

Abstract
Using simulated viral load data for a given maraviroc monotherapy study design, the feasibility of different algorithms to perform parameter estimation for a pharmacokinetic-pharmacodynamic-viral dynamics (PKPD-VD) model was assessed. The assessed algorithms are the first-order conditional estimation method with interaction (FOCEI) implemented in NONMEM VI and the SAEM algorithm implemented in MONOLIX version 2.4. Simulated data were also used to test if an effect compartment and/or a lag time could be distinguished to describe an observed delay in onset of viral inhibition using SAEM. The preferred model was then used to describe the observed maraviroc monotherapy plasma concentration and viral load data using SAEM. In this last step, three modelling approaches were compared; (i) sequential PKPD-VD with fixed individual Empirical Bayesian Estimates (EBE) for PK, (ii) sequential PKPD-VD with fixed population PK parameters and including concentrations, and (iii) simultaneous PKPD-VD. Using FOCEI, many convergence problems (56%) were experienced with fitting the sequential PKPD-VD model to the simulated data. For the sequential modelling approach, SAEM (with default settings) took less time to generate population and individual estimates including diagnostics than with FOCEI without diagnostics. For the given maraviroc monotherapy sampling design, it was difficult to separate the viral dynamics system delay from a pharmacokinetic distributional delay or delay due to receptor binding and subsequent cellular signalling. The preferred model included a viral load lag time without inter-individual variability. Parameter estimates from the SAEM analysis of observed data were comparable among the three modelling approaches. For the sequential methods, computation time is approximately 25% less when fixing individual EBE of PK parameters with omission of the concentration data compared with fixed population PK parameters and retention of concentration data in the PD-VD estimation step. Computation times were similar for the sequential method with fixed population PK parameters and the simultaneous PKPD-VD modelling approach. The current analysis demonstrated that the SAEM algorithm in MONOLIX is useful for fitting complex mechanistic models requiring multiple differential equations. The SAEM algorithm allowed simultaneous estimation of PKPD and viral dynamics parameters, as well as investigation of different model sub-components during the model building process. This was not possible with the FOCEI method (NONMEM version VI or below). SAEM provides a more feasible alternative to FOCEI when facing lengthy computation times and convergence problems with complex models.
Keywords: Maraviroc, MONOLIX, SAEM, Viral dynamics, Population PKPD
Introduction
Maraviroc (UK-427,857) is a reversible and selective antagonist of the human chemokine CCR5 receptor [1]. It has been approved for use in combination with other antiretroviral agents for the treatment of subjects infected with CCR5-tropic human immunodeficiency virus type 1 (HIV-1). Two short-term 10 days monotherapy treatment phase 2a studies (A4001007 and A4001015) were performed in asymptomatic CCR5-tropic HIV-1 infected subjects [2]. The maraviroc doses ranged from 25 mg once daily (QD) to 300 mg twice daily (BID). The mean HIV-1 viral load declined in a dose-dependent fashion with up to 1.6 log10 RNA copies ml−1 achieved (at day 11) with 300 mg BID [2].
Mathematical models have been widely used to describe the dynamics and interaction of target CD4+ cells, actively, latently, persistently and defectively infected cells and plasma virus in HIV-1 infected asymptomatic subjects after initiation of antiretroviral therapy [3–5]. These models employ sets of differential equations to describe the viral dynamics. The maraviroc monotherapy data have previously been analyzed in a two-stage approach by fitting a pharmacokinetic-pharmacodynamic-viral dynamics (PKPD-VD) model [6, 7] using nonlinear mixed-effects modelling for estimation of fixed effects, inter-individual (IIV) and residual variability implemented in the NONMEM software [8]. NONMEM estimation methods include a first-order method (FO) and first-order conditional estimation (FOCE) method, both of which involve linearization of the regression function with respect to the random effects [9–12]. Some of the practical drawbacks and/or limitations when performing PKPD-VD parameter estimations with the FOCE method with interaction (FOCEI) in older versions of NONMEM (VI and below) are:
-
(i)
very long computation times;
-
(ii)
convergence problems resulting from numerical difficulties in optimizing the linearized likelihood;
-
(iii)
model instability necessitating a two-stage approach (PK modelling followed by PKPD-VD modelling) and limitations in numbers of parameters that can be estimated;
-
(iv)
difficulties with models that have change points such as lag times.
These factors make it very difficult to develop these complex PKPD models or to perform simultaneous PKPD-VD modelling with the FOCEI method.
MONOLIX [13] implements a stochastic approximation (SA) of the standard expectation maximization (EM) (=SAEM) algorithm for nonlinear mixed-effects models without approximations. The SAEM algorithm replaces the usual estimation step of EM by a stochastic procedure which has been shown to be very efficient with improved convergence toward the maximum likelihood (ML) estimates [14]. SAEM (as well as other EM-type algorithms) performs ML estimation without any approximation of the statistical model. Then “optimal” statistical properties (consistency and minimum variance of the estimate) are expected with SAEM. In addition, implementation of SAEM in MONOLIX has been optimized; settings of the Markov Chain Monte Carlo (MCMC) used for the simulation step have been carefully defined (combination of several transition kernels, optimal acceptance rate, etc.), and a simulated annealing procedure accelerates the convergence of the algorithm toward the solution. A review of population analysis methods and software using examples for complex PK and PD methods concludes that EM methods (performed by S-ADAPT, PDx-MCPEM and MONOLIX) have greater stability in analyzing complex PKPD models and can provide accurate results with sparse and rich data [12].
The objectives of the present analysis were to assess the SAEM functionality implemented in MONOLIX for complex mechanistic models in the application of population PKPD-VD modelling of maraviroc monotherapy PK and viral load data. Three key questions are addressed (Fig. 1):
-
(i)
The feasibility of the FOCEI method (implemented in NONMEM VI) and the SAEM algorithm (implemented in MONOLIX) to perform parameter estimation for a PKPD-VD population model using simulated data;
-
(ii)
Determination of a preferred model using SAEM in looking at a lag time and an effect site delay model (ke0 model) using simulated data;
-
(iii)
Comparison of parameter estimates with sequential (two-stage) versus simultaneous modelling approaches using SAEM and observed data from maraviroc monotherapy studies.
Fig. 1.

Schematic representation of the analysis plan. Simulation model = PKPD-VD with LagE and ke0, IIV on IC50 and VD parameters (RR0, b and d 2)
The aim of this paper is thus not to directly compare parameter estimates from SAEM and FOCEI, but to explore SAEM as a tool that may allow efficient/stable estimates of PKPD and VD parameters within a nonlinear mixed effects framework with two levels of random variability.
Methods
Data
An analysis of the data from two randomized, placebo-controlled, phase 2a monotherapy studies (A4001007 and A4001015) has been performed; rich plasma maraviroc concentrations (1,250 samples) and viral load data (1,169 observations) were available from 63 asymptomatic CCR5-tropic HIV-1 infected patients. Approval from local ethics committees was obtained and written informed consent was obtained from all subjects. These studies are described in more detail elsewhere [2].
Patients were randomly assigned to the following treatment groups: maraviroc 25 mg QD, 50, 100 or 300 mg BID, or placebo under fasted conditions in study A4001007; maraviroc 150 mg BID (fed and fasted), 100 or 300 mg QD, or placebo under fasted conditions in study A4001015. In both studies, patients received treatment for 10 days and were followed up for 30 days after the last dose. The current analysis included data arising from patients who were assigned to one of the maraviroc treatment arms and excluded placebo data.
Blood samples were collected for determination of maraviroc plasma concentrations pre-morning dosing on days 1–10, and at specified times up to at least 24 h post dose on day 10. In A4001007, several additional samples up to 12 h post-morning dose on day 1, as well as 48, 72 and 120 h post dose on day 10 were collected.
Plasma samples were analysed by a centralized laboratory (Maxxam Analytics Inc, Mississauga ON, Canada) to determine maraviroc concentrations. The analytical procedure employed solid phase extraction of the analyte and internal standard with separation by high-performance liquid chromatography followed by atmospheric pressure chemical ionisation and tandem mass spectrometric detection. The same analytical method was used in both studies. The lower limit of quantification was 0.5 ng ml−1. Plasma HIV-1 RNA viral load were assessed at screening, randomization, and on days 1–13, 15, 19, 22, 25 and 40 using the Roche Amplicor v1.5 RT-PCR assay (Roche Diagnostics). Baseline viral load was computed as the mean of three log10 transformed predose values.
PKPD-viral dynamics model
The PKPD-VD model has 3 components [7]:
PK model
A basic 2-compartment disposition model (Eqs. 1–3), parameterized as clearances (total CL and intercomparmental Q) and volumes (central V2 and peripheral V3), with first-order absorption (ka), an absorption lag time (LagC), food effects on ka and F1 (relative bioavailability) and an additive residual error model was used to fit the log-transformed maraviroc concentrations. The individual Empirical Bayesian Estimates (EBE) of PK parameters were used to predict the drug concentration at the specific times when viral loads were measured for the second stage sequential PKPD-VD modelling.
Absorption compartment 1:
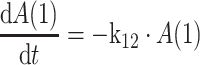 |
1 |
Central compartment 2:
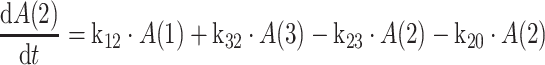 |
2 |
Peripheral compartment 3:
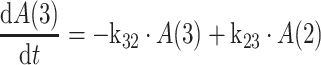 |
3 |
where k12 = ka, 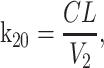
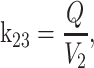
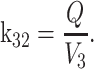 ka is the absorption rate constant; CL is the clearance; Q is the intercompartmental clearance; V
2 is the central volume; V
3 is the peripheral volume.
ka is the absorption rate constant; CL is the clearance; Q is the intercompartmental clearance; V
2 is the central volume; V
3 is the peripheral volume.
PD model
Based on the known mechanism of action of maraviroc, the effect was modelled using an inhibitory Emax model acting on the infection rate of the virus and target CD4+ cells. An area of interest, in modelling HIV-1 viral load changes in response to treatment, is exploration of the observed time delay component of viral load response to drug treatment.
After the start of antiretroviral therapy, a delay (1–2 days) in the effect on viral load is usually observed regardless of the antiretroviral agent (irrelevant of its PK and/or its class) [15–18]. The cause of this delay is probably multi-factorial, i.e. time required for drug absorption, disposition, interaction with the target receptor/enzyme, activation of subsequent cellular or intra-cellular pathways, as well as the clearance of free virus particles and the death of infected cells. Different modelling approaches can be used to describe such a delay, e.g.
-
(i)
introduction of a lag time between PK and PD;
-
(ii)
the use of an effect compartment model (Eq. 4) to describe the time lag between plasma drug concentration and drug effect with an equilibration half time parameter (ke0) [19];
Effect compartment 4: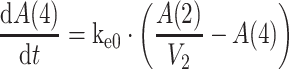
4 -
(iii)
an onset/offset model where different rate constants are allowed for the onset and offset of response.
All of the above approaches increase the complexity of the PKPD-VD model and make estimation with the FOCEI method more difficult. This offered a further opportunity for testing the functionality of SAEM (implemented in MONOLIX).
In previous analyses of the maraviroc monotherapy data using FOCEI (unpublished data), the delay in onset and offset of viral inhibition was modelled with a lag time (LagE) and ke0. This allowed for the shift of the PK profile from the onset and offset of viral inhibition.
Viral dynamics model
Details of the VD model are described elsewhere [3–7]. Briefly, the dynamics and interaction of target CD4+ cells, actively infected CD4+ cells, latently infected CD4+ cells and viruses in HIV-1 infected asymptomatic patients after initiation of antiretroviral therapy were modelled with a set of differential equations (Eqs. 5–9a, b).
Target cell (activated CD4+ cells):
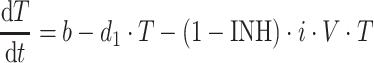 |
5 |
Actively infected cell (short-lived):
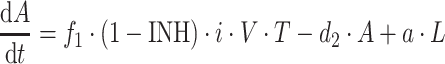 |
6 |
Latently infected resting cells (long-lived):
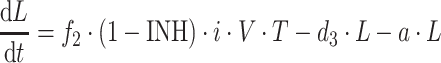 |
7 |
Infectious virus (copies HIV-1 RNA):
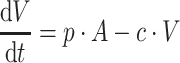 |
8 |
Viral inhibition fraction:
-
(i)driven by central compartment concentration
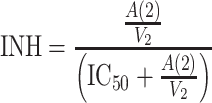
9a -
(ii)driven by effect compartment concentration
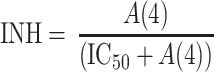
9b
where b is the birth rate constant of healthy target CD4+ cells (T); d 1 is the death rate constant of T cells; i is the infection rate of T cells; V is the number of virus particles; INH is the viral inhibition fraction driven by (central or effect compartment) maraviroc concentration with an inhibitory Emax model where maximal inhibition is fixed to 1; f 1 is the fraction of healthy T cells which become short-lived actively infected T cells (A); d 2 is the death rate constant of short-lived actively infected T cells; f 2 = (1 − f 1) is the fraction of healthy T cells which become latently infected resting cells (L); d 3 is the death rate constant of latently infected resting cells; a is the reactivation rate constant of latently infected resting cells; p is the viral production rate of short-lived actively infected T cells; c is the death rate constant of virus. The persistently and defectively infected cells with very long half-lives were excluded in the current analysis as they are not relevant to short-term (10 day) data.
The in vivo maraviroc IC50 and VD parameters (basic reproductive ratio (RR0), b and d 2) were estimated. All remaining viral dynamic inputs were fixed to values based on literature ranges [6, Table 1].
Table 1.
Selection of the preferred model using SAEM given the maraviroc monotherapy study design (n = 50)
| Model | With inter-individual variability on LagE and/or ke0? | Number of times selected as preferred model [%] |
|---|---|---|
| No lag time | No | 0 [0] |
| No effect compartment | ||
| Lag time only | No | 48 [96] |
| Lag time only | Yes | 4 [8]a |
| Effect compartment only | No | 0 [0] |
| Effect compartment only | Yes | 0 [0] |
| With lag time and effect compartment | No | 6 [12]b |
| With lag time and effect compartment | Yes | 0 [0] |
aOnly 2 coincided with the 48 runs from the lag time only model without ω[LagE]
bAll 6 coincided with the 48 runs from the lag time only model without ω[LagE]
RR0 is a derived model parameter computed as the ratio of the birth rate constants to the death rate constants (Eq. 10). RR0 gives the average number of offspring generated by a single virus particle in the absence of constraints [4]. When RR0 in the presence of an inhibitor is below 1, the system theoretically goes to extinction. When RR0 is above 1, the system adjusts to the inhibition and re-equilibrates to a new steady state.
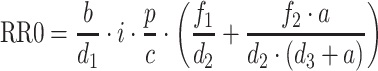 |
10 |
The reproduction minimum inhibitory concentration (RMIC) is defined as the concentration of an antiretroviral compound that decreases RR0 to below the break point of 1 which results in eradication of the disease [20]. Given that RR0 and IC50 are highly correlated during the estimation process, RMIC was computed (Eq. 11) to make more appropriate comparisons of these key parameter estimates obtained from different models or algorithms in the current analysis.
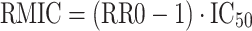 |
11 |
Software
The current analyses were performed using:
-
(i)
A nonlinear mixed-effects modelling methodology as implemented in the NONMEM software system, version VI level 1.2 [8] (patched with updated subroutines according to instructions provided from GloboMax/ICON on 30 August 2007). NM-TRAN subroutines version IV level 1.1, and the PREDPP model library (ADVAN6 TOL = 5), version V level 1.0 utilizing Intel-based PC Workstations (Pentium 4 Intel® Xeon® CPU Processor @ 3.06 or 3.20 GHz) running Red Hat Linux (3.4.6-8) operating system (GRID system) and GNU Fortran compiler (GCC 3.4.6 20060404) were used. Parameter estimation was performed using the FOCEI method. ADVAN6 TOL = 5 was chosen to be the default setting because previous experiences with NONMEM had shown that none of the other PREDPP model library or TOL settings improved computation time or convergence.
-
(ii)
The SAEM algorithm as implemented in MONOLIX version 2.4 via MATLAB version 7.5.0 on a Microsoft Windows XP operating system installed on a ThinkPad T61 with Intel® Core™ Duo CPU T7300 processors @ 2.00 GHz and 1.96 GB of RAM.
Analysis plan
Part 1: assessing fitting feasibility of the FOCEI and SAEM algorithms with simulated data
The first question to be addressed was the feasibility of the FOCEI and SAEM algorithms to perform parameter estimation for the PKPD-VD model. Simulation of 50 datasets of viral load profiles were performed in NONMEM VI using identical study designs to the maraviroc monotherapy studies (subject numbers, doses and observations). Parameter estimates from a previous PKPD-VD analysis of the data using FOCEI including IIV (on IC50, RR0, b and d 2) and residual error but not including parameter uncertainty (unpublished data, details in PKPD-Viral Dynamics Model section) were used.
The viral inhibition was driven by the predicted PK profile based on the population PK parameter estimates excluding IIV and residual error. This was done to eliminate the potential impact of maraviroc PK on the VD parameter estimation.
The simulated concentration and viral load data were then fitted separately using FOCEI and SAEM, with the true parameter values as the initial conditions. Fitting feasibility was assessed by the number of runs that terminated with successful minimization and other termination messages in NONMEM VI, as well as the precision of parameter estimates (compared with the “true” values used in simulation) obtained from FOCEI and SAEM.
If a run using FOCEI terminated due to “rounding errors”, one more attempt/run was made by using the final parameter estimates as the initial parameter estimates. If the second attempt minimized successfully, the run was classified as successful; otherwise it was classified as run terminated with “rounding errors”.
Part 2: determination of the preferred model using simulated data with SAEM
Since different modelling approaches can be used to describe the delay in onset and offset of viral inhibition, another issue was to determine if SAEM could be used to estimate parameters describing the delay for a given maraviroc monotherapy study design with simulated data. While the simulation PD model had both a lag time (LagE) and an effect compartment model (ke0) describing the delay in onset and offset of viral inhibition, the 50 simulated viral load datasets were fitted to different PD models using SAEM, i.e. inclusion/exclusion of LagE and an effect compartment, as well as with/without IIV on LagE and ke0.
The preferred model was selected based on the precision of parameter estimates, standard error of population parameters, correlation of estimates, log-likelihood (by Monte-Carlo Importance Sampling), information criteria such as Akaike information criterion (AIC) and Bayesian information criterion (BIC).
The 50 simulated viral load datasets were also fitted to the preferred model using FOCEI in order to determine if numerical difficulties occur with the less complex model. Bias and imprecision for model parameters were measured by the mean prediction error and the root mean squared prediction error [21], respectively. In addition, stability of the preferred model with the FOCEI and SAEM algorithms was assessed using Perl-speaks-NONMEM (PsN version 2.3.0) [22] with the execute function. PsN enables automatic retrial of runs using slightly perturbed initial estimates. The minimum and maximum retries were set to 2 and 5, respectively.
Part 3: comparison of parameter estimates obtained from sequential and simultaneous modelling approaches using SAEM
Finally, the observed maraviroc concentration and viral load data were fitted with the preferred model (defined in Part 2) using SAEM and 3 different modelling approaches:
-
(i)
Sequential PK and PD-VD, where PK parameters were estimated from the PK data alone, then the PD-VD parameters were estimated based on the PD data and the fixed individual EBE of PK parameters. PK data were omitted in this PD-VD parameter estimation step;
-
(ii)
Sequential PK and PD-VD with PK parameters fixed to population estimates obtained from the separate PK analysis as described in (i). The PD-VD parameters were estimated conditionally on the fitted PK model and also the PK concentration data were retained in this sequence;
-
(iii)
Simultaneous PKPD-VD, where PK and PD-VD parameters were estimated simultaneously in the presence of both concentration and viral load data.
The overall aim was to assess the performance of SAEM for parameter estimation in the presence/absence of concentrations when coupling/uncoupling the PK and PD-VD components of the PKPD-VD model.
Results
Part 1: assessing fitting feasibility of the FOCEI and SAEM algorithms
When analyzing the 50 simulated viral load data sets with FOCEI, 22 runs experienced numerical difficulties, of which 18 runs did not execute a single iteration. Six runs terminated with “rounding errors” (on the second attempt). Only 22 runs terminated with “minimization successful”, of which half failed to run a covariance step. Of the 22 successful runs, 8 had unreasonable estimates of ke0 (4 runs with 131–171 fold increases, 2 runs with 11.7 and 1,621 fold increases respectively, 2 runs with 2.7 and 5 fold decreases) and/or LagE (2 runs with two to three fold decreases). For the 2 runs with unreasonable estimates of both ke0 and LagE, ke0 (0.564 and 1.07 day−1) was 2.7 and 5.1 fold lower, while LagE (0.368 and 0.483 days) was >twofold lower than the true parameter values (ke0 = 2.86 day−1, LagE = 1.13 days).
Parameter estimates were obtained for all 50 simulated data sets using SAEM. Six runs had estimates of ke0 (4 runs with 2–2.65 fold lower estimates), RR0 (1 run with 5.5 fold higher estimate) and/or IC50 (2 runs with 2.2 and 47 fold higher estimates) inconsistent with the values used for simulation. For the run with both unreasonable estimates of RR0 and IC50, IC50 (418 ng ml−1) was 47-fold higher than the “true” parameter value of 8.66 ng ml−1. For both the FOCEI and SAEM algorithms, parameter estimation of ke0 was troublesome.
The computation time for the 22 successful runs with FOCEI ranged from 1.75 to 192.8 h (median ~ 3 h) without any post processing for diagnostics. With SAEM, computation time was approximately 2 h per PD-VD run including diagnostics using the default settings (number of simulation samples: visual predictive checks (VPC) = 100, normalized prediction distribution errors (NPDE) = 500, Monte-Carlo size used for estimating the log-likelihood (LLP) = 10,000).
Part 2: determination of the preferred model using simulated data with SAEM
A general finding when fitting the 50 simulated viral load data sets using SAEM with different PD-VD models was the difficulty in determining both ke0 and LagE parameters given the viral load sampling times. The preferred model was the lag time model without an effect compartment and without IIV on LagE. This model described 48 (96%) simulated data sets the best (Table 1) based on the criteria listed in methods. The run time for the PD-VD estimation step ranged from 1.5 to 2.5 h.
When analyzing the 50 simulated viral load data sets with FOCEI using the preferred model, 21 runs experienced numerical difficulties, of which 15 runs did not execute a single iteration; 5 runs terminated with “rounding errors” (on the second attempt). Twenty-four runs terminated with “minimization successful”, of which 4 failed to run a covariance step.
Distribution plots of the parameter estimates obtained from the successful runs for SAEM and FOCEI together with the true values used for simulation are shown in Figs. 2 and 3, respectively. In general, LagE was overestimated with both SAEM and FOCEI largely due to the exclusion of the effect compartment. Parameter estimates obtained from SAEM were more randomly spread around their true values, particularly for the random effects parameter estimates.
Fig. 2.

Distributions of fixed and random effects parameter estimates obtained by fitting the simulated viral load data sets with SAEM using the preferred model. Vertical lines represent the true values
Fig. 3.

Distributions of fixed and random effects parameter estimates obtained by fitting the simulated viral load data sets with FOCEI using the preferred model. Vertical lines represent the true values
With a minimum of 2 and maximum of 5 retries of random changes in starting estimates using PsN, 31 of the 50 simulated data sets had at least one run terminated with “minimization successful”. Ten out of the 31 simulated data sets were also analyzed using SAEM with the initial estimates perturbed exactly as in PsN. In general, the minimum to maximum variation of the fixed effects parameter estimates obtained from the 2 to 5 retries was less than twofold. A bigger variation was seen in the random effects estimates using FOCEI; 3 out of 31 had a 3–3.5 fold variation and 1 out of 31 had a 13 fold variation in IIV of d 2.
The preferred model with LagE but without IIV on LagE was taken forward for Part 3 analysis.
Part 3: comparison of parameter estimates obtained from sequential and simultaneous modelling approaches using SAEM
The PK parameter estimates obtained from sequential and simultaneous PKPD-VD modelling approaches using SAEM are presented in Table 2. Food effects on ka and F1 were modelled as fractional changes with fasted status as the reference group. Due to the limited amount of data where maraviroc was administered with food and the potential distortion of the PK model by a mis-specified PD model, the impact of the food effect on ka and F1 were very different between the sequential and simultaneous PKPD modelling approaches. However, the relative standard errors suggest that the food effect on ka, particularly for the simultaneous PKPD analyses, was not precisely estimated. Nevertheless, the structural PK parameters and associated IIV were similar between the sequential and simultaneous PKPD modelling approaches with small relative standard error (RSE).
Table 2.
Comparison of PK parameter estimates obtained from sequential and simultaneous PKPD modeling approaches with SAEM
| Definition | Parameter | PK alone (PK estimation step of the two-stage approach) | Simultaneous PKPD | ||
|---|---|---|---|---|---|
| Population estimate | RSE (%) | Population estimate | RSE (%) | ||
| Total clearance | CL (l day−1) | 5500 | 6 | 5180 | 6 |
| Central volume | V2 (l) | 274 | 12 | 349 | 10 |
| Intercompartmental clearance | Q (l day−1) | 1140 | 8 | 1290 | 8 |
| Peripheral volume | V3 (l) | 1040 | 9 | 1130 | 9 |
| Absorption rate constant | ka (day−1) | 8.11 | 10 | 9.36 | 10 |
| Food effect on ka | Food on k a | −0.755a | 36 | −0.141c | 222 |
| Relative bioavailability | F1 | 1 FIX | – | 1 FIX | – |
| Food effect on F1 | Food on F1 | −0.244b | 45 | −0.476d | 14 |
| Absorption lag time | LagC (days) | 0.0211 | 10 | 0.0178 | 13 |
| IIV on CL | ω[CL] (%) | 46.6 | 9 | 46.9 | 9 |
| IIV on V2 | ω[V2] (%) | 60.6 | 16 | 55.5 | 15 |
| IIV on Q | ω[Q] (%) | 35.1 | 20 | 47.2 | 15 |
| IIV on V3 | ω[V3] (%) | 40.0 | 22 | 51.4 | 15 |
| IIV on ka | ω[ka] (%) | 64.1 | 11 | 69.9 | 11 |
| IIV on LagC | ω[LagC] (%) | 39.0 | 26 | 60.2 | 20 |
| Residual error | Additive error (%) | 45.4 | 2 | 44.6 | 2 |
a P value = 0.0049
b P value = 0.028
c P value = 0.65
d P value < 0.0001
The PD-VD model parameter estimates, along with the computed RMIC, obtained from the sequential and simultaneous PKPD approaches are presented in Table 3. The computed RMIC, the VD parameters and their associated IIV were comparable across the 3 different modelling approaches for the given drug effect (PD) model. For the sequential method with fixed individual EBE PK (omission of PK data in the PD-VD parameter estimation step), the computation time was approximately 25% shorter than the sequential method with fixed population PK parameters (PK data retained in the PD-VD parameter estimation step). Interestingly, no computation time was saved by fixing population PK parameters in the sequential method compared with the simultaneous PKPD-VD modelling approach. Run times ranged from 10 to 16 h including diagnostics using default settings for both. The visual predictive checks for the plasma maraviroc concentrations and viral loads obtained from the final PKPD-VD model using a simultaneous PKPD modelling approach are shown in Figs. 4 and 5, respectively. The goodness-of-fit plots are presented in Fig. 6.
Table 3.
Comparison of PD and VD parameter estimates obtained from sequential and simultaneous PKPD modeling approaches with SAEM
| Definition | Parameter | Sequential PKPD with fixed individual EBEs of PK parameters (omission of PK data in PD-VD estimation step) | Sequential PKPD with fixed population PK parameters (retention of PK data in PD-VD estimation step) | Simultaneous PKPD | |||
|---|---|---|---|---|---|---|---|
| Population estimate | RSE (%) | Population estimate | RSE (%) | Population estimate | RSE (%) | ||
| Basic reproductive ratio | RR0 | 5.33 | 10 | 5.92 | 11 | 4.96 | 9 |
| Birth rate of T cells | b [day−1] | 1.22 | 15 | 1.18 | 13 | 1.36 | 14 |
| Death rate of T cells | d 2 [day−1] | 0.797 | 4 | 0.755 | 3 | 0.841 | 3 |
| In vivo potency | IC50 (ng ml−1) | 8.27 | 19 | 6.73 | 22 | 8.57 | 24 |
| Lag time of viral inhibition | LagE (days) | 1.52 | 1 | 1.35 | 3 | 1.43 | 6 |
| IIV of RR0 | ω[RR0] (%) | 78.6 | 10 | 79.4 | 10 | 64 | 10 |
| IIV of b | ω[b] (%) | 114 | 9 | 103 | 9 | 110 | 9 |
| IIV of d 2 | ω[d 2] (%) | 29.3 | 11 | 20.6 | 12 | 19.6 | 13 |
| IIV of IC50 | ω[IC50] (%) | 137 | 11 | 160 | 10 | 175 | 10 |
| Residual error | Additive error (%) | 47.9 | 2 | 48.1 | 2 | 47.9 | 2 |
| Reproduction minimum inhibitory concentration | RMIC (ng ml−1)a | 35.8 | – | 33.1 | – | 33.9 | – |
| Log-likelihood | −2 × log-likelihoodb | 291c | 2367 | 2390 | |||
| Akaike information criterion | AICb | 311c | 2387 | 2440 | |||
| Bayesian information criterion | BICb | 332.5c | 2409 | 2493 | |||
aRMIC = (RR0 − 1) × IC50
bEstimation by importance sampling
cValues can not be directly compared with other approaches due to different number of observations
Fig. 4.

Visual predictive check for plasma maraviroc concentrations stratified by treatment arm. Symbols denote observations. Dashed and solid lines represent median and the 90% prediction intervals
Fig. 5.

Visual predictive check for log10 HIV RNA viral loads stratified by treatment arm. Symbols denote observations. Dashed and solid lines represent median and the 90% prediction intervals. Vertical lines indicate treatment period (day 1–10)
Fig. 6.

Basic goodness of fit for the final PKPD-VD model using a simultaneous PKPD modeling approach with SAEM. Top panel for maraviroc concentration; bottom panel for HIV RNA. PRED is the mean prediction over the population (Pop_mean) computed by a Monte-Carlo procedure. IPRED is the individual prediction based on the computation of the conditional modes of individual parameters (Pred_ind(Cond. Mode)). WRES is the population weighted residuals (WR_pop(mean)) computed by a Monte-Carlo procedure
Discussion
NONMEM is a widely used tool for population PKPD modelling. However, as shown in the current analysis, when complex semi-mechanistic models involving the use of a differential equation solver are necessary, the FOCEI method implemented in NONMEM (versions up to VI) often experiences convergence problems resulting from numerical difficulties in performing parameter estimates. In addition, the long computation time and the model instability limit the use of FOCEI for model building as with this example of a combined PKPD-VD model. As modelling and simulation activities move towards more complex mechanistic models it becomes necessary to investigate more practical tools, with better estimation methods such as SAEM. This method was not available in NONMEM versions VI or earlier. Bauer et al. [12] have published a review benchmarking commonly available population analysis tools. This review includes a summary of the statistical theory behind the methods and testing of 4 models including one example of a more complex PKPD model and differential equations requiring numerical integration. At that time MONOLIX 2.4 was not available. However they conclude that EM methods have an advantage of greater stability in population analyses of complex PKPD models with reduced bias in assessing sparse and rich data than FOCE [23].
The SAEM algorithm has previously been used by Lavielle and Mentré [24] to perform population PK analysis of a protease inhibitor, saquinavir, with large IIV. Samson et al. [25] have also successfully applied the SAEM algorithm to describe the longitudinal decrease in log10 viral load (left censored data) after initiation of antiretroviral treatments, with a right-truncated Gaussian distribution. In addition to the ability of performing parameter estimation, the relatively shorter computation times (compared with the FOCEI method) allows exploration of different components of the PD models as well as exploring different modelling approaches to assess the impact of PK data on PD parameter estimation, and vice versa.
The fitting feasibility of the FOCEI method (implemented in NONMEM VI) and the SAEM algorithm (in MONOLIX) were compared in the first part of this work using 50 simulated data sets. In this test FOCEI experienced numerical difficulties with 44% of the data sets. In addition a further 12% of runs finished with rounding errors and 22% had a failed covariance step. The SAEM algorithm on the other hand always produced estimates of PKPD-VD parameters using either a sequential or simultaneous modelling approach, although in a minority of cases these were not very likely. Because parameter estimates for fixed effects and variances as well as standard errors are generated in all runs with SAEM, the feasibility to perform PKPD-VD parameter estimation was determined by assessing the precision of parameter estimates as in relative standard error estimates. With FOCEI, more difficulties in estimating both ke0 and LagE (with relatively little information available from the study design) were experienced. Unlikely parameter estimates for IC50 and RR0 compared with the simulated model parameters were generated only occasionally (12%) using SAEM.
Using simulated data sets, the current analysis attempted to separate the system delay from the delay of pharmacological effect using a lag time and an effect compartment. Unfortunately, for the given maraviroc monotherapy viral load sampling design, it was difficult to separate the PD lag due to drug effects from a system delay. Herz et al. [26] developed a mathematical model which incorporated an intracellular phase of the viral life-cycle to account for the virus production lag. The authors of that study concluded that plasma viral load data alone do not allow a clear distinction between the delays of pharmacological action, intracellular delays and the clearance of free virus during the transition phase. It was also suggested that frequent clinical measurements for 2 days after initiation of antiretroviral therapies are required in order to improve parameter estimates. In addition, frequent clinical measurements after the termination of antiretroviral therapies also provide very valuable information. For the model parameterization used in the current analysis, the key to improve the precision of RR0 is the clinical measurement of viral load in the rebound period upon termination of antiretroviral therapies. Thus it can be concluded that data or study design limitations rather than the estimation method in SAEM (in MONOLIX) are an issue in parameter identification for lag times.
With the PKPD-VD model with lag time only, the LagE estimate was approximately 1.5 days. This is consistent with the observed delay in viral load drop after the initiation and termination of antiviral therapy and an approximate 2 day half-time of the free virus particles and/or virus producing cells [27–29].
For good PKPD modelling practice, one should always examine the correlation between the PK of a compound and the drug effects (surrogate biomarkers or clinical measurements), as well as their interactions, i.e. the impact of one on the other. The current analysis compared parameter estimates obtained from different modelling approaches using SAEM: sequential methods with fixed individual PK parameter EBEs; fixed population PK parameters with concentration data; a simultaneous method. In general, with the maraviroc monotherapy study design and data, the structural parameters of the PK, PD and VD model components were comparable across different modelling approaches. Zhang et al. [30] have demonstrated that PK fits using a simultaneous modelling approach can be quite sensitive to the PD model, particularly when there was misspecification in the PD model. Interestingly, Zhang et al. [31] also found that with the FOCE method in NONMEM, the sequential modelling approach saved about 40% of computation time compared with the simultaneous modelling approach. In the current analysis, with the SAEM algorithm, no computation time was saved by using the sequential PKPD modelling approach with fixed population PK parameters with retention of PK data, when compared with simultaneous PKPD modelling. However, approximately 25% of computation time was gained by using the sequential method with fixed individual PK parameter EBEs and omission of PK data in the PD-VD parameter estimation step. Computation time was not directly compared between FOCEI and SAEM because different systems (parallel GRID for NONMEM VI and desktop for MONOLIX) were used. Also much time was wasted with FOCEI runs that did not terminate successfully. Due to the high failure rate and the long computation times, it was not practical to perform parameter estimation with FOCEI using the same hardware system as was used for SAEM.
Though the objective of the current analysis was not to directly compare parameter estimates from SAEM with previous FOCEI analyses, the consistency of the parameter estimate for IC50 provided confidence in the use of SAEM for this PKPD-VD model.
In conclusion, the current analyses demonstrated that the SAEM algorithm in MONOLIX is useful for fitting complex mechanistic models requiring multiple differential equations. The SAEM algorithm allowed simultaneous estimation of PKPD and VD parameters, as well as investigation of different model sub-components during the model building process. This was not possible with the FOCEI method (implemented in NONMEM version VI and below).
Acknowledgments
INRIA (Institut National de Recherche en Informatique et Automatique) was paid by Pfizer Inc. for developing, implementing and testing the model in MONOLIX. B.W., and P.J. are paid consultants to Pfizer Global Research and Development, Sandwich, Kent, UK. The authors thank Professor Mats Karlsson of Uppsala University for his advice during the analysis; Professor Karlsson was a paid consultant to Pfizer Global Research and Development, Sandwich, Kent, UK. The authors also thank Dr Raymond Miller for reviewing the draft manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Wood A, Armour D. The discovery of the CCR5 receptor antagonist, UK-427,857, a new agent for the treatment of HIV infection and AIDS. Prog Med Chem. 2005;43:239–271. doi: 10.1016/S0079-6468(05)43007-6. [DOI] [PubMed] [Google Scholar]
- 2.Fätkenheuer G, et al. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat Med. 2005;11(11):1170–1172. doi: 10.1038/nm1319. [DOI] [PubMed] [Google Scholar]
- 3.Perelson AS, et al. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 1997;387(6629):188–191. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- 4.Bonhoeffer S, et al. Virus dynamics and drug therapy. Proc Natl Acad Sci USA. 1997;94(13):6971–6976. doi: 10.1073/pnas.94.13.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Funk GA, et al. Quantification of in vivo replicative capacity of HIV-1 in different compartments of infected cells. J Acquir Immune Defic Syndr. 2001;26(5):397–404. doi: 10.1097/00126334-200104150-00001. [DOI] [PubMed] [Google Scholar]
- 6.Rosario MC, et al. A pharmacokinetic-pharmacodynamic disease model to predict in vivo antiviral activity of maraviroc. Clin Pharmacol Ther. 2005;78(5):508–519. doi: 10.1016/j.clpt.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 7.Rosario MC, et al. A pharmacokinetic-pharmacodynamic model to optimize the phase IIa development program of maraviroc. J Acquir Immune Defic Syndr. 2006;42(2):183–191. doi: 10.1097/01.qai.0000220021.64115.37. [DOI] [PubMed] [Google Scholar]
- 8.Beal SL, Sheiner LB, Boeckmann AJ (1989–2006) NONMEM users guides, icon development solutions, Ellicott City, MD, USA
- 9.Beal SL, Sheiner LB. Estimating population kinetics. Crit Rev Biomed Eng. 1982;8(3):195–222. [PubMed] [Google Scholar]
- 10.Lindstrom M, Bates D. Nonlinear mixed-effects models for repeated measure data. Biometrics. 1990;46:673–687. doi: 10.2307/2532087. [DOI] [PubMed] [Google Scholar]
- 11.Pandhard X, Samson A. Extension of the SAEM algorithm for nonlinear mixed models with 2 levels of random effects. Biostatistics. 2009;10(1):121–135. doi: 10.1093/biostatistics/kxn020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bauer RJ, Guzy S, Ng C. A survey of population analysis methods and software for complex pharmacokinetic and pharmacodynamic models with examples. AAPS J. 2007;9(1):E60–E83. doi: 10.1208/aapsj0901007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.MONOLIX 2.4 User Guide, vol. http://software.monolix.org. Accessed Oct 2010
- 14.Kuhn E, Lavielle M. Maximum likelihood estimation in nonlinear mixed effects models. Comput Stat Data Anal. 2005;49:1020–1038. doi: 10.1016/j.csda.2004.07.002. [DOI] [Google Scholar]
- 15.Gruzdev B, et al. A randomized, double-blind, placebo-controlled trial of TMC125 as 7-day monotherapy in antiretroviral naive, HIV-1 infected subjects. AIDS. 2003;17(17):2487–2494. doi: 10.1097/00002030-200311210-00011. [DOI] [PubMed] [Google Scholar]
- 16.DeJesus E, et al. Antiviral activity, pharmacokinetics, and dose response of the HIV-1 integrase inhibitor GS-9137 (JTK-303) in treatment-naive and treatment-experienced patients. J Acquir Immune Defic Syndr. 2006;43(1):1–5. doi: 10.1097/01.qai.0000233308.82860.2f. [DOI] [PubMed] [Google Scholar]
- 17.Schurmann D, et al. Antiviral activity, pharmacokinetics and safety of vicriviroc, an oral CCR5 antagonist, during 14-day monotherapy in HIV-infected adults. AIDS. 2007;21(10):1293–1299. doi: 10.1097/QAD.0b013e3280f00f9f. [DOI] [PubMed] [Google Scholar]
- 18.Golden PL et al (2007) In vitro antiviral potency and preclinical pharmacokinetics of GSK364735 predict clinical efficacy in a phase 2a study. Poster H-1047, The 47th interscience conference on antimicrobial agents and chemotherapy (ICAAC). http://img.thebody.com/confs/icaac2007/pdfs/H-1047_306%20Golden%20ICAAC%202007.pdf. Accessed Oct 2010
- 19.Holford NH, Sheiner LB. Understanding the dose–effect relationship: clinical application of pharmacokinetic-pharmacodynamic models. Clin Pharmacokinet. 1981;6(6):429–453. doi: 10.2165/00003088-198106060-00002. [DOI] [PubMed] [Google Scholar]
- 20.Jacqmin P, McFadyen L, Wade JR. Basic PK/PD principles of drug effects in circular/proliferative systems for disease modelling. J Pharmacokinet Pharmacodyn. 2010;37(2):157–177. doi: 10.1007/s10928-010-9151-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sheiner LB, Beal SL. Some suggestions for measuring predictive performance. J Pharmacokinet Biopharm. 1981;9(4):503–512. doi: 10.1007/BF01060893. [DOI] [PubMed] [Google Scholar]
- 22.Lindbom L, Ribbing J, Jonsson EN. Perl-speaks-NONMEM (PsN)—a Perl module for NONMEM related programming. Comput Methods Programs Biomed. 2004;75(2):85–94. doi: 10.1016/j.cmpb.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 23.Girard P, Mentré F (16–17 June 2005) A comparison of estimation methods in nonlinear mixed effects models using a blind analysis. Abstract 834. http://www.page-meeting.org/default.asp?id=26&keuze=abstract-view&goto=abstracts&orderby=author&abstract_id=834. Accessed Oct 2010
- 24.Lavielle M, Mentré F. Estimation of population pharmacokinetic parameters of saquinavir in HIV patients with the MONOLIX software. J Pharmacokinet Pharmacodyn. 2007;34(2):229–249. doi: 10.1007/s10928-006-9043-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Samson A, Lavielle M, Mentré F. Extension of the SAEM algorithm to left-censored data in nonlinear mixed-effects model: application to HIV dynamics model. Comput Stat Data Anal. 2006;51:1562–1574. doi: 10.1016/j.csda.2006.05.007. [DOI] [Google Scholar]
- 26.Herz AVM, et al. Viral dynamics in vivo: limitations on estimates of intracellular delay and virus decay. Proc Natl Acad Sci USA. 1996;93(14):7247–7251. doi: 10.1073/pnas.93.14.7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei X, et al. Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 1995;373(6510):117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 28.Ho DD, et al. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373(6510):123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 29.Nowak MA, et al. HIV results in the frame: results confirmed. Nature. 1995;375(6528):193. doi: 10.1038/375193a0. [DOI] [PubMed] [Google Scholar]
- 30.Zhang L, Beal SL, Sheiner LB. Simultaneous vs. sequential analysis for population PK/PD data II: robustness of methods. J Pharmacokinet Pharmacodyn. 2003;30(6):405–416. doi: 10.1023/B:JOPA.0000012999.36063.4e. [DOI] [PubMed] [Google Scholar]
- 31.Zhang L, Beal SL, Sheiner LB. Simultaneous vs. sequential analysis for population PK/PD data I: best-case performance. J Pharmacokinet Pharmacodyn. 2003;30(6):387–404. doi: 10.1023/B:JOPA.0000012998.04442.1f. [DOI] [PubMed] [Google Scholar]