surface of HEWL appear to limit the protein to four main alignment orientations. Furthermore, low levels of alignment and the protein’s interaction with some alignment media increases the experimental error. Together with heterogeneity across the alignment media arising from constraints on temperature, pH and ionic strength for some alignment media, these data are suitable for structure refinement, but not the extraction of dynamic parameters. For an analysis of protein dynamics the data must be obtained with very low errors in at least three or five independent alignment media (depending on the method used) and so far, such data have only been reported for three small 6–8 kDa proteins with identical folds: ubiquitin, GB1 and GB3. Our results suggest that HEWL is likely to be representative of many other medium to large sized proteins commonly studied by solution NMR. Comparisons with over 60 high-resolution crystal structures of HEWL reveal that the highest resolution structures are not necessarily always the best models for the protein structure in solution.
Keywords: Residual dipolar couplings, Hen egg-white lysozyme, Dynamics, Protein alignment, Ubiquitin
Introduction
Since residual dipolar couplings (RDCs) were first introduced as a means for studying protein structure, several studies have suggested methods for extracting information on protein dynamics from RDCs (Meiler et al. 2001; Tolman 2002; Deschamps et al. 2005; Bouvignies et al. 2006; Yao et al. 2008; Salmon et al. 2009). These methods require data from at least three to five independent alignment media with very low errors in the RDCs and any structural models used. Although it is possible to change the alignment of a protein by changing the alignment medium (Ramirez and Bax 1998), obtaining such data is a challenge (Ruan and Tolman 2005; Tolman 2009) and has so far only been reported for three very similar proteins: ubiquitin (Peti et al. 2002; Lakomek et al. 2006), GB3 (Ulmer et al. 2003; Yao et al. 2008) and GB1 (Ruan et al. 2008).
RDCs measured for hen egg-white lysozyme (HEWL) in three alignment media have previously been used for structure refinement (Schwalbe et al. 2001) and to characterize side-chain orientations (Higman et al. 2004). Here we report RDCs for HEWL in a further five alignment media and examine the limits on obtaining a sufficient number of independent alignments with low errors. We also examine errors arising from structural models. HEWL is a good model system because it is a medium-sized (14 kDa) protein containing both α-helical and β-sheet secondary structure. In addition, it has been studied extensively as a model protein by NMR (Buck et al. 1995; Schwalbe et al. 2001), X-ray diffraction (Vaney et al. 1996; Walsh et al. 1998) and computational methods (Smith et al. 1995; Buck and Karplus 1999; Soares et al. 2004).
Materials and methods
Sample preparation
All HEWL sample concentrations were approximately 1.4 mM. Details of the sample preparation and data collection for HEWL in media 1–3 are described elsewhere (Boyd and Redfield 1999; Higman et al. 2004).
Ether bicelles doped with La3+(medium 4)
A sample containing 3.8% w/v bicelles was prepared from a 10% stock solution of D13OPC, DHOPC, CTAB, 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-diethylenetriamine-pentaacetic acid (DMPE-DTPA) and LaCl3 in a 3:1:0.4:0.07:0.06 ratio. The pH of the solution was adjusted to 6.1 and the sample contained 5% D2O.
Cetylpyridinium bromide/NaBr/n-hexanol (medium5)
Procedures for using CpBr/NaBr/n-hexanol mixtures as an alignment medium were taken from Barrientos et al. (2000). The final sample contained 5% (w/v) CpBr/n-hexanol (1:1.33 (w/w)), 25 mM NaBr, 10 mM acetate buffer (pH 3.8) and 5% D2O.
C12E6/n-hexanol (medium 6)
Liquid crystals from n-alkyl-poly(ethylene glycols) and n-alkyl alcohols were prepared as described by Rückert and Otting (2000). To make a 600 μl sample, 12 μl of n-hexanol were added to 300 μl of a 10% C12E6 stock solution. The sample tube was shaken. When the two compounds had mixed sufficiently the solution was inspected for a bluish tinge indicative of the formation of the liquid crystalline phase. The protein was dissolved in 300 μl 90% H2O/10% D2O (pH 3.8) and added to the C12E6/n-hexanol mixture. The sample tube was shaken and again inspected for the bluish tinge. Finally the sample was briefly centrifuged to remove bubbles prior to pipetting it into the NMR sample tube.
Strained polyacrylamide gels (medium 7)
Polyacrylamide gels were made as described by Sass et al. (2000). 6, 8, 9 and 12% polyacrylamide gels were cast in 3.5 mm inner diameter tubing. Gels were washed, cut to a length of 3 cm and dried in an oven at 37°C overnight. Samples were made by placing a dried gel in a Shigemi NMR tube and then adding 475 μl of protein solution at pH 3.8 containing 10% D2O. The plunger was used to compress the gel to about 2.1 cm.
Pf1 phage (medium 8)
HEWL was dissolved in 0.1 M potassium phosphate buffer at pH 7.0 (90% H2O/10% D2O) followed by the addition of Pf1 bacteriophage from a 50 mg/ml stock solution (ASLA Biotech, Riga). This resulted in precipitation of the protein around the phage. NaCl was added from a 5 M stock solution whereupon the precipitate dissolved after shaking. The sample was then briefly centrifuged to remove bubbles. The final sample used for measurement of RDCs contained 10 mg/ml phage and 500 mM NaCl.
NMR
All NMR experiments were performed on home-built NMR spectrometers (fitted with magnets manufactured by Oxford Instruments, Eynsham, Oxfordshire) located in the Department of Biochemistry, University of Oxford. Experiments were carried out on spectrometers with 1H operating frequencies of 500, 600 and 750 MHz. The NMR data were processed and spectra were analysed using Felix 2.3 (Accelrys).
The 1H-15N residual dipolar couplings were measured from the 1JNH splitting appearing in the 15N dimension of an HSQC experiment which incorporated a S3E pulse sequence element (Meissner et al. 1997) or were recorded using the IPAP procedure (Ottiger et al. 1998). Measurements were made in an isotropic solution as well as in the anisotropic media. The temperatures and parameters used for the different samples are given in the Electronic supplementary material. The data were zero-filled once in the direct dimension and a Gaussian window function was applied. In the indirect dimension the data were zero-filled to 8192 points and a 90° shifted sine-bell window function was applied. Peak picking was carried out using an in-house FORTRAN77 program. Peaks are identified as local maxima within a defined search region with intensity above a threshold value. The peak positions (chemical shifts) in the two dimensions are interpolated using a three-point quadratic fit.
Comparison of RDCs with X-ray structures
X-ray diffraction coordinates for 56 high resolution structures (2 Å or better) of HEWL were obtained from the Brookhaven Protein Data Bank (Berman et al. 2000). The monoclinic structures contain two molecules per unit cell and were therefore divided into A and B structures. The 1IEE and 3LZT structures contained alternative conformations for parts of the backbone, thus two coordinate sets, designated a and b, were created to include these alternate conformations. This resulted in a total of 68 sets of co-ordinates. Hydrogen atoms were added to these co-ordinates using the HBUILD feature in XPLOR (Brünger 1992) followed by energy minimisation (Boyd and Redfield 1998).
The principal components (Axx, Ayy and Azz) and orientation (ϕ, θ and ψ) of the molecular alignment tensor for each X-ray structure were fitted to minimize the χ2 between experimental and calculated residual dipolar couplings using an in-house program written in FORTRAN77. Residues with heteronuclear 1H-15N NOE values of less than 0.7 were excluded from the analysis (residues 85, 100-104 and 129) (Buck et al. 1995). Residues 47 and 49 as well as 69–73 were also omitted; these regions are involved in crystal contacts in several structures. In addition, residues 88 and 118, which gave consistently high χ2 values, were omitted from the analysis.
The fitted values of the principal components and orientation of the alignment tensor were then used to calculate the residual dipolar couplings expected for the backbone N–HN groups. The N–HN bond length in the amide groups was assumed to be 1.02 Å.
Q-values were calculated during the process of fitting the structure to the experimental RDCs using the equation from Cornilescu et al. (1998):
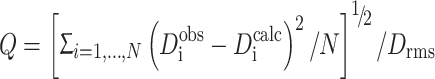 |
2.2 |
where Dobsi and Dcalci are the experimentally measured and predicted dipolar couplings, respectively, Drms is the root mean square value of all measured RDCs and the sum is over the N residues for which RDC measurements have been made.
Programs were written in FORTRAN77 to perform singular value decomposition of the RDC data matrix (Tolman 2002) and to calculate the angular RMSD of the N–HN vectors in crystal structures.
Results and discussion
15N–1HN RDC data have so far been published for HEWL in three alignment media. Two of these are positively charged, 7.5% ester bicelles doped with CTAB (medium 1) (Boyd and Redfield 1999) and 5% ether bicelles doped with CTAB (medium 2) (Higman et al. 2004), the third is either uncharged or very mildly negatively charged, 5% ester bicelles (medium 3) (Boyd and Redfield 1999). Data were acquired in a further five media: 3.8% ether bicelles doped with CTAB, the metal chelator DMPE-DTPA and La3+ (positively charged, medium 4), CpBr/hexanol/NaBr (positively charged, medium 5), C12E6/hexanol (uncharged, medium 6), compressed 6% polyacrylamide gel (uncharged, medium 7) and 10 mg/ml Pf1 bacteriophage (negatively charged, medium 8). The alignment tensors were fitted using the 193L HEWL crystal structure (Vaney et al. 1996), the fitting parameters are given in Table 1. Axis system 1 in Fig. 1a shows the relative orientations of the eight alignment tensors alongside the 193L crystal structure rotated into each of these co-ordinate frames.
Table 1.
HEWL alignment tensors determined by fitting to the 193L crystal structure (Vaney et al. 1996)
| D ba | Rc | θd | ϕd | ψd | Q-value | |
|---|---|---|---|---|---|---|
| Medium 1a | 12.09 | 0.15 | 136.5 | 156.0 | 53.4 | 0.1902 |
| Medium 2a | 11.38 | 0.18 | 132.8 | 160.3 | 91.1 | 0.1490 |
| Medium 3a | 16.03 | 0.31 | 126.5 | 153.2 | 89.0 | 0.1512 |
| Medium 4a | 14.04 | 0.48 | 121.5 | 162.9 | 75.2 | 0.1751 |
| Medium 5a | 6.88 | 0.13 | 143.1 | 169.0 | 48.0 | 0.2069 |
| Medium 6a | 10.84 | 0.39 | 122.4 | 158.8 | 93.3 | 0.1615 |
| Medium 7a | 4.86 | 0.39 | 54.8 | 112.0 | 112.7 | 0.2116 |
| Medium 8a | −9.55 | 0.17 | 40.0 | 127.7 | 57.0 | 0.2273 |
aSee text and supplementary material for composition of different media
bMagnitude of the alignment tensor
cRhombicity
dEuler angles
Fig. 1.

Principal components of the alignment tensors are depicted for a HEWL and b ubiquitin (Hus et al. 2003) along with the 193L HEWL (Vaney et al. 1996) and 1UBQ ubiquitin (Vijay-Kumar et al. 1987) crystal structures rotated into each alignment frame. Color coding: a medium 5 (red), medium 1 (orange), medium 4 (yellow), medium 2 (light green), medium 6 (dark green), medium 3 (mid blue), medium 7 (dark blue), medium 8 (purple); in axis system (2) the axes are grouped into the four main alignments (grey, pale blue, pale green and black). b C12E5/hexanol (dark red), DMPC/DHPC (red), CHAPSO/DLPC (orange), 4% CHAPSO/DLPC/CTAB (yellow), DMPC/DHPC/CTAB (light green), purple membranes (dark green), 5% CHAPSO/DLPC/CTAB (mid-blue), CHAPSO/DLPC/SDS (dark blue), CpBr/hexanol/NaBr (purple), Pf1 phage (pink)
The protein adopts four main alignments (Fig. 1a, axis system 2) dominated by its shape and charge. The elongated shape of HEWL (D||/D⊥ = 1.3) is the overriding factor in aligning the protein in both uncharged and positively charged media (media 1–7) and accounts for three of the four main alignments (Fig. 1a, grey, pale blue and pale green in axis system 2). Interestingly, the steric alignment induced by the compressed polyacrylamide gel differs significantly from that observed in the other uncharged and positively charged media, and is not simply related by a factor of −½ (Fig. 1a dark blue/pale green). Although the alignments shown in grey and pale blue in Fig. 1a appear distinct, they are still highly correlated (Table 2) and may not be strictly independent. Alignment in a strongly negatively charged medium, such as Pf1 phage, provides the fourth main alignment (Fig. 1a, purple/black). However, due to the highly positively charged surface of HEWL (pI ≈ 11), the protein strongly interacts with the negatively charged phage and a salt concentration of 500 mM was required in order to obtain even a medium quality spectrum of HEWL (Figure S1). This is undesirable, since the additional salt may influence the structure and/or dynamics of the protein. This highlights a further problem when trying to modulate the alignment tensor with different media: certain media place significant constraints on the physical conditions (temperature, pH, ionic strength). SECONDA analysis (Hus and Brüschweiler 2002) of the HEWL data across all eight media or any sub-group of seven media (Fig. 2) gives rise to a ratio between the 5th and 6th eigenvalue, ρ, of 1.1–3.4. Furthermore, the ratio between the 1st and 5th eigenvalues, an indicator for the signal to noise level, is significantly greater than 1 in all cases. Thus the error level across the different media is not sufficient for information about dynamics to be extracted.
Table 2.
Correlation coefficients between different RDC datasets measured for HEWL
| Medium 2 | Medium 3 | Medium 4 | Medium 5 | Medium 6 | Medium 7 | Medium 8 | |
|---|---|---|---|---|---|---|---|
| Medium 1 | 0.97 | 0.91 | 0.81 | 0.94 | 0.85 | −0.61 | 0.46 |
| Medium 2 | 0.95 | 0.85 | 0.87 | 0.92 | −0.70 | 0.84 | |
| Medium 3 | 0.92 | 0.78 | 0.98 | −0.73 | 0.71 | ||
| Medium 4 | 0.73 | 0.94 | −0.44 | 0.57 | |||
| Medium 5 | 0.66 | −0.52 | 0.30 | ||||
| Medium 6 | −0.67 | 0.70 | |||||
| Medium 7 | −0.85 |
Fig. 2.

SECONDA analysis (Hus and Brüschweiler 2002; Hus et al. 2003) for the eight sets of RDCs measured for HEWL. RDCs were normalised prior to the analysis as described by Lakomek et al. (2008) The eigenvalues are plotted in decreasing size and the ratio between the 5th and 6th eigenvalue, ρ, is indicated. The ratio between the 1st and 5th eigenvalues is provided in parentheses. The analysis included all eight data sets (a) or all data sets excluding medium 1 (b), medium 2 (c), medium 3 (d), medium 4 (e), medium 5 (f), medium 6 (g), medium 7 (h) or medium 8 (i)
Interestingly, the ten alignments of ubiquitin measured by Peti et al. (2002) and reported by Hus et al. (2003), show a larger number of different alignments with ρ = 11.5 (Fig. 1b). Ubiquitin appears to be more sensitive to small differences between the alignment media and the subtle interplay between steric and electrostatic forces. This is most likely because ubiquitin is a more spherical protein (D||/D⊥ = 1.17) (Tjandra et al. 1995) with a more varied surface charge distribution than HEWL (Fig. 3).
Fig. 3.

Surface representations of a HEWL (pdb code 193L (Vaney et al. 1996)) and b ubiquitin (pdb code 1UBQ (Vijay-Kumar et al. 1987), excluding the three flexible C-terminal residues) illustrating the overall shape of the proteins and their surface electrostatic potential (positive in blue, negative in red)
Further limitations to the use of RDCs to extract dynamic information come from the experimental errors, since insufficient accounting for errors can lead to misinterpretation of the RDC data. Experimental errors for ubiquitin and GB3 have been reported as lying between 0.1 and 0.5 Hz (Tolman et al. 1997; Peti et al. 2002; Clore and Schwieters 2004; Yao and Bax 2007) which is within the range of errors required for accurate extraction of dynamic parameters from RDCs (Yao et al. 2008). Although experimental errors for HEWL are within this range, the percentage error increases and Q-values clearly increase when the magnitude of the alignment tensor decreases (e.g. media 5 and 7, Table S1). In addition, the experimental error will increase in cases where the spectral quality is reduced, either due to increased protein size and longer τc values or due to excessive interactions between the protein and the alignment medium that adversely affect the spectral quality, e.g. positively charged HEWL with negatively charged Pf1 phage, or HEWL in 9–12% strained polyacrylamide gel (Figures S1, S2, Table S1 in the supplementary material) (Sass et al. 2000). Such factors will make it difficult to obtain the low errors required for the extraction of dynamic information from RDCs.
Finally, the quality of X-ray structures places a limit on the ability to extract dynamic information for those methods that use an explicit model (Bernadó and Blackledge 2004a, b). It has been shown that both structural noise within a crystal structure (Zweckstetter and Bax 2002) and the placement of hydrogen atoms on crystal structures (Ulmer et al. 2003; Higman 2004) can affect the fit between the structure and a set of RDCs. A very large number of X-ray structures is available for HEWL (Higman et al. 2004). Interestingly, we find that the correlation between crystal structure resolution and Q-value is rather low (R = 0.4, Fig. 4). All four crystal forms in which HEWL can be crystallized (tetragonal, triclinic, orthorhombic and monoclinic) have representative structures with both low and high Q-values and no crystal form represents a significantly better model for the solution structure of HEWL than the others. Particularly high Q-values, however, are observed for some monoclinic structures. The structural noise in the form of the angular RMSD of the NH vector (usually estimated to be on the order of 5°) (Bernadó and Blackledge 2004a; Ruan et al. 2008) was determined for the 20 highest resolution structures (0.92-1.64 Å) and found to be 4.5° in secondary structure, but 7.6° when including loops. This rise is not surprising, since the 20 crystal structures represent several crystal forms. The largest variations between these are seen in the loops which are often involved in crystal contacts. This highlights the fact that protein crystal structures cannot be assumed to represent the average solution structure observed by NMR accurately. It also provides an explanation for the observation made by Bernadó et al. that the inclusion of a dynamic model into the fit between the 193L HEWL crystal structure (which has a consistently low Q-value across all eight alignment media, Table S1) and RDC data is statistically not significant (Bernadó and Blackledge 2004a). This structure appears to be a good model for the solution structure and a static interpretation of the RDCs is sufficient within experimental error. The danger when using an X-ray structure to model dynamics explicitly is that real differences between the solution and crystal structures can be interpreted as dynamics. In the light of this, it is important that there are several methods which are not reliant on prior structural models (Tolman 2002; Lakomek et al. 2008; Yao et al. 2008; Salmon et al. 2009).
Fig. 4.

Plot of crystal structure resolution vs. average Q-value across all eight RDC data sets for HEWL. Panel B shows an expansion of part of panel A. The crystal form is indicated as tetragonal (red square), monoclinic (black circle), orthorhombic (blue triangle) or triclinic (gold diamond). A correlation coefficient (R) of only 0.4 is obtained between the average Q-values and X-ray structure resolution
Conclusion
We have shown firstly, using HEWL as an example, that the physical attributes of a protein may limit the number of independent alignments which it can adopt. Ubiquitin, GB1 and GB3 may be uniquely suited to undergoing modulations in their alignment tensors in alignment media with only small differences in their steric and electrostatic properties, while the limitations we have identified with HEWL could be typical of many larger or more complex proteins. Indeed, in a study on the alignment of an RNA construct, it was found that it was very difficult to modulate the alignment tensor, since the elongated structure and high negative charge dominated the alignment and prevented the use of positively charged media (Latham et al. 2008). Lanthanide binding tags (Wöhnert et al. 2003), which can provide alternative modes of alignment, may provide the best solution to this problem, but involve additional molecular biology steps. Conservative mutagenesis as a means to alignment tensor modulation provides a further possibility (Yao and Bax 2007), but may be less effective with proteins which are highly charged or significantly elongated. Secondly, the size of the protein and the properties of the alignment media used will have a significant impact on the experimental errors and thus on the ability to extract dynamic information. Finally, when using X-ray structures as models, not only does structural noise at a level of 5–8° have to be considered, but also the accuracy with which the model reflects the solution structure probed by NMR. Although it has been possible to obtain data in four independent alignment media for HEWL, the homogeneity across these media and/or the errors for this medium-sized protein are only sufficient to allow the extraction of structural, and not dynamic, parameters from the RDCs.
Acknowledgments
V. A. H. was funded by the BBSRC and St. Peter’s College, Oxford. C. R. was funded by the Wellcome Trust (Grant number 079440).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Contributor Information
Lorna J. Smith, Phone: +44-1865-272600, FAX: +44-1865-272690, Email: lorna.smith@chem.ox.ac.uk
Christina Redfield, Phone: +44-1865-275330, FAX: +44-1865-613201, Email: christina.redfield@bioch.ox.ac.uk.
References
- Barrientos LG, Dolan C, Gronenborn AM. Characterization of surfactant liquid crystal phases suitable for molecular alignment and measurement of dipolar couplings. J Biomol NMR. 2000;16:329–337. doi: 10.1023/A:1008356618658. [DOI] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernadó P, Blackledge M. Anisotropic small amplitude peptide plane dynamics in proteins from residual dipolar couplings. J Am Chem Soc. 2004;126:4907–4920. doi: 10.1021/ja036977w. [DOI] [PubMed] [Google Scholar]
- Bernadó P, Blackledge M. Local dynamic amplitude on the protein backbone from dipolar couplings: toward the elucidation of slower motions in biomolecules. J Am Chem Soc. 2004;126:7760–7761. doi: 10.1021/ja048785m. [DOI] [PubMed] [Google Scholar]
- Bouvignies G, Markwick P, Brüschweiler R, Blackledge M. Simultaneous determination of protein backbone structure and dynamics from residual dipolar couplings. J Am Chem Soc. 2006;128:15100–15101. doi: 10.1021/ja066704b. [DOI] [PubMed] [Google Scholar]
- Boyd J, Redfield C. Defining the orientation of the 15N shielding tensor using 15N NMR relaxation data for a protein in solution. J Am Chem Soc. 1998;120:9692–9693. doi: 10.1021/ja9815733. [DOI] [Google Scholar]
- Boyd J, Redfield C. Characterization of 15N chemical shift anisotropy from orientation-dependent changes to 15N chemical shifts in dilute bicelle solutions. J Am Chem Soc. 1999;121:7441–7442. doi: 10.1021/ja9911216. [DOI] [Google Scholar]
- Brünger AT. XPLOR version 3.1: a system for X-ray crystallography and NMR. New Haven, CT: Yale University Press; 1992. [Google Scholar]
- Buck M, Karplus M. Internal and overall peptide group motion in proteins: molecular dynamics simulations for lysozyme compared with results from X-ray and NMR spectroscopy. J Am Chem Soc. 1999;121:9645–9658. doi: 10.1021/ja991309p. [DOI] [Google Scholar]
- Buck M, Boyd J, Redfield C, MacKenzie DA, Jeenes DJ, Archer DB, Dobson CM. Structural determinants of protein dynamics: analysis of 15N NMR relaxation measurements for main-chain and side-chain nuclei of hen egg white lysozyme. Biochemistry. 1995;34:4041–4055. doi: 10.1021/bi00012a023. [DOI] [PubMed] [Google Scholar]
- Clore GM, Schwieters CD. Amplitudes of protein backbone dynamics and correlated motions in a small alpha/beta protein: correspondence of dipolar coupling and heteronuclear relaxation measurements. Biochemistry. 2004;43:10678–10691. doi: 10.1021/bi049357w. [DOI] [PubMed] [Google Scholar]
- Cornilescu G, Marquardt JL, Ottiger M, Bax A. Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. J Am Chem Soc. 1998;120:6836–6837. doi: 10.1021/ja9812610. [DOI] [Google Scholar]
- Deschamps M, Campbell ID, Boyd J. Residual dipolar couplings and some specific models for motional averaging. J Magn Res. 2005;172:118–132. doi: 10.1016/j.jmr.2004.09.023. [DOI] [PubMed] [Google Scholar]
- Higman VA (2004) The use of bicelles and other ordered media to study protein structure and dynamics. DPhil Thesis, University of Oxford
- Higman VA, Boyd J, Smith LJ, Redfield C. Asparagine and glutamine side-chain conformations in solution and crystal: a comparison for hen egg-white lysozyme using residual dipolar couplings. J Biomol NMR. 2004;30:327–346. doi: 10.1007/s10858-004-3218-y. [DOI] [PubMed] [Google Scholar]
- Hus J-C, Brüschweiler R. Principal component method for assessing structural heterogeneity across multiple alignment media. J Biomol NMR. 2002;24:123–132. doi: 10.1023/A:1020927930910. [DOI] [PubMed] [Google Scholar]
- Hus J-C, Peti W, Griesinger C, Brüschweiler R. Self-consistency analysis of dipolar couplings in multiple alignments of ubiquitin. J Am Chem Soc. 2003;125:5596–5597. doi: 10.1021/ja029719s. [DOI] [PubMed] [Google Scholar]
- Lakomek NA, Carlomagno T, Becker S, Griesinger C, Meiler J. A thorough dynamic interpretation of residual dipolar couplings in ubiquitin. J Biomol NMR. 2006;34:101–115. doi: 10.1007/s10858-005-5686-0. [DOI] [PubMed] [Google Scholar]
- Lakomek NA, Walter KFA, Farès C, Lange OF, de Groot BL, Grubmüller H, Brüschweiler R, Munk A, Becker S, Meiler J, Griesinger C. Self-consistent residual dipolar coupling based model-free analysis for the robust determination of nanosecond to microsecond protein dynamics. J Biomol NMR. 2008;41:139–155. doi: 10.1007/s10858-008-9244-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latham MP, Hanson P, Brown DJ, Pardi A. Comparison of alignment tensors generated for native tRNA(Val) using magnetic fields and liquid crystalline media. J Biomol NMR. 2008;40:83–94. doi: 10.1007/s10858-007-9212-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiler J, Prompers JJ, Peti W, Griesinger C, Brüschweiler R. Model-free approach to the dynamic interpretation of residual dipolar couplings in globular proteins. J Am Chem Soc. 2001;123:6098–6107. doi: 10.1021/ja010002z. [DOI] [PubMed] [Google Scholar]
- Meissner A, Duus JØ, Sørensen OW. Spin-state-selective excitation. Application for E.COSY-type measurement of JHH coupling constants. J Magn Res. 1997;128:92–97. doi: 10.1006/jmre.1997.1213. [DOI] [Google Scholar]
- Ottiger M, Delaglio F, Bax A. Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J Magn Res. 1998;131:373–378. doi: 10.1006/jmre.1998.1361. [DOI] [PubMed] [Google Scholar]
- Peti W, Meiler J, Brüschweiler R, Griesinger C. Model-free analysis of protein backbone motion from residual dipolar couplings. J Am Chem Soc. 2002;124:5822–5833. doi: 10.1021/ja011883c. [DOI] [PubMed] [Google Scholar]
- Ramirez BE, Bax A. Modulation of the alignment tensor of macromolecules dissolved in a dilute liquid crystalline medium. J Am Chem Soc. 1998;120:9106–9107. doi: 10.1021/ja982310b. [DOI] [Google Scholar]
- Ruan K, Tolman JR. Composite alignment media for the measurement of independent sets of NMR residual dipolar couplings. J Am Chem Soc. 2005;127:15032–15033. doi: 10.1021/ja055520e. [DOI] [PubMed] [Google Scholar]
- Ruan K, Briggman KB, Tolman JR. De novo determination of internuclear vector orientations from residual dipolar couplings measured in three independent alignment media. J Biomol NMR. 2008;41:61–76. doi: 10.1007/s10858-008-9240-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rückert M, Otting G. Alignment of biological macromolecules in novel nonionic liquid crystalline media for NMR experiments. J Am Chem Soc. 2000;122:7793–7797. doi: 10.1021/ja001068h. [DOI] [Google Scholar]
- Salmon L, Bouvignies G, Markwick P, Lakomek NA, Showalter S, Li D-W, Walter KFA, Griesinger C, Brüschweiler R, Blackledge M. Protein conformational flexibility from structure-free analysis of NMR dipolar couplings: quantitative and absolute determination of backbone motion in ubiquitin. Angew Chem Int Ed. 2009;48:4154–4157. doi: 10.1002/anie.200900476. [DOI] [PubMed] [Google Scholar]
- Sass H-J, Musco G, Stahl SJ, Wingfield PT, Grzesiek S. Solution NMR of proteins within polyacrylamide gels: diffusional properties and residual alignment by mechanical stress or embedding of oriented purple membranes. J Biomol NMR. 2000;18:303–309. doi: 10.1023/A:1026703605147. [DOI] [PubMed] [Google Scholar]
- Schwalbe H, Grimshaw SB, Spencer A, Buck M, Boyd J, Dobson CM, Redfield C, Smith LJ. A refined solution structure of hen lysozyme determined using residual dipolar coupling data. Prot Sci. 2001;10:677–688. doi: 10.1110/ps.43301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LJ, Mark AE, Dobson CM, van Gunsteren WF. Comparison of MD simulations and NMR experiments for hen lysozyme. Analysis of local fluctuations, cooperative motions, and global changes. Biochemistry. 1995;34:10918–10931. doi: 10.1021/bi00034a026. [DOI] [PubMed] [Google Scholar]
- Soares TA, Daura X, Oostenbrink C, Smith LJ, van Gunsteren WF. Validation of the GROMOS force-field parameter set 45A3 against nuclear magnetic resonance data of hen egg lysozyme. J Biomol NMR. 2004;30:407–422. doi: 10.1007/s10858-004-5430-1. [DOI] [PubMed] [Google Scholar]
- Tjandra N, Feller SE, Pastor RW, Bax A. Rotational diffusion anisotropy of human ubiquitin from 15N NMR relaxation. J Am Chem Soc. 1995;117:12562–12566. doi: 10.1021/ja00155a020. [DOI] [Google Scholar]
- Tolman JR. A novel approach to the retrieval of structural and dynamic information from residual dipolar couplings using several oriented media in biomolecular NMR spectroscopy. J Am Chem Soc. 2002;124:12020–12030. doi: 10.1021/ja0261123. [DOI] [PubMed] [Google Scholar]
- Tolman JR. Protein dynamics from disorder. Nature. 2009;459:1063–1064. doi: 10.1038/4591063a. [DOI] [PubMed] [Google Scholar]
- Tolman JR, Flanagan JM, Kennedy MA, Prestegard JH. NMR evidence for slow collective motions in cyanometmyoglobin. Nat Struct Biol. 1997;4:292–297. doi: 10.1038/nsb0497-292. [DOI] [PubMed] [Google Scholar]
- Ulmer TS, Ramirez BE, Delaglio F, Bax A. Evaluation of backbone proton positions and dynamics in a small protein by liquid crystal NMR spectroscopy. J Am Chem Soc. 2003;125:9179–9191. doi: 10.1021/ja0350684. [DOI] [PubMed] [Google Scholar]
- Vaney MC, Maignan S, Riès-Kautt M, Ducruix A. High-resolution structure (1.33 Å) of a HEW lysozyme tetragonal crystal grown in the APCF apparatus. Data and structural comparison with a crystal grown under microgravity from SpaceHab-01 mission. Acta Crystallogr D. 1996;52:505–517. doi: 10.1107/S090744499501674X. [DOI] [PubMed] [Google Scholar]
- Vijay-Kumar S, Bugg CE, Cook WJ. Structure of ubiquitin refined at 1.8 Å resolution. J Mol Biol. 1987;194:531–544. doi: 10.1016/0022-2836(87)90679-6. [DOI] [PubMed] [Google Scholar]
- Walsh MA, Schneider TR, Sieker LC, Dauter Z, Lamzin VS, Wilson KS. Refinement of triclinic hen egg-white lysozyme at atomic resolution. Acta Crystallogr D. 1998;54:522–546. doi: 10.1107/S0907444997013656. [DOI] [PubMed] [Google Scholar]
- Wöhnert J, Franz KJ, Nitz M, Imperiali B, Schwalbe H. Protein alignment by coexpressed lanthanide-binding tag for the measurement of residual dipolar couplings. J Am Chem Soc. 2003;125:13338–13339. doi: 10.1021/ja036022d. [DOI] [PubMed] [Google Scholar]
- Yao L, Bax A. Modulating protein alignment in a liquid-crystalline medium through conservative mutagenesis. J Am Chem Soc. 2007;129:11326–11327. doi: 10.1021/ja073937+. [DOI] [PubMed] [Google Scholar]
- Yao L, Vögeli B, Torchia DA, Bax A. Simultaneous NMR study of protein structure and dynamics using conservative mutagenesis. J Phys Chem B. 2008;112:6045–6056. doi: 10.1021/jp0772124. [DOI] [PubMed] [Google Scholar]
- Zweckstetter M, Bax A. Evaluation of uncertainty in alignment tensors obtained from dipolar couplings. J Biomol NMR. 2002;23:127–137. doi: 10.1023/A:1016316415261. [DOI] [PubMed] [Google Scholar]