

Abstract
Ebola virus maturation occurs at the plasma membrane of infected cells and involves the clustering of the viral matrix protein VP40 at the assembly site as well as its interaction with the lipid bilayer. Here we report the X-ray crystal structure of VP40 from Ebola virus at 2.0 Å resolution. The crystal structure reveals that Ebola virus VP40 is topologically distinct from all other known viral matrix proteins, consisting of two domains with unique folds, connected by a flexible linker. The C-terminal domain, which is absolutely required for membrane binding, contains large hydrophobic patches that may be involved in the interaction with lipid bilayers. Likewise, a highly basic region is shared between the two domains. The crystal structure reveals how the molecule may be able to switch from a monomeric conformation to a hexameric form, as observed in vitro. Its implications for the assembly process are discussed.
Keywords: crystal structure/Ebola virus/matrix protein/membrane association/VP40
Introduction
Filoviridae are negative-stranded non-segmented viruses, which are classified into two species: Marburg virus and Ebola virus, the latter containing four subspecies (Ivory Coast, Reston, Sudan and Zaire). Ebola virus is the causative agent of a severe haemorrhagic fever in humans, which is associated with high mortality (Siegert et al., 1967; Feldmann and Klenk, 1996). Filoviruses enter cells such as monocytes, macrophages, hepatocytes and endothelial cells by employing their surface glycoprotein (Schnittler et al., 1993; Becker et al., 1995;Feldmann et al., 1996; Takada et al., 1997; Yang et al., 1998) in an infectious process most probably established through receptor-mediated endocytosis (Geisbert and Jahrling, 1995; Wool-Lewis and Bates, 1998). Subsequent fusion of viral and cellular membranes is mediated by the viral fusion protein (Gallaher, 1996; Volchkov, 1998; Weissenhorn et al., 1998). Replication of Ebola virus occurs in the cytoplasm (Feldmann and Kiley, 1998) and the assembly and budding of new viral particles takes place at the plasma membrane (Feldmann et al., 1996).
The matrix protein VP40 is the most abundant protein in viral particles, and is positioned beneath the viral bilayer to ensure the structural integrity of the particle (Geisbert and Jahrling, 1995). Matrix proteins have been shown to play an important role in virus assembly and budding. During this process, the matrix proteins associate with cellular membranes and are clustered at the site of assembly through interactions with the C-terminal tails of viral glycoproteins, an event that is thought to trigger assembly (reviewed by Garoff et al., 1998). Numerous matrix proteins, including Ebola virus VP40 itself, have been shown to interact preferentially with bilayers with elevated levels of negatively charged phospholipids in vitro (Zakowski et al., 1980; Ruigrok et al., 2000a,b). Notably, the viral envelope seems to possess a defined lipid composition that plays a crucial role during assembly (Lenard and Compans, 1974). Lipid rafts have been implicated in the acquisition of specific lipids into the viral envelope (Scheiffele et al., 1999; Manié et al., 2000), a process that may provide the special lipid environment required for matrix-protein membrane interactions.
The structures of a number of matrix proteins from members of the Retroviridae have been solved by NMR and X-ray crystallography. Although their sequences differ considerably, their three-dimensional structures are quite similar (Massiah et al., 1994; Matthews et al., 1994; Rao et al., 1995; Christensen et al., 1996; Hill et al., 1996; Matthews et al., 1996; Conte et al., 1997). Matrix proteins from SIV-1 and HIV-1 crystallize as trimers which display a large basic surface proposed to mediate binding to the lipid bilayer (Rao et al., 1995; Hill et al., 1996). A similar basic surface is present in the structures of retroviral matrix proteins from HTLV-II (Christensen et al., 1996), Mason-Pfizer monkey virus (Conte et al., 1997) and bovine leukaemia virus (Matthews et al., 1996). Furthermore, a basic surface was suggested to mediate membrane targeting of influenza virus matrix protein M1 (Sha and Luo, 1997). However, the structural and functional properties of matrix proteins of retroviruses and negative-stranded RNA viruses are likely to differ significantly. The former surrounds a spherical nucleocapsid that originates from a polyprotein and mediates budding at the basolateral side of polarized cells. The latter, on the other hand, are not processed by proteolytic cleavage, enclose a helical nucleocapsid and often trigger budding at apical cell surfaces.
We have shown previously that the C-terminal region comprising residues 213–326 of Ebola virus VP40 is absolutely required for membrane association in vitro. Notably, trypsinized VP40 lacking this region assembles into hexameric ring-like structures, as seen in biochemical studies and electron microscopy images, which may play a role during viral assembly and budding (Ruigrok et al., 2000b).
Here we report the three-dimensional structure of the matrix protein VP40 from Ebola virus, the first matrix protein structure from a negative-stranded non-segmented RNA virus. Both the N- and C-terminal domains fold into similar β-sandwich structures, suggesting that they might have been generated by gene duplication. Few interactions between the domains and the position of the trypsin cleavage site indicate the possibility of a conformational change or suggest a domain movement that might play a role in the function of VP40.
Results and discussion
Structure determination and architecture
A protease-resistant core of VP40 was identified by factor Xa and trypsin cleavage from full-length VP40, expressed in Escherichia coli (Dessen et al., 2000; Ruigrok et al., 2000b). The crystal structure of VP40 (residues 31–326) was determined by the multiwavelength anomalous diffraction (MAD) method (Hendrickson and Ogata, 1997) using selenomethionine-substituted VP40 (data and phasing statistics are presented in Table I and in Materials and methods). The experimental maps calculated after SHARP, WARP and SOLVE/DM procedures (see Materials and methods) were of excellent quality (Figure 1) and allowed unequivocal tracing of the chain. The refined model consists of residues 44–321 and has been refined to 2.0 Å resolution with a crystallographic R-value of 22.0% (free R-value of 25.0%).
Table I. Crystallographic statistics.
| Data collection |
|
Remote |
Peak |
Inflection |
|
| Wavelength (Å) | λ1 = 0.8855 | λ2 = 0.97888 | λ3 = 0.97907 | ||
| Max. resolution (Å) | 2.0 | 2.0 | 2.0 | ||
| Rmergea | 3.9 (22.4)b | 5.8 (19.8) | 3.6 (19.7) | ||
| % complete | 93.7 (86.8) | 97.9 (92.9) | 93.8 (86.1) | ||
| No. of reflections | 166 358 | 159 796 | 164 655 | ||
| No. of unique reflections | 19 062 | 19 188 | 19 185 | ||
| <I/σ(I)> | 10.6 (3.8) | 15.2 (5.0) | 10.7 (3.2) | ||
| f ′ (e-) | –3.02 | –6.20 | –10.20 | ||
| f ′′ (e-) | 4.40 | 7.41 | 2.82 | ||
| MAD phasing (SHARP) | |||||
| Phasing powerc | RCullisd | Figure of merite | |||
| (ano) | (iso) | (ano) | (iso) | (acentric) | (centric) |
| 2.14 | 1.81 | 0.63 | 0.51 | 0.77 | 0.59 |
| Refinement | |||||
| No. of reflections in working set | 17 007 | ||||
| Rcrystf | 0.220 (0.278)b | ||||
| Rfreef | 0.250 (0.337) | ||||
| Average B | 28.3 Å2 | ||||
| Residues | 260 (out of 296) | ||||
| Bond length r.m.s.d. | 0.006 Å | ||||
| Bond angles r.m.s.d. | 1.47° | ||||
| Ramachandran plotg % in most favoured and additionally allowed regions | 98.6 | ||||
| Water molecules | 121 |
aRmerge = ΣhΣi|Ii(h) – <I(h>)|/ΣhΣiIi(h), where Ii(h) is the ith measurement and <I(h)> is the weighted mean of all measurements of I(h).
bNumbers in parentheses are for the final shell.
cPhasing power = <|FH|>/E, where <|FH|> is the r.m.s. structure factor amplitude for the heavy atom and E is the estimated lack-of-closure error.
dRCullis = mean lack of closure divided by mean isomorphous difference.
eFigure of merit = <|ΣP(α)eiα/Σ|P(α)|>, where α is the phase and P(α) is the phase probability distribution.
fRcryst and Rfree = Σh||F(h)obs| – |F(h)calc||/Σh|F(h)obs| for reflections in the working and test sets, respectively.
gAs defined in PROCHECK (Laskowski et al., 1993).

Fig. 1. Stereo view of the experimental map generated with MAD phases obtained from six selenium sites identified by SOLVE and subsequently solvent flattened. The map is contoured at the 1σ level, and focuses on a conserved loop region in domain 2 connecting β-strands 7 and 8.
The VP40 monomer is an elongated, two-domain assembly, with dimensions of 40 × 50 × 25 Å (Figure 2A). The N-terminal domain is folded into a β-sandwich consisting of six antiparallel strands arranged in two β-sheets of three strands each (N-terminus: β1–β6; Figure 2B). Three loops lie arranged in the form of open jaws on the apex of the N-terminal domain that is farthest from the C-terminal region. The fold of the N-terminal domain is loosely reminiscent of classic membrane-targeting C2 domains identified in many signal transduction molecules (Rizo and Südhof, 1999), although the latter display one additional β-strand per sheet. The C-terminal domain (C-terminus: β7–β12; Figure 2) consists of one antiparallel triple-stranded β-sheet and an opposite set of three β-strands which form a strongly bent β-sheet. In both domains, the β-strand arrangements manifest a concave twist, and an α-helix packs laterally with respect to the β-sheets. The domains are connected by a flexible linker (residues 195–200) that is not traceable in the experimental maps, and make an approximate angle of 60° to one another. Although the longest loop in the C-terminal domain which connects strand β11 to β12 (residues 293–303) is traceable, two other loops (222–226 and 274–280) are not, suggesting higher flexibility in this region of the protein. In addition, no clear density was observed corresponding to residues 31–43 at the N-terminus as well as to residues 322–326 at the C-terminus, substantiating their lack of order.

Fig. 2. (A) Ribbon diagram of Ebola VP40. Two domains are shown in different colours. Secondary structure elements coloured yellow and dark blue lie on a plane below those coloured orange and light blue. Flexible loop regions, which were not traceable in the experimental maps, including the interdomain connecting loop (horizontal arrow) are drawn as dashed lines. The trypsin cleavage site, after Lys212, is highlighted by a vertical arrow. Trypsinization at this site causes complete disengagement of the C-terminal domain from the N-terminus, and subsequent hexamerization of the N-terminal domain. (B) Topology diagram of the two domains of Ebola virus VP40. β-strands are represented as arrows, while α-helices are rectangles. Short helical turns are represented as small squares. The domains display structural resemblance.
New topology
Using complete VP40 as well as the N- and C-terminal domains separately, a search for structurally homologous proteins (DALI; Holm and Sander, 1993) yielded no matches of statistical significance. However, superposition of the domains themselves revealed a similar architecture. Most differences reside on the lengths of individual β-strands, which are longer in the N-terminal domain, and the positions of the corresponding loops. Their similarity can also be observed on the VP40 topology diagram (Figure 2B). The spatial positioning and direction of the β-strands is similar, displaying main chain r.m.s. deviations of 1.6 Å upon fitting of all β-strands as well as α2 and α6, which connect the different levels of sheets in both domains (Figure 2B). A notable difference between the two domains is the strongly bent β-sheet arrangement of β-strands 7, 8 and 12. Nevertheless, it is conceivable that both domains arose from a common ancestor by gene duplication, although we cannot detect any significant sequence homology.
Sequence comparison
Sequence comparison of Ebola virus (strain Zaire) and Marburg virus (strain Popp) VP40 reveals a similarity of 29% (VP40 sequences from other Ebola virus subtypes currently are not available) (Bukreyev et al., 1993) (Figure 3). The conservation of the primary sequence is most prominent in the N-terminal region that corresponds to the secondary structure elements. Notably, there is an especially high sequence identity comprising the region around β-strand 4, α-helix 3 and β-strand 5. The C-terminal domain shows only a single continuous stretch of sequence conservation located in the region of β-strands 7 and 8 (Figure 3).

Fig. 3. Sequence alignment of VP40 from Ebola virus (strain Zaire, DDBJ/EMBL/GenBank accession No. AF086833) and from Marburg virus (strain Popp, DDBJ/EMBL/GenBank accession No. Z29337). Identical residues are shown in red letters and conservative substitutions in blue. The sequence present in the structure is shown schematically with the assignment of secondary structure elements (based on the structure of Ebola virus VP40). The black arrows at positions 31 and 326 indicate the first and last residues present in the construct used for crystallization. The yellow arrow at position 212 indicates the position of the trypsin cleavage site that induces hexamerization of VP40 in solution in vitro (Ruigrok et al., 2000b). The numbering is according to the Ebola virus VP40 sequence.
A number of matrix proteins have been proposed to interact with cellular proteins via WW domain-binding motifs (Garnier et al., 1996; Macias et al., 1996; Xiang et al., 1996; Harty et al., 1999; Yasuda et al., 1999), an instrumental event for successful assembly and budding. Both Ebola virus VP40 (10-PPEY-13) and Marburg virus VP40 (16-PPPY-19) contain potential WW-binding motifs at their N-termini (Harty et al., 1999; Figure 3). However, this part is not present in the structure due to the sensitivity to protease digestion (Dessen et al., 2000; Ruigrok et al., 2000b); its flexibility and potential availability for interaction is underlined further by the lack of traceable electron density for residues 31–43.
Filoviruses have been grouped in the order Mononegavirales together with the families of Bornaviridae, Paramyxoviridae and Rhabdoviridae, based on their non-segmented negative-stranded RNA genome and their conserved genome organization. Sequence comparisons of VP40 with matrix proteins from members of the other families did not reveal any significant homology. In fact, only members of the Paramyxoviridae express a matrix protein with a size comparable to that of Ebola virus VP40, while matrix proteins from Rhabdoviridae and Bornaviridae are generally ∼100 amino acids shorter than VP40, indicating major structural differences.
Similarity to other known viral matrix protein structures
The structure of Ebola VP40 displays no resemblance to structures of retroviral matrix proteins (reviewed by Conte and Matthews, 1998) which can be attributed to major differences in the assembly/maturation processes of these viruses (Garoff et al., 1998). The HIV-1 matrix protein consists of five α-helices, two short 310 helical stretches and a three-stranded mixed β-sheet which form a single compact globular domain (Massiah et al., 1994; Rao et al., 1995; Hill et al., 1996). The other known viral matrix protein structure from influenza virus also shows no structural homology to Ebola virus VP40. An N-terminal protease-resistant domain of influenza virus M1 protein folds into a compact α-helical structure (Sha and Luo, 1997). Proteolysis of influenza M1 indirectly suggests that this matrix protein may be also composed of two domains, like Ebola virus VP40.
The interdomain connecting region
The VP40 domains are not tightly packed against each other, which becomes evident from the lack of order observed in the interconnecting loop (residues 195–200, Figure 4A, yellow dotted lines) as well as the absence of major interdomain side chain interactions (Figure 4A). Such contributions are made uniquely by Gln184, which hydrogen bonds to Asp253 and Lys256, Gln155 which hydrogen bonds to Ser319, and Ser316 which hydrogen bonds to the backbone carbonyl of Pro97 (Figure 4A).
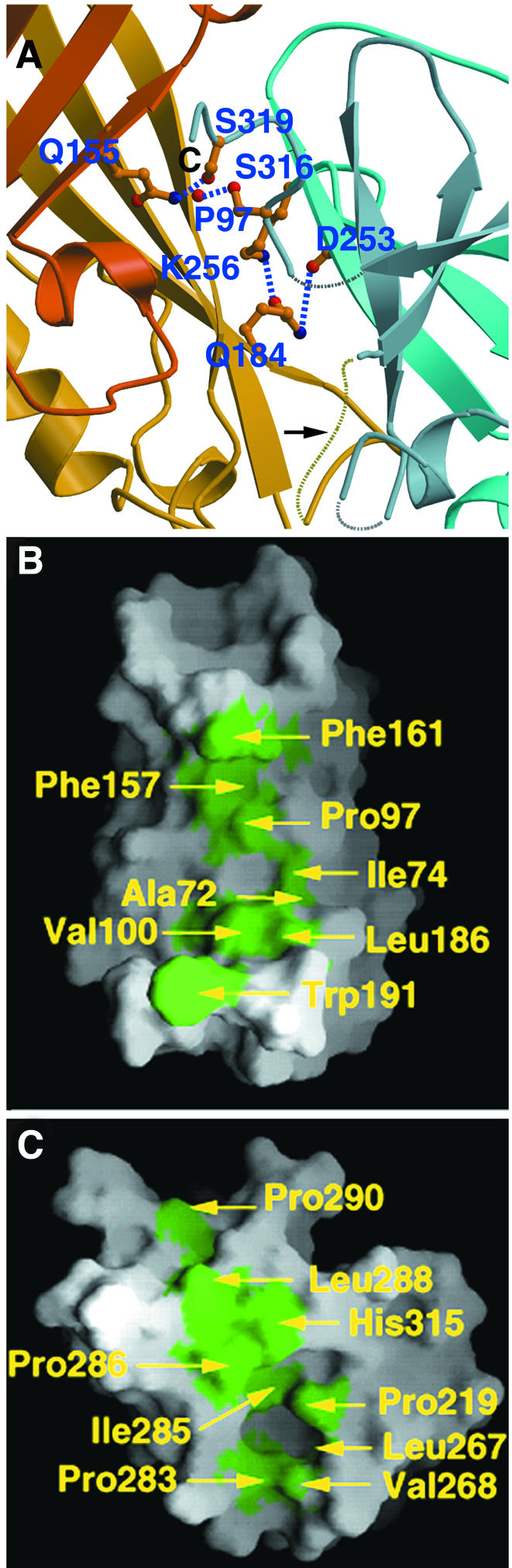
Fig. 4. Interdomain interactions. (A) Close up of polar interactions between the N- and C-terminal domains. Residues involved in salt bridges and hydrogen bonds are shown. For clarity, the connection between residue 307 and 310 is shown as a grey dashed line. The loop connecting both domains is indicated with an arrow. (B) Surface representation of the N-terminal domain (residues 44–194) and (C) of the C-terminal domain (residues 201–321). Hydrophobic residues lining the interface on the N- and C-terminal domains are shown in green.
In addition, the interdomain region contains a zipper of hydrophobic residues (Figure 4B and C). Most of the residues corresponding to this hydrophobic stretch are conserved between Ebola virus and Marburg virus VP40 sequences (Figure 3). The most prominent feature is Trp191 of the N-terminal domain, which lies in the traceable region that precedes the flexible interconnecting loop, and fits into a hydrophobic cleft composed of conserved C-terminal residues Pro219, Leu267, Val268, Pro283 and Ile285 (Figure 4B and C).
Trypsin proteolysis of full-length VP40 results in cleavage after Lys212 (Ruigrok et al., 2000b), located between β-strands 7 and 8 in the C-terminal domain (Figure 2A). This leads to the spontaneous dissociation of the C-terminal domain in solution, underlining their weak interaction. It is of note that the collective interactions provided by the aforementioned hydrogen bonds, as well as the stabilization added by the hydrophobic zipper including the insertion of Trp191 into a C-terminal hydrophobic cleft (Figure 4), are insufficient to maintain the domain association after Lys212 cleavage. The removal of this C-terminal part induces the hexamerization of VP40 in vitro (Ruigrok et al., 2000b), indicating that the C-terminal domain may sterically prevent the formation of hexamers through its interactions with the N-terminal domain. This suggests that the conformation observed in the crystal structure may be metastable, and the removal of the C-terminus is sufficient to induce a change in quaternary structure. The biochemical data combined with the observation of the loose interdomain packing in the X-ray structure strongly indicate that the C-terminal domain may be able to move with respect to the N-terminal domain and thus facilitate such a change in its oligomeric state. A similar flexibility has been speculated to be important for the matrix protein of influenza virus (Sha and Luo, 1997) as well as for vesicular stomatitis virus (Gaudin et al., 1997), mostly to accommodate the different functions attributed to the matrix protein during the viral life cycle. However, we cannot exclude the possibility that the hexamerization of VP40 may be induced by conformational change(s) other than the removal or movement of the C-terminal domain.
Membrane-targeting surfaces
We have shown previously that VP40 interacts preferentially with membranes containing negatively charged phospholipids (Ruigrok et al., 2000b), a feature observed and suggested for a number of viral matrix proteins (Zakowski et al., 1980; Rao et al., 1995; Hill et al., 1996; Provitera et al., 2000; Ruigrok et al., 2000a). The C-terminal truncation of VP40 (trypsin cleavage after Lys212, Figures 2A and 4) abolishes membrane interaction (Ruigrok et al., 2000b), indicating a crucial role for the C-terminal domain of VP40 in lipid bilayer targeting. In addition, membrane association of VP40 in vitro was shown to involve electrostatic interactions (Ruigrok et al., 2000b).
The C-terminal domain exhibits large solvent-exposed hydrophobic surface regions (Figure 5A and B) which also involve residues in β-strands 7 and 8, the most conserved linear sequence between Ebola and Marburg virus VP40 within the C-terminal domain (Figure 3). However, the location of basic residues possibly involved in membrane targeting is not as clear. The only significant basic patch is observed at the interface of the two domains, involving residues from β-strands 4 and 5 and α-helix 3 of the N-terminal domain as well as β-strands 7 and 8 from the C-terminal domain (Figure 5C and D). This orientation of the molecule also shows a hydrophobic bulk (Pro321) exposed to the solvent and surrounded by basic residues Lys127, Arg134, Arg148, Lys212 and Arg214. The combined role of basic and hydrophobic residues in membrane interactions has been discussed for a number of proteins with varied interface-related functionalities (Macedo-Ribeiro et al., 1999; Pratt et al., 1999; Dessen, 2000; Provitera et al., 2000). However, further studies are necessary to locate the exact membrane-targeting sequence.

Fig. 5. Distribution of hydrophobic and electrostatic surfaces. (A and B) Surface representation of VP40 in two orientations, rotated by 90° with respect to each other. The N-terminal domain is shown in white and the C-terminal domain in yellow. Hydrophobic residues are depicted in green. (C and D) Charge distribution of Ebola virus VP40 (two orientations, rotated by 90°). Surface potential representation of VP40 with regions where the electrostatic potential is less than –10 kBT are red, while those more than +10 kBT are blue (kB, Boltzmann constant; T, absolute temperature). A basic patch is clearly visible in the central region of the molecule and encompasses residues from both domains.
Implications for assembly and budding
Viral matrix proteins are thought to play an important role in viral assembly and budding (reviewed by Garoff et al., 1998). Marburg virus and probably Ebola virus assembly take place at the plasma membrane (Feldmann et al., 1996) where an interaction with the cytoplasmic tail of the glycoprotein may occur in a fashion comparable to those shown for a number of other viral glycoproteins and their matrix proteins (Sanderson et al., 1994; Metsikkö and Simons, 1986; Cosson, 1997; Jin et al., 1997; Schnell et al., 1998; Mebatsion et al., 1999; Schmitt et al., 1999). This process may cluster the matrix proteins and the glycoproteins at the initial starting site of assembly.
Biochemical data and electron microscopy studies show that the removal of most of the C-terminal domain of VP40 from Ebola virus is sufficient to induce hexamer formation (Ruigrok et al., 2000b). Based on the X-ray crystal structure of VP40, we conclude that the N-terminal domain of VP40 is most likely to be responsible for the hexamerization observed for trypsinized VP40 in solution, while the C-terminal domain probably harbours most of the membrane-targeting sequence. The formation of a hexameric matrix protein lattice has been speculated to play a role in the assembly of HIV and SIV. Low-resolution electron microscopy studies (Nermut et al., 1993) as well as the observation of a crystalline trimeric HIV matrix protein (Hill et al., 1996) and the arrangement of trimeric SIV matrix protein in a hexameric crystal lattice (Rao et al., 1995) indicated a role for hexamerization in retrovirus assembly. However, the icosahedral symmetry of retroviral particles suggested by these data was not confirmed by others (Fuller et al., 1997; Yeager et al., 1998). In contrast, biochemical data on VP40 (Ruigrok et al., 2000b) clearly indicate the possibility of hexamerization of a truncated VP40 matrix protein from Ebola virus in vitro, and the crystal structure of VP40 suggests how such a transition from monomer to hexamer could possibly be achieved in vivo. Although we cannot exclude that hexamerization of VP40 may occur at a step other than virus assembly during the life cycle of Ebola virus, it is tempting to speculate that hexamerization may play a role in assembly, and we therefore propose the following model for VP40-induced assembly. (i) VP40 contacts the lipid bilayer through a combination of hydrophobic and electrostatic interactions, mostly contributed by its C-terminal domain. (ii) VP40 undergoes a conformational change(s) in a process that most probably includes a movement of the C-terminal domain with respect to the N-terminal unit, which is achievable due to the lack of major interdomain contacts. The N-terminal domains then hexamerize. (iii) The hexameric building blocks may form a lattice underlying the plasma membrane in a macro-oligomerization process induced by contacts between bridging C-terminal domains as well as interactions with the cytoplasmic tails of the glycoprotein. This chain of events may initiate assembly by further involving the participation of the ribonucleoprotein particle in a process that will ultimately lead to budding of viral particles.
Materials and methods
Data collection and processing
Expression, purification and crystallization of VP40(31–326) and selenomethionine-derivatized VP40(31–326) (Ebola virus strain Zaire) were reported elsewhere (Dessen et al., 2000). At the stage of model building, we detected a His269Leu mutation through a new round of DNA sequencing. His269 is solvent-exposed on α-helix 7 and does not affect membrane association. Crystals were grown with unit cell dimensions a = 64.64 Å, b = 91.09 Å, c = 49.23 Å, β = 97.3° and are in space group C2, with one molecule per asymmetric unit. MAD data were collected to 2.0 Å at the beamline BM14 of the European Synchrotron Radiation Facility (ESRF, Grenoble, France), using a MAR CCD detector. MAD data, at three wavelengths, were measured from a single flash-cooled selenomethionyl crystal. The wavelengths, near the K-shell edge of selenium, were chosen from an X-ray fluorescence spectrum taken from the crystal. A total of 160° were collected at each wavelength to optimize completeness and Bijvoet pair accumulation. Integration, scaling and merging of data were performed with the programs Denzo and Scalepack (Otwinowski and Minor, 1997).
Phase determination, model building, refinement and analysis
Six selenium sites were identified with the program SOLVE (Terwillinger et al., 1987) and further refinement was carried out with the program SHARP (de la Fortelle and Bricogne, 1997). Density modification was performed by SOLOMON (CCP4, 1994), as implemented in SHARP, considering a solvent content of 32%. The final phases were employed by WARP (Perrakis et al., 1999) in the tracing of a polyalanine chain with 210 residues (out of a total of 296). Two additional experimental maps, including one produced by WARP and a third one generated after density modification of phases calculated from the SOLVE procedure with DM (Cowtan and Main, 1996), were employed in order to resolve ambiguity in positioning of four solvent-exposed loop regions in the C-terminal domain. Sequence assignment and further model building, which included the identification of 50 more residues, were performed manually with the program O (Jones and Kjeldgaard, 1997). The selenium sites provided markers for the methionine side chains and facilitated the assignment of the amino acid sequence. Atomic refinement of Ebola VP40 was performed with CNS (Brünger et al., 1998) employing all measured reflections of the data set (Table I) using positional and B-factor refinement interspersed with rounds of manual rebuilding. Statistics for data collection, phasing and refinement are included in Table I. The final structure includes residues 44–194, 201–221, 227–273 and 281–321, and 121 water molecules, and has an R-factor of 22.0% for all reflections. An Rfree of 25.0% was obtained from a subset of 10% of the data which were excluded from the refinement. The model has good stereochemistry and structural quality, with an average bond length and bond angle deviation of 0.006 Å and 1.47°, respectively. A total of 98.6% of all residues are in most favourable and additionally allowed regions of a Ramachandran plot. The structure factors of the λ1 data set (Remote) and the atomic coordinates have been deposited in the RCSB Protein Data Bank under accession No. 1ES6.
Superposition of the N- and C-terminal domains
The fitted regions comprise ∼33% of all residues. β1 residues 69–80 were overlaid with β7 residues 203–209, β2 96–98 with β8 216–218, α2 108–117 with α6 236–246, β3 120–126 with β9 247–253, β4 131–138 with β10 257–264, β5 154–158 with β11 284–288, and β6 residues 175–183 were superimposed with β12 residues 301–309.
Preparation of figures
Figures were generated with the programs O (Figure 1; Jones and Kjeldgaard, 1997), Molscript (Figures 2A and 4A; Kraulis, 1991) and Grasp (Figures 4B and C and 5; Nicholls et al., 1991).
Acknowledgments
Acknowledgements
We thank Dr G.Leonard (ESRF, Grenoble) and members of the ESRF/EMBL Joint Structural Biology Group for support at the ESRF beamlines, and S.Scianimanico for excellent technical assistance. This work was supported in part by Deutsche Forschungsgemeinschaft SFB 286.
References
- Becker S., Spiess,M. and Klenk,H.D. (1995) The asialoglycoprotein receptor is a potential liver-specific receptor for Marburg virus. J. Gen. Virol., 76, 393–399. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallographic and NMR system (CNS): a new software system for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Bukreyev A.A., Volchkov,V.E., Blinov,V.M. and Netesov,S.V. (1993) The Vp35 and VP40 proteins of filoviruses. Homology between Marburg and Ebola viruses. FEBS Lett., 322, 41–46. [DOI] [PubMed] [Google Scholar]
- Christensen A.M., Massiah,M.A., Turner,B.G., Sundquist,W.I. and Summers,M.F. (1996) Three-dimensional structure of the HTLV-II matrix protein and comparative analysis of matrix proteins from different classes of pathogenic human retroviruses. J. Mol. Biol., 264, 1117–1131. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–776. [DOI] [PubMed] [Google Scholar]
- Conte M.R. and Matthews,S. (1998) Retroviral matrix proteins: a structural perspective. Virology, 246, 191–198. [DOI] [PubMed] [Google Scholar]
- Conte M.R., Klikova,M., Hunter,E., Ruml,T. and Matthews,S. (1997) The three-dimensional solution structure of the matrix protein from the type D retrovirus, the Mason-Pfizer monkey virus, and implications for the morphology of retroviral assembly. EMBO J., 16, 5819–5826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosson P. (1997) Direct interaction between the envelope and the matrix proteins of HIV. EMBO J., 15, 5783–5788. [PMC free article] [PubMed] [Google Scholar]
- Cowtan K.D. and Main,P. (1996) Phase combination and cross validation in iterated density modification calculations. Acta Crystallogr. D, 42, 43–48. [DOI] [PubMed] [Google Scholar]
- Dessen A. (2000) Phospholipase A2 enzymes: structural diversity in lipid messenger metabolism. Structure, 8, 15–22. [DOI] [PubMed] [Google Scholar]
- Dessen A., Forest,E., Volchkov,V., Dolnik,O., Klenk,H.-D. and Weissenhorn,W. (2000) Crystallization and preliminary X-ray analysis of the matrix protein from Ebola virus. Acta Crystallogr. D, 56, 758–760. [DOI] [PubMed] [Google Scholar]
- Feldmann H. and Kiley,M.P. (1998) Classification, structure, and replication of filoviruses. Curr. Top. Microbiol. Immunol., 235, 1–21. [DOI] [PubMed] [Google Scholar]
- Feldmann H. and Klenk,H.-D. (1996) Marburg and Ebola viruses. Adv. Virus Res., 47, 1–52. [DOI] [PubMed] [Google Scholar]
- Feldmann H., Bugany,H., Mahner,F., Klenk,H.-D., Drenckhahn,D. and Schnittler,H.J. (1996) Filovirus induced endothelial leakage triggered by infected monocytes/macrophages. J. Virol., 70, 2208–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fortelle E. and Bricogne,G. (1997) Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol., 276, 472–494. [DOI] [PubMed] [Google Scholar]
- Fuller S.D., Wilk,T., Gowen,B.E., Krausslich,H.-G. and Vogt,V.M. (1997) Cryo-electron microscopy reveals ordered domains in the immature HIV-1 particle. Curr. Biol., 7, 729–738. [DOI] [PubMed] [Google Scholar]
- Gallaher W.R. (1996) Similar structural models of the transmembrane proteins of Ebola and avian sarcoma viruses. Cell, 85, 477–478. [DOI] [PubMed] [Google Scholar]
- Garnier L., Wills,J.W., Verderame,M.F. and Sudol,M. (1996) WW domains and retrovirus budding. Nature, 381, 744–745. [DOI] [PubMed] [Google Scholar]
- Garoff H., Hewson,R. and Opstelten,D.-J.E. (1998) Virus maturation by budding. Microbiol. Mol. Biol. Rev., 62, 1171–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudin Y., Sturgis,L., Doumith,M., Barge,A., Robert,B. and Ruigrok,R.W.H. (1997) Conformational flexibility and polymerization of vesicular stomatitis virus matrix protein. J. Mol. Biol., 274, 816–825. [DOI] [PubMed] [Google Scholar]
- Geisbert T.W. and Jahrling,P.B. (1995) Differentiation of filoviruses by electron microscopy. Virus Res., 39, 129–150. [DOI] [PubMed] [Google Scholar]
- Harty R.N., Paragas,J., Sudol,M. and Palese,P. (1999) A proline-rich motif within the matrix protein of vesicular stomatitis virus and rabies virus interacts with WW domains of cellular proteins: implications for viral budding. J. Virol., 73, 2921–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson W.A., and Ogata,C.M. (1997) Phase determination from multiwavelength anomalous diffraction measurements. Methods Enzymol., 276, 494–523. [DOI] [PubMed] [Google Scholar]
- Hill C.P., Worthylake,D., Bancroft,D.P., Christensen,A.M. and Sundquist,W.I. (1996) Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc. Natl Acad. Sci. USA, 93, 3099–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L. and Sander,C. (1993) Protein structure comparison by alignment of distance matrices. J. Mol. Biol., 233, 123–138.. [DOI] [PubMed] [Google Scholar]
- Jin H., Leser,G.P., Zhang,J. and Lamb,R.A. (1997) Influenza virus hemagglutinin and neuraminidase cytoplasmic tails control particle shape. EMBO J., 16, 1236–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T.A. and Kjeldgaard,M. (1997) Electron-density map interpretation. Methods Enzymol., 277, 173–208. [DOI] [PubMed] [Google Scholar]
- Kraulis P. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 924–950. [Google Scholar]
- Laskowski R.A., MacArthur,M.W., Moss,D.S. and Thornton,J.M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr., 26, 283–290. [Google Scholar]
- Lenard J. and Compans,R.W. (1974) The membrane structure of lipid-containing viruses. Biochim. Biophys. Acta, 344, 51–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macedo-Ribeiro S. et al. (1999) Crystal structures of the membrane binding C2 domain of human coagulation factor V. Nature, 420, 434–439. [DOI] [PubMed] [Google Scholar]
- Macias M.J., Hyvonem,M., Baraldi,E., Schultz,J., Sudol,M., Saraste,M. and Oschkinat,H. (1996) Structure of the WW domain of a kinase-associated protein complexed with a proline-rich peptide. Nature, 382, 646–649. [DOI] [PubMed] [Google Scholar]
- Manié S.N., Debreyne,S., Vincent,S. and Gerlier,D. (2000) Measles virus structural components are enriched into lipid raft microdomains: a potential cellular location for virus assembly. J. Virol., 74, 305–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massiah M., Starich,M.R., Paschall,C.M., Summers,M.F., Christensen,A.M. and Sundquist,W.I. (1994) Three-dimensional structure of the human immunodeficiency virus type 1 matrix protein. J. Mol. Biol., 244, 198–223. [DOI] [PubMed] [Google Scholar]
- Matthews S. et al. (1994) Structural similarity between the p17 matrix protein of HIV-1 and interferon-γ. Nature, 370, 666–668. [DOI] [PubMed] [Google Scholar]
- Matthews S., Mikhailov,M., Burny,A. and Roy,P. (1996) The solution structure of the bovine leukaemia virus matrix protein and similarity with lentiviral matrix proteins. EMBO J., 15, 3267–3274. [PMC free article] [PubMed] [Google Scholar]
- Mebatsion T., Weiland,F. and Conzelmann,K.K. (1999) Matrix protein of rabies virus is responsible for the assembly and budding of bullet-shaped particles and interacts with the transmembrane spike glycoprotein G. J. Virol., 73, 242–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metsikkö K. and Simons,K. (1986) The budding mechanism of spikeless vesicular stomatitis virus particles. EMBO J., 5, 1913–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nermut M.V., Grief,C., Hashmi,S. and Hockley,D.J. (1993) Further evidence of icosahedral symmetry in human and simian immunodeficiency virus. AIDS Res. Hum. Retroviruses, 9, 929–938. [DOI] [PubMed] [Google Scholar]
- Nicholls A., Sharp,K.A. and Honig,B. (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins: Struct. Funct. Genet., 11, 281–296. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray data collected in oscillation mode. Methods Enzymol., 276, 307–325. [DOI] [PubMed] [Google Scholar]
- Perrakis A., Morris,R. and Lamzin,V.S. (1999) Automated protein model building combined with iterative structure refinement. Nature Struct. Biol., 6, 458–463. [DOI] [PubMed] [Google Scholar]
- Pratt K.P., Shen,B.W., Takeshima,K., Davie, E.W., Fujikawa,K. and Stoddard,B.L. (1999) Structure of the C2 domain of human factor VIII at 1.5 Å resolution. Nature, 402, 439–442. [DOI] [PubMed] [Google Scholar]
- Provitera P., Bouamr,F. Murray,D., Carter,C. and Scarlata,S. (2000) Binding of equine infectious anemia virus matrix protein to membrane bilayers involves multiple interactions. J. Mol. Biol., 296, 887–898. [DOI] [PubMed] [Google Scholar]
- Rao Z., Belyaev,A.S., Fry,E., Roy,P., Jones,I.A. and Stuart,D.I. (1995) Crystal structure of SIV matrix antigen and implications for viral assembly. Nature, 378, 743–747. [DOI] [PubMed] [Google Scholar]
- Rizo J. and Südhof,T.C. (1999) C2-domains, structure and function of a universal Ca2+-binding domain. J. Biol. Chem., 273, 15879–15882. [DOI] [PubMed] [Google Scholar]
- Ruigrok R.W.H., Barge,A., Durrer,P., Brunner,J., Ma,K. and Whittaker,G.R. (2000a) Membrane interaction of influenza virus M1 protein. Virology, 267, 289–298. [DOI] [PubMed] [Google Scholar]
- Ruigrok R.W.H., Schon.G., Dessen,A., Forest,E., Volchkov,V., Dolnik,O., Klenk,H.-D. and Weissenhorn,W. (2000b) Structural characterization and membrane binding properties of the matrix protein VP40 of Ebola. J. Mol. Biol., 300, 103–112. [DOI] [PubMed] [Google Scholar]
- Sanderson C.M., Wu,H.H. and Nayak,D.P. (1994) Sendai virus M protein binds independently to either the F or the HN glycoprotein in vivo. J. Virol., 68, 69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffele P., Rietveld,A., Wilk,T. and Simons,K. (1999) Influenza virus select ordered lipid domains during budding from the plasma membrane. J. Biol. Chem., 274, 2038–2044. [DOI] [PubMed] [Google Scholar]
- Schmitt A.P., He,B. and Lamb,R.A. (1999) Involvement of the cytoplasmic domain of the hemagglutinin-neuraminidase protein in assembly of the paramyxovirus simian virus 5. J. Virol., 73, 8703–8712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell M.J., Buonocore,L, Boritz,E., Ghosh,H.P., Chernish,R. and Rose,J.K. (1998) Requirement for a non-specific glycoprotein cytoplasmic domain sequence to drive efficient budding of vesicular stomatitis virus. EMBO J., 10, 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnittler H.J., Mahner,F., Drenckhahn,D., Klenk,H.-D. and Feldmann,H. (1993) Replication of Marburg virus in human endothelial cells. A possible mechanism for the development of viral hemorrhagic disease. J. Clin. Invest., 91, 1301–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha B. and Luo,M. (1997) Structure of a bifunctional membrane–RNA binding protein, influenza virus matrix protein M1. Nature Struct. Biol., 4, 239–244. [DOI] [PubMed] [Google Scholar]
- Siegert R., Shu,H.-L., Slenczka,W., Peters,D. and Müller,G. (1967) Zur Ätiologie einer unbekannten von Affen ausgegangenen Infektionskrankheit. Dtsch. Med. Wochenschr., 92, 2341–2343. [DOI] [PubMed] [Google Scholar]
- Takada A., Robison,C., Goto,H., Sanchez,A., Murti,K.G., Whitt,M.A. and Kawaoka,Y. (1997) A system for functional analysis of Ebola virus glycoprotein. Proc. Natl Acad. Sci. USA, 94, 14764–14769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwillinger T.C., Kim,S.-H. and Eisenberg,D. (1987) Generalized method of determining heavy atom positions using the difference Patterson function. Acta Crystallogr. A, 43, 1–5. [Google Scholar]
- Volchkov V. (1998) Processing of the Ebola virus glycoprotein. Curr. Top. Microbiol. Immunol., 235, 35–47. [DOI] [PubMed] [Google Scholar]
- Weissenhorn W., Carfi,A., Lee,K.-H., Skehel,J.J. and Wiley,D.C. (1998) Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein subunit ectodomain. Mol. Cell, 2, 605–616. [DOI] [PubMed] [Google Scholar]
- Wool-Lewis R.J. and Bates,P. (1998) Characterization of Ebola virus entry by using pseudotyped viruses: identification of receptor-deficient cell lines. J. Virol., 72, 3155–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y., Cameron,C.E., Wills,J.W. and Leis,J. (1996) Fine mapping and characterization of the Rous sarcoma virus Pr76gag late assembly domain. J. Virol., 70, 5695–5700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Delgado,R., Xu,L., Todd,R.F., Nabel,E.G., Sanchez,A. and Nabel,G.J. (1998) Distinct cellular interactions of secreted and transmembrane Ebola virus glycoproteins. Science, 279, 1034–1037. [DOI] [PubMed] [Google Scholar]
- Yasuda J. and Hunter,E. (1999) A proline-rich motif (PPPY) in the Gag polyprotein of Mason-Pfizer monkey virus plays a maturation-independent role in virion release. J. Virol., 72, 4095–4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeager M., Wilson-Kubalek,E.M., Weiner,S.G., Brown,P.O. and Rein,A. (1998) Supramolecular organization of immature and mature murine leukemia virus revealed by electron cryo-microscopy: implications for retroviral assembly mechanisms. Proc. Natl Acad. Sci. USA, 95, 7299–7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakowski J.J., Petri,W.A. and Wagner,R.R. (1980) Role of matrix protein in assembling the membrane of vesicular stomatitis virus: reconstitution of matrix protein with negatively charged phospholipid vesicles. Biochemistry, 20, 3902–3907. [DOI] [PubMed] [Google Scholar]