

Abstract
Cryopyrin-associated periodic syndrome (CAPS) is a rare hereditary inflammatory disorder encompassing a continuum of three phenotypes: familial cold autoinflammatory syndrome, Muckle-Wells syndrome, and neonatal-onset multisystem inflammatory disease. Distinguishing features include cutaneous, neurological, ophthalmologic, and rheumatologic manifestations. CAPS results from a gain-of-function mutation of the NLRP3 gene coding for cryopyrin, which forms intracellular protein complexes known as inflammasomes. Defects of the inflammasomes lead to overproduction of interleukin-1, resulting in inflammatory symptoms seen in CAPS. Diagnosis is often delayed and requires a thorough review of clinical symptoms. Remarkable advances in our understanding of the genetics and the molecular pathway that is responsible for the clinical phenotype of CAPS has led to the development of effective treatments. It also has become clear that the NLRP3 inflammasome plays a critical role in innate immune defense and therefore has wider implications for other inflammatory disease states.
Keywords: Cryopyrin-associated periodic syndrome (CAPS), Familial cold autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS), Neonatal-onset multisystem inflammatory disease (NOMID), Autoinflammatory, Inflammasome, Interleukin-1, NLRP3, Anakinra, Rilonacept, Canakinumab
Introduction
Clinical Features
Cryopyrin-associated periodic syndrome (CAPS) is a rare hereditary inflammatory disorder encompassing a continuum of three phenotypes: familial cold autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS), and neonatal-onset multisystem inflammatory disease (NOMID). These cryopyrinopathies were once thought to be distinct conditions but now are used to describe disease severity. FCAS is of the mildest phenotype and was first reported in 1940 [1]. It is characterized by recurrent urticaria, arthralgia, and fever after general exposure to cold, not necessarily by touch [2, 3]. MWS is of the intermediate phenotype and was first described in 1962 by Muckle and Wells [4], who determined the condition to be inherited in an autosomal dominant fashion. Inflammation can result spontaneously without apparent provocation as well as from triggers such as cold, stress, or exercise. In addition to the characteristics seen in FCAS, MWS is characterized by renal amyloidosis, sensorineural hearing loss, and conjunctivitis [4]. The most severe is NOMID and was first described in 1973 [5]. The hallmark of NOMID is neonatal onset of cutaneous symptoms along with end-organ damage. These include the “triad” of arthropathy, chronic urticaria, and central nervous system (CNS) involvement [6, 7]. CNS involvement can range from hearing loss to chronic aseptic meningitis and mental retardation [8]. NOMID more commonly arises from de novo mutations resulting from the early debilitating nature of this condition.
Distinctive features in CAPS can aid in identification. Cutaneous manifestations include urticaria that is atypical. It tends not to be pruritic, and patients instead describe the affected skin as feeling “tight” and/or “warm.” The rash usually appears on the trunk and limbs, with individual lesions that are migratory and last less than 24 h (Fig. 1). The morphology of the lesions includes faint pink figurate erythema to juicy urticated papules and plaques, and there is commonly a diurnal pattern, with rash severity increasing in the evening [9], although in NOMID patients, the rash may be present persistently. Neurological manifestations can arise from increased inflammation, including headaches, hearing loss, raised intracranial pressure, and meningitis [3]. Sensorineural deafness is progressive and can occur in adolescence or adulthood [10, 11]. Examination reveals cochlear inflammation without any observed amyloidosis in the Corti apparatus or cochlear nerve [12, 13]. In addition, autopsy of MWS patients with deafness revealed atrophy of the cochlear nerve and a lack of the Corti organ, suggesting the inflammation causes irreversible damage, and pointing out the importance of early identification and treatment [11]. Ophthalmologic manifestations include conjunctivitis, uveitis, and papilledema. Rheumatologic manifestations, more specifically in NOMID, include characteristic arthropathy affecting large joints such as the knees, resulting in joint enlargement and functional disability. This arises from abnormal endochondral ossification, which causes the formation of calcified masses in the joints [14]. Lastly, systemic AA amyloidosis can occur in about 25% of CAPS patients, commonly affecting the kidneys [8].
Fig 1.
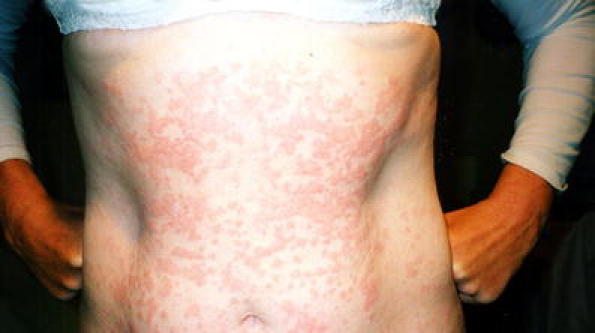
Typical cryopyrin-associated periodic syndrome rash showing figurate erythematous macules and urticated papules
Pathogenesis
In all three phenotypes of CAPS, there is a gain-of-function mutation of the NLRP3 gene (also known as CIAS1), which is located on chromosome 1q44 and codes for cryopyrin [15, 16]. However, as many as 40% of patients with clinical NOMID do not show mutations on this gene, implicating the involvement of other autoinflammatory genes or the involvement of genetic mosaicism [17, 18]. Cryopyrin is among the nucleotide-binding domain, leucine rich–containing (NLR) family of proteins, which serve an important role in the regulation of the innate immune response [19, 20]. In addition, cryopyrin forms intracellular protein complexes known as inflammasomes that assist in activating caspase-1 to cleave pro–interleukin (IL)-1β into its mature form [21]. The defect in this pathway induces an overproduction of IL-1β, leading to the inflammatory symptoms seen in CAPS, and the success of IL-1 blockade has helped confirm the role of IL-1β. Cryopyrin is expressed in monocytes and neutrophils, and is also expressed in human chondrocytes, accounting for the arthropathy seen in NOMID [15, 22]. This is evident in the finding that monocytes in CAPS patients have been shown to secrete more IL-1β [21]. This increase can be attributed to the monocytes of CAPS patients that have impaired redox homeostasis and response to oxidative stress, resulting in accelerated IL-1β secretion [23]. However, the correlation of these findings with mutations in NLRP3 requires further investigation.
Diagnostic Procedures
The rarity of CAPS and the overlap of symptoms with other conditions often results in a delay in making the diagnosis. Because of the varying degrees of severity in CAPS, a thorough review of clinical symptoms is necessary, and a combination of diagnostic procedures should be considered. The following include laboratory assessments, skin biopsy, and genetic testing.
Laboratory Assessments
Acute-phase protein levels should be monitored, including C-reactive protein (CRP) and, if available, serum amyloid A (SAA). Although no cutaneous signs may be present, these inflammatory markers are normally elevated, oftentimes greater than five times the reference range. Normal CRP and SAA levels are rarely seen in CAPS, but if there is doubt, serial measurements should be taken. Complete blood counts typically reveal a slightly reduced hematocrit and mild neutrophilia. Renal function should be recorded, including a urinalysis to check for evidence of proteinuria. If proteinuria is found, patients should be assessed for nephrotic syndrome, a late complication of systemic amyloidosis. For NOMID patients with neurological symptoms, cerebrospinal fluid (CSF) can be another diagnostic tool. It has been reported in one patient with a novel NLRP3 mutation that cytokine levels in the CSF were elevated, but serum levels remained normal [24]. In another case report, a NOMID patient had normal inflammatory markers in the CSF and serum; however, neopterin levels in the CSF were shown to correlate with presentation of symptoms, suggesting another possible marker of disease [25].
Skin Biopsy
Histologic examination of affected skin can assist in confirming an early diagnosis of CAPS. A common characteristic feature is neutrophilic dermal infiltrate in the reticular dermis [9]. The infiltrate tends to be perivascular and also may be peri-eccrine. This is consistent with the atypical urticaria seen in CAPS patients, as the cellular infiltrate does not consist of mast cells (Fig. 2).
Fig 2.
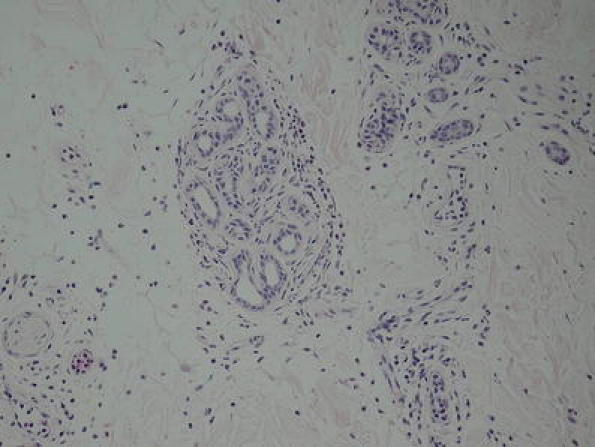
Skin biopsies showing perivascular and periadnexal neutrophilic infiltration within the dermis
Genetic Testing
A family of 14 NALP proteins has been identified, of which NALP3 is included [26]. The NLRP3 gene contains nine exons, with CAPS mutations predominantly localizing to missense changes in exon 3 of the NACHT domain [16, 27, 28]. A total of 121 sequence variants have been identified [29]. Conversely, CAPS patient have been identified who do not have a mutation in NLRP3, and the clinical presentations of CAPS patients with and without mutations are very similar [7, 28]. The importance of sequencing the whole NLRP3 gene is evidenced by missense mutations in exon 4 (G755R, G755A) and exon 6 (Y859C) that have been identified in CAPS patients [30, 31]. However, the currently available commercial test by GeneDx (Gaithersburg, MD) only sequences exon 3. It is likely that in the future, more extensive sequencing analysis will be available to patients diagnostically, as the cost will be reduced considerably due to the availability of newer technologies.
Differential Diagnosis
Cold Contact Urticaria
Cold contact urticaria is the second most common physical urticaria subtype, with as-yet-unknown etiology [32]. The distinguishing feature is the development of urticaria and/or angioedema on areas of the skin exposed to cold. This is normally localized to sites of contact, but in some cases in which a large area of skin is in contact (eg, swimming), greater systemic involvement can occur, including generalized urticaria, headache, dyspnea, hypotension, and loss of consciousness [33]. A positive cold provocation test (ice cube test) can confirm a diagnosis of cold contact urticaria. An ice cube placed in a glove or plastic bag is applied to the skin, and the test is positive if a wheal appears within 5 min. The ice cube test is negative in CAPS, although cold exposure may precipitate generalized rash and febrile symptoms a few hours after contact [32].
Systemic-Onset Juvenile Idiopathic Arthritis/Adult-Onset Still’s Disease
Systemic-onset juvenile idiopathic arthritis (SOJIA) and adult-onset Still’s disease (AOSD) are rare, related inflammatory disorders distinguished by age at onset, with AOSD patients being 16 years of age or older [34]. SOJIA and AOSD have characteristic features of polyarthritis/polyarthralgia, high fevers, leukocytosis with neutrophilia, and evanescent skin rash that is nonpruritic and salmon colored [35, 36]. Although the etiology is unknown, the effective use of IL-1 antagonists in reducing symptoms helps provide evidence for the role of IL-1 [34, 37]. Furthermore, SOJIA/AOSD have similar symptoms and signs as CAPS patients, as demonstrated by the multisystem inflammation and by the raised levels of acute-phase proteins [38, 39]. SOJIA and AOSD are frequently diagnoses of exclusion once underlying autoimmune, infectious, or paraneoplastic disorders have been ruled out [40]. It is important to consider CAPS, although patients with CAPS typically present in infancy or childhood and do not have a relapsing/remitting course.
Deficiency in Interleukin-1 Receptor Antagonist
Deficiency in IL-1 receptor antagonist (DIRA) is a rare autoinflammatory disease in which a deletion in the IL1RN gene, located on chromosome 2, results in a loss of production of the naturally occurring IL-1 receptor antagonist (RA) [41]. This loss of IL-1 RA results in a functional excess of the proinflammatory cytokines IL-1α and IL-1β. The clinical phenotype is similar to that of severe CAPS (NOMID spectrum). DIRA presents early in childhood with joint pain and enlargement, neutrophilic dermal infiltrate on histologic examination, and elevated levels of acute-phase reactants such as CRP. Clinically, the most important distinguishing feature in DIRA is a pustular rash similar to pustular psoriasis, as opposed to the urticarial eruption of CAPS. DIRA patients who have had a therapeutic trial with anakinra have shown a dramatic and sustained improvement in their clinical signs and a normalization of their acute-phase response [42].
Schnitzler Syndrome
Schnitzler syndrome is a rare disorder characterized by chronic urticarial rash and monoclonal gammopathy, along with fever, arthralgia, or bone pain [43]. Although the pathophysiology is presently unknown, the clinical improvement seen in these patients treated with IL-1 antagonists implies an autoinflammatory process [44]. In many respects, the clinical presentation is very similar to that of CAPS. Both conditions show chronic, nonpruritic urticarial lesions, and upon histologic examination show neutrophilic inflammation. The age at onset differs, with Schnitzler syndrome presenting at a much older age (mean age at onset, 60 years) [45]. In addition, by definition, Schnitzler syndrome patients have a monoclonal gammopathy that has not been reported in CAPS.
Treatment
Conventional Anti-inflammatories
Many anti-inflammatories, immunosuppressants, and antihistamines have been tried to control the symptoms and signs seen in CAPS. These include methotrexate, cyclosporine, azathioprine, cyclophosphamide, and tumor necrosis factor blockers, all of which have been disappointing. Modest improvements with high-dose oral corticosteroids and thalidomide have offered some patients partial symptom control but have not been tolerated due to their adverse side effect profile [8]. The lack of effective treatments necessitated the need for development of more targeted therapies.
Interleukin-1 Targeted Therapy
The identification of the NLRP3 gene responsible for FCAS and MWS and the finding that IL-1β production is increased in NOMID has created an important therapeutic target for CAPS [16, 28]. In addition, as IL-1 induces an increase in SAA levels, alleviating high levels of IL-1 can help prevent the development of systemic amyloidosis and lessen the potential for end-organ disease [46]. In the past 7 years, three drugs have shown efficacy in targeting IL-1β for CAPS patients. Each is unique in its strategy, but all have been shown to be effective in alleviating clinical symptoms.
Anakinra: Interleukin-1 Receptor Antagonist
IL-1 is a ubiquitous proinflammatory cytokine that has been implicated in many diseases (eg, septic shock, rheumatoid arthritis, and gout) [47–49]. It consists of two isoforms (α and β), and its effects are moderated by the competitive binding of the IL-1 receptor by naturally occurring IL-1 RA. Anakinra is a recombinant form of IL-1 RA that has a short half-life of 4 to 6 h [50]. Anakinra was originally developed as a treatment for septic shock [51]. An open-label study involving 99 patients suggested that anakinra might be beneficial in the treatment of sepsis; however, a subsequent, randomized, double-blind, placebo-controlled study with 893 patients found no significant difference between the groups [52, 53]. However, preliminary studies in patients with rheumatoid arthritis found that daily injections of anakinra were effective in alleviating symptoms, and double-blind, placebo-controlled studies demonstrated its efficacy and safety [48, 54, 55]. It was approved by the US Food and Drug Administration for the treatment of rheumatoid arthritis in 2001. In the first case report for anakinra and CAPS, two MWS patients were given daily 100-mg injections and showed a reduction in symptoms within hours [56]. Following this, several case reports and studies demonstrated the efficacy of anakinra in CAPS [57–60]. These findings provided the basis for larger studies.
In a prospective study, 18 NOMID patients were recruited to receive daily, subcutaneous, 1-mg/kg of body weight injections of anakinra [12]. All patients showed two of the following symptoms: urticarial rash, CNS involvement, or epiphyseal or patellar overgrowth. Patients ranged from 4 to 32 years of age. Efficacy assessments were made at months 1, 3, and 6 from the first injection. At month 3, patients who showed treatment response discontinued the anakinra until they presented with a clinical flare, although this withdrawal phase was subsequently discontinued due to the severity of disease flares seen in the initial patients. After the appearance of clinical flares, patients recommenced anakinra and were followed up to 24 months. Primary outcomes were CRP and SAA levels as well as daily ratings of symptoms. All patients showed a disappearance of urticarial rash within 3 days, and CRP/SAA levels showed dramatic decreases after 1 month that were sustained. In total, 8 patients showed remission of inflammatory symptoms after 3 months, with the remaining 10 patients showing remission after 6 months. The 11 patients who underwent withdrawal of anakinra relapsed after a median of 5 days. These patients all achieved remission when recommencing the anakinra. Furthermore, treatment with anakinra reduced progressively both spontaneous and stimulated secretion of IL-1β from cultured patients’ peripheral blood mononuclear cells after 6 months of therapy. Transcript levels of IL-1β and genes downstream of IL-1β also fell while the patients were on treatment. No patient discontinued treatment, and the most common adverse events were injection site reactions and upper respiratory infections.
These results were corroborated by a retrospective review of 22 patients with CAPS [8]. Primary outcomes consisted of monthly measurements of CRP/SAA levels, as well as review of clinical symptoms. Of these patients, 15 were treated with anakinra, starting with daily 100-mg subcutaneous injections and then reduced to 20 to 50 mg in order to determine efficacy at lower doses. The two children involved in the study continued to receive 100 mg daily. All 15 patients showed complete remission of disease activity within 12 h of anakinra injections, with 6 patients showing remission after 4 h. Levels of CRP and SAA returned to normal after 1 week in all patients. During this time, five patients briefly stopped treatment and showed a reappearance of symptoms after only 36 to 48 h, providing more evidence for the importance of daily injections. All patients tolerated the treatment well, except for injection site reactions.
Anakinra was a major advancement in the management of CAPS, as no other treatment had been effective previously. However, despite the success of anakinra, the short half-life requires daily injections, leading to sometimes-painful injection site reactions. Although it was never US Food and Drug Administration approved for use in CAPS, anakinra continues to be used effectively as an off-label treatment. In addition, the efficacy of anakinra is being explored in other disorders, such as rheumatoid arthritis (NCT00213538), renal disease (NCT00420290, NCT00897715), type 1 diabetes (NCT00711503), SOJIA (NCT00339157), and atopic dermatitis (NCT01122914). Treatment with anakinra has been reported to be beneficial for other disorders, such as Schnitzler syndrome, adult-onset Still’s disease, and even in the prevention of heart failure after acute myocardial infarction [61–63].
Rilonacept: Interleukin-1 Trap
Cytokine inhibition has been effective with certain cytokines (eg, with tumor necrosis factor-α blockers such as etanercept); however, inhibition of IL-1 was much more difficult because of the complexity of its receptor system [64]. The development of cytokine trap technology has allowed for successful inhibition because these multicomponent “traps” contain binding sites for the cytokine as well as their accessory proteins. Thus, cytokine traps are useful in autoinflammatory conditions because they have been shown to effectively bind to their respective cytokines with high affinity [64].
Rilonacept was created using cytokine trap technology. It is a dimeric fusion protein that contains binding regions for the type 1 receptor and the IL-1 receptor accessory protein, and it is fused to the Fc portion of human IgG1 [65]. In 2008, it was the first US Food and Drug Administration–approved drug therapy for CAPS, specifically for FCAS and MWS in adults and children 12 years of age and older. Rilonacept has a circulating half-life of 8.6 days [66]. This makes weekly subcutaneous injections of rilonacept a major improvement over the daily injections of anakinra; however, the monthly cost of anakinra is still much lower than that of rilonacept.
An open-label pilot study of rilonacept was performed, with five FCAS patients having confirmed mutations [67]. At baseline, all patients were given a 300-mg loading dose of rilonacept and were not dosed again until disease flares occurred. Disease flares were defined as a greater than 50% increase in acute-phase protein levels. After a flare occurred, patients were again dosed with 300 mg, followed by weekly 100-mg doses. If complete remission of symptoms, defined by levels of CRP and SAA, had not occurred, the patients would then be eligible for dose escalation. The first dose increase was 160 mg. If remission did not occur after 4 weeks at this dose, patients were eligible for another increase, to 320 mg. All patients who reached a dose of 160 mg were then monitored for 2 years. In response to the baseline dose, all five patients showed improvement of symptoms, with maximum improvement at day 10 for four patients and at day 6 for one patient. Disease flares occurred within a range of 10 to 28 days from the initial loading dose. All five patients continued to the extension phase and were dosed at 160 mg weekly. Remission did not occur in four patients, and they were then dosed at 320 mg. These four patients reported subjective improvement of symptoms; however, acute-phase reactant levels were not significantly different as compared with the 160-mg dose. All five patients tolerated the drug very well with no injection site reactions. Adverse events reported by patients were only mild to moderate.
The first randomized, double-blind consecutive studies of any treatment for CAPS were completed in 2008, with 47 patients having confirmed NLRP3 mutations [68]. The first study consisted of a 6-week, randomized, double-blind, controlled period, with 23 patients receiving 160-mg weekly injections and 24 patients serving as controls. At the end of this 6-week treatment period, patients receiving rilonacept had a remarkable reduction in symptoms, including lower levels of CRP and SAA. The second study immediately followed the first and consisted of two parts. Part A included 46 patients from the first study and consisted of a single-blind, 9-week period with 160-mg weekly injections. Part B was a randomized, double-blind, 9-week withdrawal period. Patients receiving placebo in the first study showed rapid and significant improvement during part A of the second study. In part B, patients receiving placebo showed a gradual return of symptoms, while patients receiving rilonacept continued to show a decrease in symptoms. These studies demonstrated that rilonacept was generally well-tolerated; the most frequent adverse events were injection site reactions, upper respiratory tract infections, headache, arthralgia, and diarrhea. One older adult patient died after developing sinusitis and meningitis during the open-label extension phase, but the study investigators thought that the death was unrelated to rilonacept. The authors concluded that there may be potential risk of serious infections for patients on targeted IL-1 therapy.
In addition to CAPS, rilonacept has been investigated for the treatment of other indications. A single-blind pilot study has been completed among 10 patients with gouty arthritis, showing that rilonacept could be an effective treatment [69]. Currently, a larger, randomized, double-blind study is being conducted to ascertain the effectiveness of rilonacept in gout (NCT00856206). Other clinical trials are currently investigating the effectiveness of rilonacept in diabetes mellitus type 1 (NCT00962026) and juvenile idiopathic arthritis (NCT00534495).
Canakinumab: Interleukin-1β Monoclonal Antibody
Canakinumab is an IL-1β fully human, monoclonal antibody that is specific for IL-1β and not other members of the IL-1 family. The development of this compound has allowed for the improved detection of circulating levels of IL-1β in vivo and, more importantly, the elucidation of the biology of IL-1β. The use of canakinumab in CAPS patients has shown that the release of IL-1β is driven by a positive feedback loop triggered by IL-1β, and that the level of IL-1β production is correlated with disease severity [70]. Canakinumab has a mean half-life of 26 days, which allows a dosing schedule of a subcutaneous injection every 8 weeks [71]. This dosing schedule compares favorably with that of anakinra (daily) and rilonacept (weekly). It received US Food and Drug Administration approval for use in CAPS patients—adults and children older than 4 years of age with FCAS and MWS—in June 2009.
The first open-label clinical trial for canakinumab was completed in 2008 with seven mutation-confirmed CAPS patients [70]. The first four patients were dosed at 10 mg/kg intravenously, and time until relapse was noted. After relapse, another dose of 1 mg/kg intravenously was administered, and time to relapse was again noted. From this point, each patient was dosed 150 mg subcutaneously after each relapse. An additional three patients were enrolled and initially treated with 150 mg and again at relapse. Levels of circulating canakinumab were determined using enzyme-linked immunosorbent assay. The first four patients dosed had their urticarial symptoms disappear after just 1 day, and a complete clinical response—as defined by a lack of skin rash and joint pain, improvement of arthralgia, and normal laboratory values—was achieved after 1 week. At the 10-mg/kg intravenous dose, relapse did not occur until a median time of 185 days. At the 1-mg/kg intravenous dose, relapse did not occur until a median of 90.5 days. With the 150-mg subcutaneous dose, the median duration of clinical remission was 127 days (range, 55–230 days). In these patients, IL-1β production fell to levels similar to those seen in unaffected healthy controls after 8 weeks of therapy. These exploratory findings provided the foundation for the next trial.
The first double-blind, randomized trial was completed in 2008 [72•]. It consisted of a 3-part, 48-week, withdrawal study with 35 CAPS patients. Part one of the study consisted of an 8-week period with a single open-label dose of canakinumab; adults received 150 mg and children were dosed at 2 mg/kg body weight if they weighed less than 40 kg. Part two consisted of a 24-week, double-blind withdrawal period in which patients who completed part one received canakinumab or placebo every 8 weeks. At the end of part two or at clinical relapse, patients entered part three, which consisted of an open-label administration of canakinumab every 8 weeks for a minimum of 16 weeks. All patients had confirmed NLRP3 mutations, were between 4 and 75 years of age, weighed at least 15 kg, and were allowed to enroll immediately after discontinuation of other CAPS treatments. Of the 35 patients who entered into the study at part one, 34 had a complete response, defined by physician assessments of disease activity and rash, as well as CRP and SAA levels. Most patients achieved a complete response by day 8. For part two, 31 patients were continued on study, with 15 patients receiving drug and 16 receiving placebo. All patients in the treatment group continued in remission of symptoms, while 13 in the placebo group had disease flares, including elevated levels of CRP and SAA. All 31 patients continued on to part three of the study, with 29 continuing to completion. The results showed sustained clinical remission with minimal disease activity, absence of rash, and levels of CRP and SAA within the reference range. Canakinumab was tolerated well, with only two patients reporting serious adverse events, which included an episode of vertigo followed by acute close-angle glaucoma related to complications of CAPS, and a recurrent lower urinary tract infection and sepsis. The most commonly reported adverse events among those on drug were nasopharyngitis, rhinitis, nausea, diarrhea, and vertigo. Most patients did not report any injection site reactions.
Canakinumab has shown efficacy as a treatment for other conditions, including rheumatoid arthritis and gout [73, 74]. The effectiveness of this drug is currently being explored in other inflammatory conditions, including SOJIA (NCT00886769) and osteoarthritis (NCT01160822), as well as in metabolic disorders such as type 2 diabetes mellitus (NCT00900146).
Caspase-1 Targeted Therapy
Caspase-1 inhibitors act upstream in this IL-1β production pathway. The drug VX-765 (Vertex Pharmaceuticals, Cambridge, MA) is a powerful selective inhibitor of caspase-1. In vitro and animal models have shown VX-765 to reduce levels of IL-1β in a dose-dependent manner [75]. Another study showed that IL-1β secretion in peripheral blood mononuclear cells isolated from FCAS patients was greatly inhibited by VX-765 [76]. Despite these findings, no clinical trials have been registered involving CAPS patients; however, a phase 2 trial of VX-765 for the treatment of psoriasis (NCT00205465) was completed in 2005, and another phase 2 trial for the treatment of epilepsy is currently being conducted (NCT01048255). Further investigation is required to determine efficacy in CAPS patients.
Quality-of-Life Assessments
Traditional quality-of-life surveys have been used to assess CAPS patients, and they are useful in understanding the full impact of the disease on the patient, as well as assessing response to treatment. These include the Medical Outcomes Survey Short Form 36, Health Assessment Questionnaire, Functional Assessment of Chronic Illness Therapy Fatigue Scale, and the Child Health Questionnaire-Parent Form. Because of the variable nature of symptoms and severity in CAPS, it is often difficult to accurately assess the quality of life of CAPS patients. Daily health assessment forms have been developed that reliably capture disease severity or symptoms and can be used to measure treatment efficacy [77].
Conclusions
Remarkable advances have been made in our understanding of the genetics and the molecular pathway that is responsible for the clinical phenotype of CAPS. The discovery of mutations in the NLRP3 gene coupled with elucidation of IL-1 biology has led to rational targeted therapy for this rare disease. Therapy offers rapid and sustained clinical remissions for the vast majority of CAPS patients. All three agents appear to be well-tolerated with a relatively safe side effect profile. It also has become clear that the NLRP3 inflammasome plays a critical role in innate immune defense and therefore has wider implications for other inflammatory disease states. Mutations in NLRP3 are not unique to CAPS patients, as they have been identified in other diseases (eg, gout) [78]. In addition, involvement of the inflammasome in disease is suspected in a variety of conditions, such as pyoderma gangrenosum, type 2 diabetes mellitus, and atherogenesis, which offers other clinical targets for IL-1 directed therapy [79–81].
Acknowledgments
Disclosure
Dr. Leslie has served as a consultant for Novartis and as an investigator for a Novartis-sponsored study. Mr. Yu reported no potential conflicts of interest relevant to this article.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Contributor Information
Justin R. Yu, Email: yuj@derm.ucsf.edu
Kieron S. Leslie, Email: lesliek@derm.ucsf.edu
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
- 1.Kile RL, Rusk HA. A case of cold urticaria with an unusual family history. J Am Med Assoc. 1940;114(12):1067–1068. [Google Scholar]
- 2.Stych B, Dobrovolny D. Familial cold auto-inflammatory syndrome (FCAS): characterization of symptomatology and impact on patients’ lives. Curr Med Res Opin. 2008;24(6):1577–1582. doi: 10.1185/03007990802081543. [DOI] [PubMed] [Google Scholar]
- 3.Montealegre Sanchez GA, Hashkes PJ. Neurological manifestations of the Mendelian-inherited autoinflammatory syndromes. Dev Med Child Neurol. 2009;51(6):420–428. doi: 10.1111/j.1469-8749.2009.03336.x. [DOI] [PubMed] [Google Scholar]
- 4.Muckle TJ, Wells M. Urticaria, deafness, and amyloidosis: a new heredo-familial syndrome. Q. J. Med. 1962;31:235–248. [PubMed] [Google Scholar]
- 5.Lorber J. Syndrome for diagnosis: dwarfing, persistently open fontanelle; recurrent meningitis; recurrent subdural effusions with temporary alternate-sided hemiplegia; high-tone deafness; visual defect with pseudopapilloedema; slowing intellectual development; recurrent acute polyarthritis; erythema marginatum, splenomegaly and iron-resistant hypochromic anaemia. Proc. R. Soc. Med. 1973;66(11):1070–1071. [PMC free article] [PubMed] [Google Scholar]
- 6.Huttenlocher A, Frieden IJ, Emery H. Neonatal onset multisystem inflammatory disease. J. Rheumatol. 1995;22(6):1171–1173. [PubMed] [Google Scholar]
- 7.Kilcline C, Shinkai K, Bree A, et al. Neonatal-Onset Multisystem Inflammatory Disorder: The Emerging Role of Pyrin Genes in Autoinflammatory Diseases. Arch Dermatol. 2005;141(2):248–253. doi: 10.1001/archderm.141.2.248. [DOI] [PubMed] [Google Scholar]
- 8.Leslie KS, Lachmann HJ, Bruning E, et al. Phenotype, Genotype, and Sustained Response to Anakinra in 22 Patients With Autoinflammatory Disease Associated With CIAS-1/NALP3 Mutations. Arch Dermatol. 2006;142(12):1591–1597. doi: 10.1001/archderm.142.12.1591. [DOI] [PubMed] [Google Scholar]
- 9.Shinkai K, McCalmont TH, Leslie KS. Cryopyrin-associated periodic syndromes and autoinflammation. Clin. Exp. Dermatol. 2008;33(1):1–9. doi: 10.1111/j.1365-2230.2007.02540.x. [DOI] [PubMed] [Google Scholar]
- 10.Biswas D, Stafford N. Otolaryngological manifestations of ‘Muckle-Wells syndrome’. Int. J. Pediatr. Otorhinolaryngol. 2010;74(5):553–555. doi: 10.1016/j.ijporl.2010.02.023. [DOI] [PubMed] [Google Scholar]
- 11.Neven B, Marvillet I, Terrada C, et al. Long-term efficacy of the interleukin-1 receptor antagonist anakinra in ten patients with neonatal-onset multisystem inflammatory disease/chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum. 2010;62(1):258–267. doi: 10.1002/art.25057. [DOI] [PubMed] [Google Scholar]
- 12.Goldbach-Mansky R, Dailey NJ, Canna SW, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N. Engl. J. Med. 2006;355(6):581–592. doi: 10.1056/NEJMoa055137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mirault T, Launay D, Cuisset L, et al. Recovery from deafness in a patient with Muckle-Wells syndrome treated with anakinra. Arthritis Rheum. 2006;54(5):1697–1700. doi: 10.1002/art.21807. [DOI] [PubMed] [Google Scholar]
- 14.Hill SC, Namde M, Dwyer A, et al. Arthropathy of neonatal onset multisystem inflammatory disease (NOMID/CINCA) Pediatr Radiol. 2007;37(2):145–152. doi: 10.1007/s00247-006-0358-0. [DOI] [PubMed] [Google Scholar]
- 15.Feldmann J, Prieur A, Quartier P, et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am. J. Hum. Genet. 2002;71(1):198–203. doi: 10.1086/341357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoffman HM, Mueller JL, Broide DH, et al. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001;29(3):301–305. doi: 10.1038/ng756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldbach-Mansky R, Kastner DL. Autoinflammation: The prominent role of IL-1 in monogenic autoinflammatory diseases and implications for common illnesses. Journal of Allergy and Clinical Immunology. 2009;124(6):1141–1149. doi: 10.1016/j.jaci.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aróstegui JI, Lopez Saldaña MD, Pascal M, et al. A somatic NLRP3 mutation as a cause of a sporadic case of chronic infantile neurologic, cutaneous, articular syndrome/neonatal-onset multisystem inflammatory disease: Novel evidence of the role of low-level mosaicism as the pathophysiologic mechanism underlying mendelian inherited diseases. Arthritis Rheum. 2010;62(4):1158–1166. doi: 10.1002/art.27342. [DOI] [PubMed] [Google Scholar]
- 19.Henderson C, Goldbach-Mansky R. Monogenic IL-1 mediated autoinflammatory and immunodeficiency syndromes: Finding the right balance in response to danger signals. Clinical Immunology. 2010;135(2):210–222. doi: 10.1016/j.clim.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ye Z, Ting JP. NLR, the nucleotide-binding domain leucine-rich repeat containing gene family. Curr. Opin. Immunol. 2008;20(1):3–9. doi: 10.1016/j.coi.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 21.Agostini L, Martinon F, Burns K, et al. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20(3):319–325. doi: 10.1016/S1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 22.Dowds TA, Masumoto J, Zhu L, et al. Cryopyrin-induced interleukin 1beta secretion in monocytic cells: enhanced activity of disease-associated mutants and requirement for ASC. J. Biol. Chem. 2004;279(21):21924–21928. doi: 10.1074/jbc.M401178200. [DOI] [PubMed] [Google Scholar]
- 23.Tassi S, Carta S, Delfino L, et al. Altered redox state of monocytes from cryopyrin-associated periodic syndromes causes accelerated IL-1beta secretion. Proc. Natl. Acad. Sci. U.S.A. 2010;107(21):9789–9794. doi: 10.1073/pnas.1000779107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stojanov S, Weiss M, Lohse P, Belohradsky BH. A Novel CIAS1 Mutation and Plasma/Cerebrospinal Fluid Cytokine Profile in a German Patient With Neonatal-Onset Multisystem Inflammatory Disease Responsive to Methotrexate Therapy. Pediatrics. 2004;114(1):e124–127. doi: 10.1542/peds.114.1.e124. [DOI] [PubMed] [Google Scholar]
- 25.Serrano M, Ormazábal A, Antón J, et al. Cerebrospinal Fluid Neopterin and Cryopyrin-Associated Periodic Syndrome. Pediatric Neurology. 2009;41(6):448–450. doi: 10.1016/j.pediatrneurol.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 26.Watanabe H, Gaide O, Pétrilli V, et al. Activation of the IL-1beta-processing inflammasome is involved in contact hypersensitivity. J. Invest. Dermatol. 2007;127(8):1956–1963. doi: 10.1038/sj.jid.5700819. [DOI] [PubMed] [Google Scholar]
- 27.Aganna E, Martinon F, Hawkins PN, et al. Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis Rheum. 2002;46(9):2445–2452. doi: 10.1002/art.10509. [DOI] [PubMed] [Google Scholar]
- 28.Aksentijevich I, Nowak M, Mallah M, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): A new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis & Rheumatism. 2002;46(12):3340–3348. doi: 10.1002/art.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoffman, H. Infevers - NLRP3 Sequence Variants. http://fmf.igh.cnrs.fr/ISSAID/infevers/search.php?n=4. Accessed September 20, 2010.
- 30.Aksentijevich ID, Putnam C, Remmers EF, et al. The clinical continuum of cryopyrinopathies: Novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum. 2007;56(4):1273–1285. doi: 10.1002/art.22491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jesus A, Silva C, Segundo G, et al. Phenotype–Genotype Analysis of Cryopyrin-Associated Periodic Syndromes (CAPS): Description of a Rare Non-Exon 3 and a Novel CIAS1 Missense Mutation. Journal of Clinical Immunology. 2008;28(2):134–138. doi: 10.1007/s10875-007-9150-7. [DOI] [PubMed] [Google Scholar]
- 32.Krause K, Zuberbier T, Maurer M. Modern approaches to the diagnosis and treatment of cold contact urticaria. Curr Allergy Asthma Rep. 2010;10(4):243–249. doi: 10.1007/s11882-010-0121-3. [DOI] [PubMed] [Google Scholar]
- 33.Siebenhaar F, Weller K, Mlynek A, et al. Acquired cold urticaria: clinical picture and update on diagnosis and treatment. Clin. Exp. Dermatol. 2007;32(3):241–245. doi: 10.1111/j.1365-2230.2007.02376.x. [DOI] [PubMed] [Google Scholar]
- 34.Lequerré T, Quartier P, Rosellini D, et al. Interleukin-1 receptor antagonist (anakinra) treatment in patients with systemic-onset juvenile idiopathic arthritis or adult onset Still disease: preliminary experience in France. Ann. Rheum. Dis. 2008;67(3):302–308. doi: 10.1136/ard.2007.076034. [DOI] [PubMed] [Google Scholar]
- 35.Fortna RR, Gudjonsson JE, Seidel G, et al. Persistent pruritic papules and plaques: a characteristic histopathologic presentation seen in a subset of patients with adult-onset and juvenile Still’s disease. J. Cutan. Pathol. 2010;37(9):932–937. doi: 10.1111/j.1600-0560.2010.01570.x. [DOI] [PubMed] [Google Scholar]
- 36.Frieri M. Identification of masqueraders of autoimmune disease in the office. Allergy Asthma Proc. 2003;24(6):421–9. [PubMed] [Google Scholar]
- 37.Ohlsson V, Baildam E, Foster H, et al. Anakinra treatment for systemic onset juvenile idiopathic arthritis (SOJIA) Rheumatology (Oxford). 2008;47(4):555–556. doi: 10.1093/rheumatology/ken030. [DOI] [PubMed] [Google Scholar]
- 38.Pascual V, Allantaz F, Arce E, et al. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J. Exp. Med. 2005;201(9):1479–1486. doi: 10.1084/jem.20050473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gwyther M, Schwarz H, Howard A, Ansell BM. C-reactive protein in juvenile chronic arthritis: an indicator of disease activity and possibly amyloidosis. Ann. Rheum. Dis. 1982;41(3):259–262. doi: 10.1136/ard.41.3.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Efthimiou P, Paik PK, Bielory L. Diagnosis and management of adult onset Still’s disease. Ann. Rheum. Dis. 2006;65(5):564–572. doi: 10.1136/ard.2005.042143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reddy S, Jia S, Geoffrey R, et al. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N. Engl. J. Med. 2009;360(23):2438–2444. doi: 10.1056/NEJMoa0809568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aksentijevich I, Masters SL, Ferguson PJ, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N. Engl. J. Med. 2009;360(23):2426–2437. doi: 10.1056/NEJMoa0807865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Asahina A, Sakurai N, Suzuki Y, Narushima K. Schnitzler’s syndrome with prominent neutrophil infiltration misdiagnosed as Sweet’s syndrome: a typical example of urticarial neutrophilic dermatosis. Clin. Exp. Dermatol. 2010;35(4):e123–126. doi: 10.1111/j.1365-2230.2009.03746.x. [DOI] [PubMed] [Google Scholar]
- 44.de Koning HD, Bodar EJ, Simon A, et al. Beneficial response to anakinra and thalidomide in Schnitzler’s syndrome. Ann. Rheum. Dis. 2006;65(4):542–544. doi: 10.1136/ard.2005.045245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soubrier M. Schnitzler syndrome. Joint Bone Spine. 2008;75(3):263–266. doi: 10.1016/j.jbspin.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 46.Gillmore JD, Lovat LB, Persey MR, et al. Amyloid load and clinical outcome in AA amyloidosis in relation to circulating concentration of serum amyloid A protein. Lancet. 2001;358(9275):24–29. doi: 10.1016/S0140-6736(00)05252-1. [DOI] [PubMed] [Google Scholar]
- 47.Fischer E, Van Zee KJ, Marano MA, et al. Interleukin-1 receptor antagonist circulates in experimental inflammation and in human disease. Blood. 1992;79(9):2196–2200. [PubMed] [Google Scholar]
- 48.Campion GV, Lebsack ME, Lookabaugh J, et al. Dose-range and dose-frequency study of recombinant human interleukin-1 receptor antagonist in patients with rheumatoid arthritis. The IL-1Ra Arthritis Study Group. Arthritis Rheum. 1996;39(7):1092–1101. doi: 10.1002/art.1780390704. [DOI] [PubMed] [Google Scholar]
- 49.So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res. Ther. 2007;9(2):R28. doi: 10.1186/ar2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kineret [package insert]. Thousand Oaks, CA: Amgen Manufacturing, Limited, 2001-2003.
- 51.Fischer E, Marano MA, Van Zee KJ, et al. Interleukin-1 receptor blockade improves survival and hemodynamic performance in Escherichia coli septic shock, but fails to alter host responses to sublethal endotoxemia. J. Clin. Invest. 1992;89(5):1551–1557. doi: 10.1172/JCI115748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fisher CJ, Slotman GJ, Opal SM, et al. Initial evaluation of human recombinant interleukin-1 receptor antagonist in the treatment of sepsis syndrome: a randomized, open-label, placebo-controlled multicenter trial. Crit. Care Med. 1994;22(1):12–21. doi: 10.1097/00003246-199401000-00267. [DOI] [PubMed] [Google Scholar]
- 53.Fisher CJ, Dhainaut JF, Opal SM, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. JAMA. 1994;271(23):1836–1843. doi: 10.1001/jama.271.23.1836. [DOI] [PubMed] [Google Scholar]
- 54.Jiang Y, Genant HK, Watt I, et al. A multicenter, double-blind, dose-ranging, randomized, placebo-controlled study of recombinant human interleukin-1 receptor antagonist in patients with rheumatoid arthritis: radiologic progression and correlation of Genant and Larsen scores. Arthritis Rheum. 2000;43(5):1001–1009. doi: 10.1002/1529-0131(200005)43:5<1001::AID-ANR7>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 55.Bresnihan B, Alvaro-Gracia JM, Cobby M, et al. Treatment of rheumatoid arthritis with recombinant human interleukin-1 receptor antagonist. Arthritis Rheum. 1998;41(12):2196–2204. doi: 10.1002/1529-0131(199812)41:12<2196::AID-ART15>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 56.Hawkins PN, Lachmann HJ, McDermott MF. Interleukin-1-receptor antagonist in the Muckle-Wells syndrome. N. Engl. J. Med. 2003;348(25):2583–2584. doi: 10.1056/NEJM200306193482523. [DOI] [PubMed] [Google Scholar]
- 57.Hoffman HM, Rosengren S, Boyle DL, et al. Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet. 2004;364(9447):1779–1785. doi: 10.1016/S0140-6736(04)17401-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hawkins PN, Lachmann HJ, Aganna E, McDermott MF. Spectrum of clinical features in Muckle-Wells syndrome and response to anakinra. Arthritis Rheum. 2004;50(2):607–612. doi: 10.1002/art.20033. [DOI] [PubMed] [Google Scholar]
- 59.Ramos E, Aróstegui JI, Campuzano S, et al. Positive clinical and biochemical responses to anakinra in a 3-yr-old patient with cryopyrin-associated periodic syndrome (CAPS) Rheumatology (Oxford). 2005;44(8):1072–1073. doi: 10.1093/rheumatology/keh652. [DOI] [PubMed] [Google Scholar]
- 60.Boschan C, Witt O, Lohse P, et al. Neonatal-onset multisystem inflammatory disease (NOMID) due to a novel S331R mutation of the CIAS1 gene and response to interleukin-1 receptor antagonist treatment. Am. J. Med. Genet. A. 2006;140(8):883–886. doi: 10.1002/ajmg.a.31148. [DOI] [PubMed] [Google Scholar]
- 61.Cascavilla N, Bisceglia M, D’Arena G. Successful treatment of Schnitzler’s syndrome with anakinra after failure of rituximab trial. Int J Immunopathol Pharmacol. 2010;23(2):633–636. doi: 10.1177/039463201002300226. [DOI] [PubMed] [Google Scholar]
- 62.Lahiri M, Teng G. A case of refractory adult-onset Still's disease treated with anakinra. Int J Rheum Dis. 2010;13(3):e36–41. doi: 10.1111/j.1756-185X.2010.01474.x. [DOI] [PubMed] [Google Scholar]
- 63.Abbate A, Kontos MC, Grizzard JD, et al. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study) Am. J. Cardiol. 2010;105(10):1371–1377. doi: 10.1016/j.amjcard.2009.12.059. [DOI] [PubMed] [Google Scholar]
- 64.Economides AN, Carpenter LR, Rudge JS, et al. Cytokine traps: multi-component, high-affinity blockers of cytokine action. Nat. Med. 2003;9(1):47–52. doi: 10.1038/nm811. [DOI] [PubMed] [Google Scholar]
- 65.Stahl N, Radin A, Mellis S. Rilonacept–CAPS and beyond. Ann. N. Y. Acad. Sci. 2009;1182:124–134. doi: 10.1111/j.1749-6632.2009.05074.x. [DOI] [PubMed] [Google Scholar]
- 66.Arcalyst [package insert]. Tarrytown, NY: Regeneron Pharmaceuticals, Inc; 2008.
- 67.Goldbach-Mansky R, Shroff SD, Wilson M, et al. A pilot study to evaluate the safety and efficacy of the long-acting interleukin-1 inhibitor rilonacept (interleukin-1 Trap) in patients with familial cold autoinflammatory syndrome. Arthritis Rheum. 2008;58(8):2432–2442. doi: 10.1002/art.23620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hoffman HM, Throne ML, Amar NJ, et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum. 2008;58(8):2443–2452. doi: 10.1002/art.23687. [DOI] [PubMed] [Google Scholar]
- 69.Terkeltaub R, Sundy JS, Schumacher HR, et al. The interleukin 1 inhibitor rilonacept in treatment of chronic gouty arthritis: results of a placebo-controlled, monosequence crossover, non-randomised, single-blind pilot study. Ann. Rheum. Dis. 2009;68(10):1613–1617. doi: 10.1136/ard.2009.108936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lachmann HJ, Lowe P, Felix SD, et al. In vivo regulation of interleukin 1beta in patients with cryopyrin-associated periodic syndromes. J. Exp. Med. 2009;206(5):1029–1036. doi: 10.1084/jem.20082481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ilaris [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corp; 2009.
- 72.Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N. Engl. J. Med. 2009;360(23):2416–2425. doi: 10.1056/NEJMoa0810787. [DOI] [PubMed] [Google Scholar]
- 73.Alten R, Gram H, Joosten LA, et al. The human anti-IL-1 beta monoclonal antibody ACZ885 is effective in joint inflammation models in mice and in a proof-of-concept study in patients with rheumatoid arthritis. Arthritis Res. Ther. 2008;10(3):R67. doi: 10.1186/ar2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.So A, De Meulemeester M, Pikhlak A, et al. Canakinumab for the treatment of acute flares in difficult-to-treat gouty arthritis: Results of a multicenter, phase II, dose-ranging study. Arthritis Rheum. 2010;62(10):3064–3076. doi: 10.1002/art.27600. [DOI] [PubMed] [Google Scholar]
- 75.Wannamaker W, Davies R, Namchuk M, et al. (S)-1-((S)-2-{[1-(4-amino-3-chloro-phenyl)-methanoyl]-amino}-3, 3-dimethyl-butanoyl)-pyrrolidine-2-carboxylic acid ((2R, 3 S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/caspase-1 inhibitor, exhibits potent anti-inflammatory activities by inhibiting the release of IL-1beta and IL-18. J. Pharmacol. Exp. Ther. 2007;321(2):509–516. doi: 10.1124/jpet.106.111344. [DOI] [PubMed] [Google Scholar]
- 76.Stack JH, Beaumont K, Larsen PD, et al. IL-converting enzyme/caspase-1 inhibitor VX-765 blocks the hypersensitive response to an inflammatory stimulus in monocytes from familial cold autoinflammatory syndrome patients. J. Immunol. 2005;175(4):2630–2634. doi: 10.4049/jimmunol.175.4.2630. [DOI] [PubMed] [Google Scholar]
- 77.Hoffman HM, Wolfe F, Belomestnov P, Mellis SJ. Cryopyrin-associated periodic syndromes: development of a patient-reported outcomes instrument to assess the pattern and severity of clinical disease activity. Curr Med Res Opin. 2008;24(9):2531–2543. doi: 10.1185/03007990802297495. [DOI] [PubMed] [Google Scholar]
- 78.So A. Developments in the scientific and clinical understanding of gout. Arthritis Res. Ther. 2008;10(5):221. doi: 10.1186/ar2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brenner M, Ruzicka T, Plewig G, et al. Targeted treatment of pyoderma gangrenosum in PAPA (pyogenic arthritis, pyoderma gangrenosum and acne) syndrome with the recombinant human interleukin-1 receptor antagonist anakinra. Br. J. Dermatol. 2009;161(5):1199–1201. doi: 10.1111/j.1365-2133.2009.09404.x. [DOI] [PubMed] [Google Scholar]
- 80.Maedler K, Dharmadhikari G, Schumann DM, Størling J. Interleukin-1 beta targeted therapy for type 2 diabetes. Expert Opin Biol Ther. 2009;9(9):1177–1188. doi: 10.1517/14712590903136688. [DOI] [PubMed] [Google Scholar]
- 81.Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]